

Abstract
The primary aim of this investigation was to determine whether binding sites corresponding to the 5-HT7 receptor could be detected in smooth muscle of the rat jejunum. Binding studies in rat brain (whole brain minus cerebellum) and guinea-pig ileal longitudinal muscle were also undertaken in order to compare the binding characteristics of these tissues. Studies were performed using [3H]-mesulergine, as it has a high affinity for 5-HT7 receptors.
In the rat brain and guinea-pig ileum, pKD values for [3H]-mesulergine of 8.0±0.04 and 7.9±0.11 (n=3) and Bmax values of 9.9±0.3 and 21.5±4.9 fmol mg−1 protein were obtained respectively, but no binding was detected in the rat jejunum. [3H]-mesulergine binding in the rat brain and guinea-pig ileum was displaced with the agonists 5-carboxamidotryptamine (5-CT)>5-hydroxytryptamine (5-HT)⩾5-methoxytryptamine (5-MeOT)>sumatriptan and the antagonists risperidone⩾LSD⩾metergoline>ritanserin>>pindolol.
Despite the lack of [3H]-mesulergine binding in the rat jejunum, functional studies undertaken revealed a biphasic contractile response to 5-HT which was partly blocked by ondansetron (1 μM). The residual response was present in over 50% of tissues studied and was found to be inhibited by risperidone>LSD>metergoline>mesulergine=ritanserin>pindolol, but was unaffected by RS 102221 (3 μM), cinanserin (30 nM), yohimbine (0.1 μM) and GR 113808 (1 μM). In addition, the agonist order of potency was 5-CT>5-HT>5-MeOT>sumatriptan.
In conclusion, binding studies performed with [3H]-mesulergine were able to detect 5-HT7 sites in rat brain and guinea-pig ileum, but not in rat jejunum, where a functional 5-HT7-like receptor was present.
Keywords: 5-HT7 receptor, [3H]-mesulergine, rat jejunum, guinea-pig ileum, rat brain
Introduction
Recent advances in receptor classification using molecular cloning techniques have resulted in the identification of additional 5-HT receptor subtypes such as 5-ht5, 5-ht6 and 5-HT7 to the four major subtypes already present. The 5-HT7 receptor has been cloned from rat (Lovenberg et al., 1993; Ruat et al., 1993), mouse (Plassat et al., 1993), human (Bard et al., 1993) and guinea-pig (Tsou et al., 1994). Whilst evidence for the endogenous expression and function of the gene products which encode the 5-ht5 and 5-ht6 receptors is yet to be obtained, the 5-HT7 receptor is now recognized as the receptor previously classified as being 5-HT1-like (Tsou et al., 1994; Eglen et al., 1997).
The 5-HT7 receptor is positively coupled to adenylate cyclase and although there are no selective 5-HT7 receptor ligands available at present, the identification of 5-HT7 sites has been established using the operational criteria of rank order of agonist and antagonist potency at recombinant 5-HT7 receptors (Eglen et al., 1994; Sleight et al., 1995b). The expressed 5-HT7 receptor has a high affinity (pKi 8.0–10.0) for 5-carboxamidotryptamine (5-CT), 5-hydroxytryptamine (5-HT), 5-methoxytryptamine (5-MeOT), clozapine, LSD, and mesulergine, moderate affinity (pKi 6–7.9) for (±)-2-dipropyl-amino - 8 - hydroxy - 1, 2, 3, 4, - tetrahydronaphthalene (8 - OH-DPAT), methysergide, ergotamine and spiperone and low affinity (pKi<6.0) for pindolol, cyanopindolol, and buspirone (Hoyer et al., 1994; To et al., 1995). These attributes comprise a unique pharmacological profile for the 5-HT7 receptor, which distinguishes it from other closely related receptors such as the 5-HT1A receptor.
The 5-HT7 receptor has been identified in regions of rat and guinea-pig brain (Tsou et al., 1994; Shenker et al., 1987), in particular the limbic system and thalamocortical regions, where it has been suggested that it may have a role in affective behaviours (To et al., 1995; Gustafson et al., 1996). Functional studies have identified 5-HT7 receptors in porcine vena cava and myometrium (Sumner et al., 1989; Kitazawa et al., 1998), canine coronary artery (Cushing & Cohen, 1992), marmoset aorta (Dyer et al., 1994) and in guinea-pig ileum (Feniuk et al., 1984; Kalkman et al., 1986; Carter et al., 1995).
Until recently, the pharmacological profile of the smooth muscle 5-HT7 receptor was thought to be consistent across species, where activation of the receptor has been shown to result in relaxation. However, smooth muscle contraction by a postjunctional 5-HT7-like receptor in the rat jejunum has also been reported (McLean & Coupar, 1996), raising the possibility of either a different transduction mechanism or further subtypes of the 5-HT7 receptor. The latter is supported by the finding of cDNA in the rat which differed in the carboxy terminal of the seven transmembrane structure of the expressed 5-HT7 receptor (Lovenberg et al., 1993; Ruat et al., 1993). The generation of receptors differing in the carboxy regions can result in differential coupling to G-proteins (Lucas & Hen, 1995). In addition there have been isolated reports of an apparent species difference in the 5-HT7 receptor. For instance, the affinity of clozapine for the rat 5-HT7 receptor was found to be almost two orders of magnitude greater than the affinity of the compound at the mouse 5-HT7 receptor (Sleight et al., 1995b).
Hence, the primary aim of this investigation was to determine whether binding sites corresponding to 5-HT7 receptors could be detected in smooth muscle homogenates of rat jejunum. In addition, binding studies in rat brain and guinea-pig ileum were undertaken to identify 5-HT7 sites, in order to compare the binding characteristics in different tissues. 5-HT7 sites have been previously labelled in the brain using the agonists [3H]-5-CT and [3H]-5-HT (Sleight et al., 1995a; To et al., 1995), but often binding with an agonist radioligand may be complicated by the presence of multiple affinity states for the ligand, whereas this is generally not a problem with the use of an antagonist radioligand (Kenakin, 1984).
Methods
Radioligand binding experiments
Membrane preparations Rat brain
Hooded Wistar rats of either sex were decapitated followed by rapid removal of the brain. The cerebellum was removed, the remaining mass weighed, and homogenized with 10 volumes of 50 mM Na2HPO4 phosphate buffer pH 7.4, using 20 strokes of a glass homogenizer. Following centrifugation at 1000×g for 10 min at −2°C, the supernatant was re-centrifuged at 40,000×g for 20 min at −2°C. The resulting pellet was then washed with a further 10 volumes of buffer before the final centrifugation at 40,000×g at −2°C for 20 min. The final pellet was collected and frozen at −80°C until required for radioligand binding studies.
Guinea-pig ileum
Homogenates were prepared according to the method of Kalkman et al. (1986), who detected a 5-HT7-like receptor using [125I]-LSD. Briefly, guinea-pigs of either sex were stunned and exsanguinated before removal of the ileum and flushing out of the intra-luminal contents with phosphate buffer. The segments (ca. 20 cm) were stretched over a glass rod, to enable separation of the longitudinal and circular muscle layers by gentle rubbing and peeling of the top layer. The wet mass was then collected, weighed, mixed with 10 volumes of buffer, homogenized in an Ultra Turrax set at 0.75×maximum speed for 10 s, and centrifuged at 700×g for 10 min at −2°C. After centrifugation the buffy coat was skimmed off the surface, and the remaining supernatant and pellet re-homogenized, passed through a double layer of coarse mesh gauze and re-centrifuged at 14,000× g for 30 min. The pellet was collected, then washed with 10 volumes of buffer and re-centrifuged as previously. The final pellet was collected and stored as above.
Rat jejunum
Homogenates were prepared according to a similar method employed by Pinkus et al. (1990) in an investigation of 5-HT3 binding sites using [3H]-zacopride. Briefly, hooded Wistar rats of either sex were stunned and exsanguinated before removal of about 20 cm of jejunum, measured from the ligament of Trietz. Phosphate buffer was used to flush out the intra-luminal contents. The intestine was cut vertically and the mucosa scraped off with a glass slide. The remaining tissue was mixed with 50 ml of buffer, homogenized in an Ultra Turrax set at 0.75×maximum speed for 10 s, and centrifuged at 500×g for 5 min at −2°C. The supernatant was collected and centrifuged at 45,000× g for 20 min. The pellet was then collected, washed with 20 ml of buffer and re-centrifuged as previously. The final pellet was collected and stored as above.
Receptor binding assays
Binding studies were performed using the ligand [3H]-mesulergine. Mesulergine has a high affinity for 5-HT7 receptors as well as an affinity for 5-HT2A and 5-HT2C receptors, dopamine D2 receptors and α1/α2 adrenoceptors (Closse, 1983; Rinne, 1983; Pazos et al., 1985; Hoyer et al., 1994). In order to overcome the binding of mesulergine to receptors other than 5-HT7, masking drugs consisting of cinanserin (30 nM), RS 102221 (3 μM), raclopride (1 μM), prazosin (0.1 μM) and yohimbine (0.1 μM) were used, respectively. The concentrations of the masking drugs were chosen so that there was a theoretical occupancy of at least 90% of their respective receptor with little effect (<20% in total) on 5-HT7 receptor binding.
In addition, phosphate buffer (Na2HPO4 50 mM) was chosen instead of Tris-HCl (Tris 50 mM, NaCl 140 mM and MgCl2 5 mM) as it has been reported that the specific binding of [3H]-mesulergine is reduced in the presence of 100 mM or greater of sodium (Closse, 1983). Sodium ascorbate (1 μM) and pargyline (10 μM) were added to the buffer with experiments involving 5-HT.
Saturation curves were constructed in rat brain, jejunum and guinea-pig ileum. In each case, an aliquot of 200 μl containing between 1–1.8, 2–2.5 and 0.8–1.2 mg of protein, respectively, determined by the Bradford method (Bradford, 1976), was used with various concentrations of the radioligand ranging from 0.3–30 nM (final concentration) and buffer containing the masking drugs to give a total volume of 500 μl. Non-specific binding was defined by risperidone (1 μM) and experiments were performed in triplicate.
The assay composition for competition studies in rat brain was as follows: 100 μl of the membrane preparation, 50 μl of the radioligand (10 nM, final concentration), 50 μl of the displacing agent at various concentrations, and buffer containing the masking drugs to give a total volume of 350 μl. Displacing agents were added in 13 different concentrations and the experiment performed in duplicate. In guinea-pig ileum and rat jejunum this was varied slightly to conserve radioligand. The assay medium was as follows: 150 μl of the membrane preparation, 25 μl of the radioligand (10 nM, final concentration), 25 μl of the displacing agent at various concentrations, and buffer containing the masking drugs to give a total volume of 250 μl. Displacing agents were added in at least five different concentrations and the experiment performed in quadruplicate.
In both saturation and competition studies, the drugs and buffer were incubated initially for 10 min at 37°C before addition of the homogenate and incubation at 37°C for a further 60 min. Preliminary studies showed no significant difference in specific binding (P>0.05) between experiments incubated for 30, 60 and 90 min. Termination of the experiment was performed by vacuum filtration through Whatman GF/B filters presoaked in 0.5% polyethylene glycol and risperidone (1 μM). Each filter was placed in a plastic vial with 5 ml of Filtercount (Packard), vortexed, and the radioactivity determined by a scintillation counter (Packard Tricarb 2000, CA, U.S.A.) after at least 5 h had elapsed.
Functional studies
Hooded Wistar rats of either sex were stunned and exsanguinated before removal of about 20 cm of jejunum, measured from the ligament of Trietz (McLean & Coupar, 1996). The intraluminal contents were flushed out using warm modified Krebs-Henseleit solution of the following composition (mM): NaCl 116, KCl 5.4, MgSO4 0.6, CaCl2 2.5, NaH2PO4 1.2, NaHCO3 25, glucose 11.1. Four segments of jejunum (3–4 cm in length), were set up separately on tissue holders under 1 g weight tension and equilibrated in 20 ml organ baths containing Krebs-Henseleit solution gassed with 95% O2: 5% CO2 and maintained at 37°C. Responses were measured using an isotonic transducer (HUgo Basile) connected to a Grass model 79D polygraph recorder. Following equilibration, non-cumulative concentration-response curves were constructed for the agonists to avoid desensitization, as reported by McLean & Coupar (1996). Preliminary experiments showed non-reproducible curves, in agreement with previous findings in the rat jejunum (McLean & Coupar, 1996), therefore only one concentration-response curve was recorded in each tissue segment. Curves were established as described by addition of increasing concentrations of the agonist to the bathing solution at 10 min intervals, and agonists left in contact with the tissues for<30 s. Paired segments were used in order to measure the effect of antagonists, one always serving as control and the others to compare the response of the agonist in the presence of different antagonist concentrations. The effect of the agonists in the absence and presence of antagonist were expressed as a percentage of a maximum contraction obtained with acetylcholine (1 μM) in each segment at the end of the experiment. Antagonists were incubated for 40 min before testing of agonists.
Drugs
The following drugs were used: 5-carboxamidotryptamine, cinanserin, lysergic acid diethylamide (LSD), metergoline, 5-methoxytryptamine, (−) pindolol, raclopride L-tartrate, risperidone, tetrodotoxin citrate (Research Biochemicals International, Natick, MA, U.S.A.), acetylcholine chloride, atropine hydrochloride, 5-hydroxytryptamine creatinine sulphate, pargyline hydrochloride (Sigma, Poole, U.K.), prazosin hydrochloride, yohimbine hydrochloride (ICN Biomedicals, Aurora, OH, U.S.A.), haloperidol (Searle, Melbourne, Australia), [3H]-mesulergine (Amersham, Amersham, U.K.), RS 102221 8-[5-(2,4-dimethoxy-5-(4-trifluromethylphenylsulphonamido)phenyl-5-oxopentyl]-1,3,8 triazaspiro[4.5]decane-2,4-dione (Tocris Cookson, Bristol, U.K.), ritanserin (Janssen-Cilag, Sydney, Australia), GR 113808 ({1-[2-(methyl-sulphonylamino)ethyl]-4-piperidinyl}methyl 1-methyl-1H-indole-3-carboxylate, ondansetron hydrochloride, sumatriptan succinate (Glaxo, Melbourne, Australia). Ritanserin, pindolol and cinanserin were dissolved in methanol, ondansetron, risperidone and metergoline were dissolved in ethanol and RS 102221 was dissolved in dimethylsulphoxide (DMSO) before dilution to the required concentrations. The volumes of solvent used constituted less than 0.02% of the final concentration in both functional and binding studies and had no effect in either study. All other drugs were dissolved in distilled water.
Data analysis
Equilibrium dissociation constants (KD) for saturation studies were obtained using non-linear regression analysis and competition data were analysed according to one or two-site binding models. The equation used for fitting one site competition curves was:
where min is the apparent minimum, max is the apparent maximum and Log IC50 is the logarithm of the concentration of the competing drug,×, required to inhibit the binding of the radioligand by 50%. The equation used for fitting two-site competition curves was:
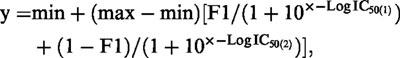 |
where min is the apparent minimum, max is the apparent maximum, F1 is the fraction of receptors with an affinity described by Log IC50(1) (the logarithm of the concentration of the competing drug,×, required to inhibit the binding of the radioligand by 50%) and the remaining receptors have an affinity described by Log IC50(2). Ki values were obtained using the Cheng & Prusoff equation (1973).
Contractions to serotonergic agonists were expressed as a percentage of the maximum contraction obtained to acetylcholine (1 μM). The EC50 values, concentrations producing 50% of the maximum response elicited by the agonists, were estimated from non-linear regression plots of single curves. The data was analysed by fitting a logistic curve of the form:
where i is apparent number of curves, max is the apparent maximum, min is the minimum point, Log EC50 is the logarithm of the concentration,×, producing 50% of the maximum response of each individual curve and nH is the slope factor. Antagonist potencies were expressed as either a pKB or an apparent pKB value. Initially, pA2 values were calculated from experiments in which three concentrations of antagonist were tested, using the method of Arunlakshana & Schild (1959). The slope of the Schild plot was calculated and compared to a slope of unity as expected for a competitive antagonist, using Student's unpaired t-test. When the slope did not differ significantly from unity (P<0.05), an estimate of the pKB value was made by fitting a regression with the slope constrained to unity. Apparent pKB values were calculated from the relationship:
where CR is the concentration ratio of the agonist in the presence and absence of antagonist (B), (Furchgott, 1972).
Student's t-test was used for comparison of individual means and Dunnett's test was used when multiple means were compared to a common control; the criterion for statistical significance for both tests was set at P<0.05. Arithmetic and geometric means are given with mean±s.e.mean or mean with 95% confidence intervals, respectively. Pearson correlation coefficients were calculated using the computer program Graph Pad Prism 2.0 (GraphPad Software, San Diego, CA, U.S.A.). All other calculations and graphics were also performed using Graph Pad Prism 2.0.
Results
Saturation curves in the rat brain were performed in the presence of raclopride (1 μM) to inhibit the binding of [3H]-mesulergine to dopamine D2 receptors. The additional inhibition of α-adrenoceptors with prazosin (0.1 μM) and yohimbine (0.1 μM) and 5-HT2A receptors using cinanserin (30 nM) had little effect on the specific binding of the radioligand. However, inhibition of 5-HT2C receptors using RS 102221 was found to significantly (P<0.05) decrease the specific binding of [3H]-mesulergine (0.3–30 nM), indicating the possibility of non-homogeneous binding sites recognized by the radioligand (Figure 1).
Figure 1.
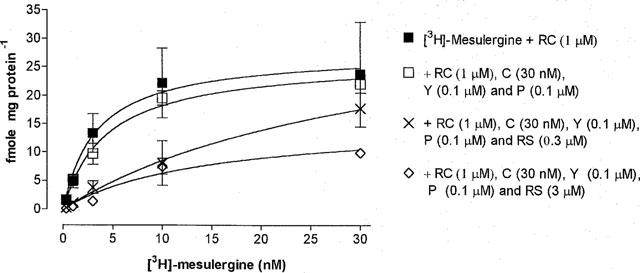
Saturation curve of [3H]-mesulergine binding in the rat brain in the absence and presence of yohimbine (Y: 0.1 μM), prazosin (P: 0.1 μM) cinanserin (C: 30 nM) and RS 102221 (RS: 0.3 and 3 μM). All experiments were performed in the presence of raclopride (RC: 1 μM). Abscissa: Concentration of [3H]-mesulergine in nM; Ordinate: fmole mg of protein−1. Each point is the mean±s.e.mean from three experiments.
At a concentration of 0.3 μM (100×KD value at 5-HT2C receptors), RS 102221 did not sufficiently block 5-HT2C receptors when 30 nM of [3H]-mesulergine was used. This is illustrated in Figure 1, where an increase in specific binding occurred when 30 nM of [3H]-mesulergine was used. To overcome this problem, the concentration of RS 102221 was increased 10 fold to 3.0 μM. This concentration was found to be sufficient in reducing the counts obtained when using 30 nM of [3H]-mesulergine, and the curve reached a plateau as previously. A pKD value of 8.0±0.04 and a Bmax of 9.9±0.3 fmol mg protein−1 (n=3) was calculated.
The rank order of potency of the displacing drugs studied in the rat brain was as follows: (agonists) 5-CT>5-HT⩾5-MeOT>sumatriptan and (antagonists) risperidone⩾LSD⩾metergoline>ritanserin>pindolol (Table 1). All agonists best fitted a two-site competition model (Figure 2a, Table 1) and all antagonist ligands best fitted a one-site competition model except for ritanserin which best fitted a two-site model (Figure 2b, Table 1). In a preliminary experiment, haloperidol (0.01 nM to 1 μM), a compound with weak affinity at 5-HT7 receptors, did not displace [3H]-mesulergine binding. The correlation between the pKi values obtained in the rat brain and previously published values from recombinant 5-HT7 receptors obtained from rat or mouse and expressed in transfected cells was significant (P<0.05); a Pearson correlation factor of 0.90 was obtained (Figure 3).
Table 1.
Comparison of ligand affinities in guinea-pig ileum, rat brain and isolated rat jejunum. All data are expressed as mean values with 95% confidence intervals noted in parentheses. High and low affinity values as well as the per cent fraction are shown for compounds displaying two-site competition
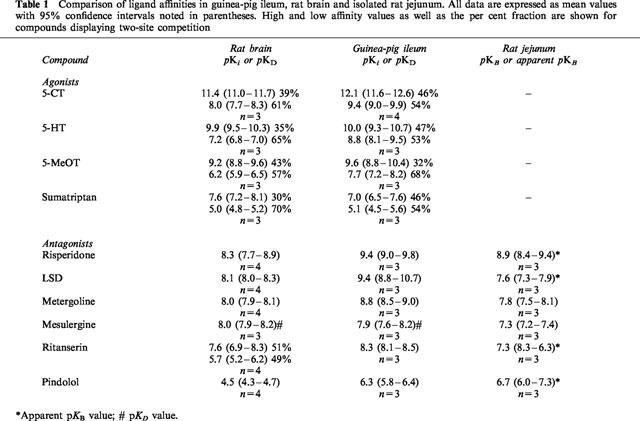
Figure 2.
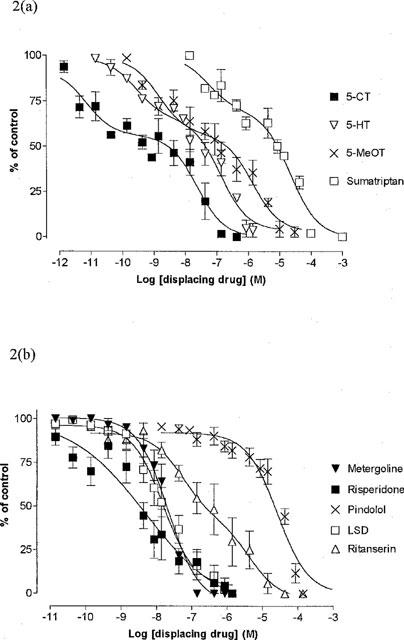
(a, b) inhibition of [3H]-mesulergine binding to rat brain and (c, d) guinea-pig ileum by various ligands. Abscissa: Log molar concentration of the displacing drug, Ordinate: % of specific binding. The data represent the mean±s.e.mean per cent of maximum specific binding (defined with 1 μM risperidone) of at least three experiments.
Figure 3.
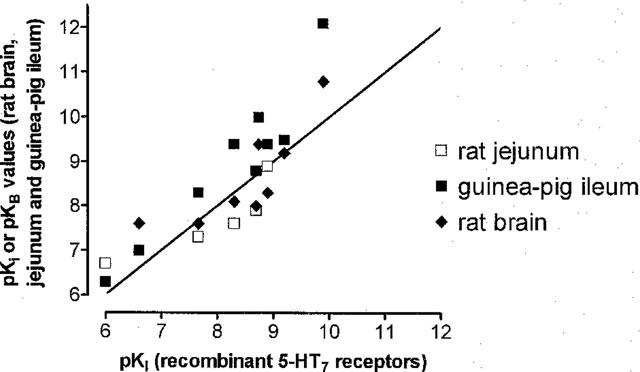
Correlation between pKi values obtained in recombinant 5-HT7 receptors from rat or mouse and expressed in transfected cells (Roth et al., 1994; Plassat et al., 1993; Ruat et al., 1993; Shen et al., 1993) and pKi or pKB values obtained in rat brain, guinea-pig ileum and rat jejunum. Abscissa: pKi values for recombinant 5-HT7 receptors, Ordinate: pKi or pKB values obtained in rat brain and jejunum and guinea-pig ileum. The continuous line represents a line of identity.
Saturation studies in the guinea-pig ileum were performed in the presence of the concentrations of masking drugs found to be optimum for 5-HT7 receptor binding in the rat brain, under these conditions (see Methods). The pKD value of [3H]-mesulergine was calculated to be 7.9±0.11 and the Bmax was 21.5±4.9 fmol mg protein−1 (n=3).
The order of affinity of the agonist and antagonist displacers studied was the same as in the rat brain with some slightly higher pKi values, see Table 1. All agonist ligands best fitted a two-site model whilst all antagonist ligands best fitted a one-site competition model (Figure 2c and d). The correlation between the pKi values obtained in the guinea-pig ileum and recombinant rat or mouse 5-HT7 receptors was significant (P<0.05); a Pearson correlation factor of 0.94 was obtained (Figure 3).
Similar binding studies to those performed in the rat brain and guinea-pig ileum using [3H]-mesulergine were undertaken in the rat jejunum, but no binding to the 5-HT7 receptor was detected.
Effect of 5-HT in the absence and presence of ondansetron in the isolated rat jejunum
In agreement with the findings of McLean & Coupar (1996) a biphasic concentration-response curve for 5-HT (10 nM–3 μM) induced contractions were obtained in the rat jejunum (Figure 4). A biphasic model better described the interaction of 5-HT in the rat jejunum than a monophasic model with R2 values of 0.83 and 0.79 respectively. The pEC50 value of the first phase of the curve was 7.58±0.12 with a maximum of 34.1±4.4% of the response to acetylcholine (1 μM). The pEC50 value of the second phase of the curve was 6.54±0.17 with a maximum of 69.5±8.5 of the response to acetylcholine (1 μM). However, in the presence of ondansetron (0.1, 1 and 10 μM) the curve flattened and was better defined by a monophasic curve, (pEC50=6.79±0.07). There was no significant difference (P>0.05) between the curves obtained in the presence of 0.1, 1 and 10 μM of ondansetron. In each case ondansetron significantly reduced the maximum response of 5-HT by ca. 32% (P<0.05, n=4). Thus ondansetron (1 μM) was included in all further experiments involving 5-HT in order to investigate the non-5-HT3 receptor-mediated response. However, a response to 5-HT in the rat jejunum after blockade of 5-HT3 receptors with ondansetron was not always present, ca. 50% of tissues failed to respond to the agonist, even though a response to acetylcholine was present.
Figure 4.
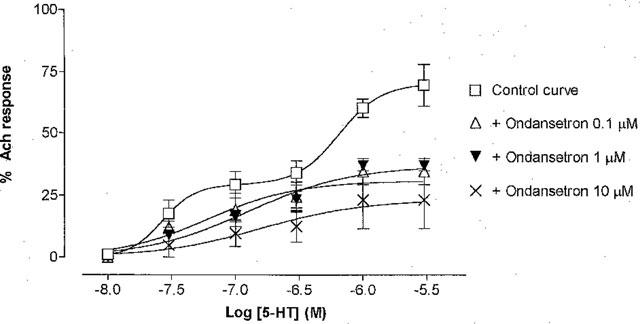
Concentration-response curve for the contractile effect of 5-HT in the absence and presence of ondansetron (0.1, 1 and 10 μM) in isolated rat jejunum. Abscissa: Log molar concentration of 5-HT, Ordinate: % of maximum contraction obtained with acetylcholine (1 μM). The data represent the mean±s.e.mean of four paired experiments.
Effect of 5-CT, 5-MeOT and sumatriptan
5-CT (3 nM–3 μM) resulted in a concentration-response curve similar to the monophasic curve of 5-HT. The pEC50 of 5-CT (7.03±0.07) was significantly higher (P<0.05) than that of 5-HT (6.79±0.07) in the presence of ondansetron (1 μM), however the maximum contraction produced (37.1±2.7% of the contraction to Ach, 1 μM) was not significantly different to that produced by 5-HT (36.9±1.8%).
The pEC50 of 5-MeOT (5.33±0.11) was significantly lower (P<0.05) than that of 5-HT (6.79±0.07) in the presence of ondansetron (1 μM), however the maximum contraction produced (46.7±7.2% of the contraction to Ach, 1 μM) was greater but not significantly different to that produced by 5-HT (P>0.05).
A pEC50 value of<5 was obtained for sumatriptan, with a maximum contraction of only 11.3±3.1% of the contraction to Ach, (1 μM).
Effect of 5-HT in the absence and presence of various antagonists
The effect of 5-HT in the presence of risperidone (10–100 nM), ritanserin (100 nM–1 μM), LSD (10–100 nM), metergoline (10–100 nM), mesulergine (10–100 nM) and pindolol (1 and 3 μM) was investigated. At a concentration of 10 nM, risperidone and ritanserin caused both a shift in the control curve to 5-HT and a depression of the maximum response to the agonist. However, at higher concentrations of the antagonists, neither the depression of the maximum nor the shift in the control curve was increased. LSD also resulted in a depression of the maximum response with no further shift of the 5-HT curve when used at 30 nM, and this effect was increased at a concentration of 100 nM of LSD. Thus apparent pKB values were reported for risperidone, ritanserin and LSD (Table 1). Schild slopes of 1.0±0.04 (data points; 9) and 0.8±0.09 (9) were obtained for mesulergine and metergoline (Figure 5) respectively. These values were not significantly different (P<0.05) from unity. The correlation between the KB and apparent pKB values obtained in the rat jejunum and recombinant 5-HT7 receptors from rat or mouse expressed in transfected cells (Hoyer et al., 1994) was significant (P<0.05); a Pearson correlation factor of 0.87 was obtained (Figure 3).
Figure 5.
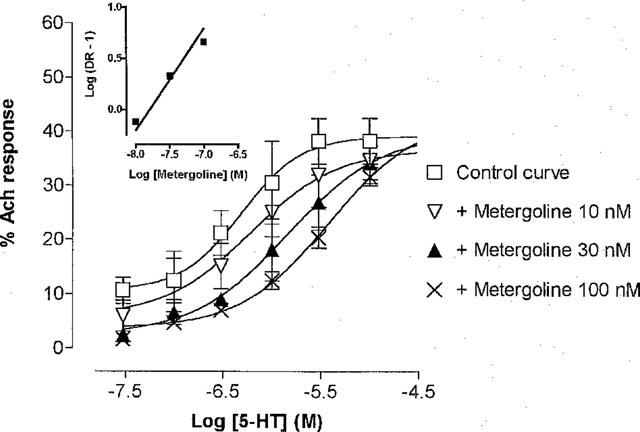
Concentration-response curves for 5-HT in the absence and presence of metergoline (10, 30 and 100 nM). Abscissa: Log molar concentration of 5-HT, Ordinate: % of maximum contraction obtained with acetylcholine (1 μM). The data represent the mean±s.e.mean of three paired experiments. All experiments were conducted in the presence of ondansetron (1 μM). The slope of the Schild regression (inset) was 0.8±0.09.
Preliminary studies showed RS 102221 (3 μM), yohimbine (0.1 μM), cinanserin (30 nM) and GR 113808 (1 μM) to have no effect on the contractile response to 5-HT in the rat jejunum, with pEC50 values of 5-HT in the presence of the antagonists not significantly different to that of the control curve, (P>0.05). Atropine (1 μM) and tetrodotoxin (1 μM) failed to significantly modify (P>0.05) the non 5-HT3-mediated response.
Discussion
The results from the present study suggest the presence of 5-HT7 receptor binding sites in rat brain and guinea-pig ileum, but not in rat jejunum where a functional 5-HT7-like receptor was detected.
Receptor characterization
Due to the lack of selective and specific ligands currently available for studying the 5-HT7 receptor, the use of rank order of agonist and antagonist affinity was used to characterize the 5-HT7 site, as has been undertaken previously in studies using expressed and native 5-HT7 receptors (Kalkman et al., 1986; Plassat et al., 1993; Monsma et al., 1993; Erlander et al., 1993; To et al., 1995; Carter et al., 1995; McLean & Coupar, 1996).
The presently described results obtained in rat brain are in agreement with findings in both guinea-pig and rat brain of 5-HT7 sites, radiolabelled using serotonin receptor agonists (Sleight et al., 1995a; To et al., 1995; Gustafson et al., 1996).
A number of masking drugs were used in the present study in order to inhibit the binding of [3H]-mesulergine to other receptors for which it has affinity. Mesulergine has been reported to have affinity for 5-HT2A and 5-HT2C receptors as well as some affinity for dopamine D2 and α1/α2-adrenoceptors (Closse, 1983; Rinne, 1983; Hoyer et al., 1994), (see Methods). The masking drugs, some of which also have affinity for 5-HT7 receptors, were chosen at concentrations which would theoretically occupy at least 90% of their targetted receptor populations (Kalkman et al., 1986; Hoyer et al., 1994; Sleight et al., 1995a; Bonhaus et al., 1997), without significantly affecting the binding of the radioligand to the 5-HT7 receptor.
Saturation studies performed in rat brain and guinea-pig ileum provided pKD values of 8.0±0.04 and 7.9±0.11 respectively for [3H]-mesulergine, which were in accordance with a previously reported pKB value of 7.8 (Carter et al., 1995) and a pKi value of 8.15 (Hoyer et al., 1994; To et al., 1995) at the 5-HT7 receptor. However, no evidence of specific binding to a 5-HT7 site was apparent in the rat jejunum.
Displacement of [3H]-mesulergine binding in rat brain and guinea-pig ileum was observed using the agonists 5-CT (pKi=11.4, 12.1), 5-HT (9.9, 10.0), 5-MeOT (9.2, 9.6) as well as sumatriptan (7.6, 7.0) and the antagonists risperidone (8.3, 9.4), ritanserin (7.6, 8.3), metergoline (8.0, 8.8), LSD (8.1, 9.4) and pindolol (4.5, 6.3), respectively. The pKi values of these compounds correlated well with those from other studies of the 5-HT7 receptor in both order and magnitude (Kalkman et al., 1986; Bard et al., 1993; Plassat et al., 1993; Ruat et al., 1993; Shen et al., 1993; Roth et al., 1994; To et al., 1995).
5-CT, 5-HT, 5-MeOT and sumatriptan displayed two-site displacement binding in the rat brain and guinea-pig ileum, which is not unusual for agonists, since they can bind to both a high and low affinity state of the receptor (Kenakin, 1984). However, the two-site binding displayed by ritanserin in the rat brain is more difficult to explain, and was not detected in the guinea-pig ileum nor reported in guinea-pig brain using [3H]-5-CT (To et al., 1995).
The possibility of [3H]-mesulergine binding to non-serotonergic receptors, such as dopamine receptors, were ruled out by the addition of masking drugs such as raclopride (Rinne, 1983) and by the use of the agonists 5-CT, 5-HT and 5-MeOT which are specific for serotonin receptors. In addition, a preliminary experiment performed with haloperidol (0.01 nM to 1 μM), a potent D2 receptor antagonist (pKi=9.3), with little affinity for 5-HT7 receptors (pKi=6.6; Roth et al., 1994), failed to displace mesulergine binding in rat brain.
The binding site identified in both rat brain and guinea-pig ileum is unlikely to be of the 5-HT1A or 5-HT1B (rat brain) subtype, since pindolol was found to have a low (micromolar) affinity in this study, consistent with 5-HT7 but not 5-HT1 receptors, where it has nanomolar affinity (Lovenberg et al., 1993; Ruat et al., 1993; Hoyer et al., 1994; Carter et al., 1995; McLean & Coupar, 1996). The other 5-HT1 receptor subtypes; 5-HT1D (guinea-pig ileum), 5-ht1E, and 5-ht1F were ruled out by the high affinity of 5-CT in both the rat brain (pKi=11.4) and guinea-pig ileum (pKi=12.1, Hoyer et al., 1994).
The receptor is also unlikely to be the 5-HT2A or 5-HT2C subtype, firstly because these receptors were excluded with cinanserin and RS 102221, respectively, and more importantly the antagonist order of affinity of risperidone (pKi=8.7, 9.4), metergoline (8.0, 8.8) and ritanserin (7.7, 8.3) in rat brain and guinea-pig ileum, respectively did not correlate with either 5-HT2A (risperidone>ritanserin>metergoline) or 5-HT2C (metergoline>ritanserin>risperidone) receptors (Hoyer et al., 1994, Sleight et al., 1995a). In addition, 5-CT was found to have nanomolar affinity in this study whilst it has micromolar affinity or lower at both 5-HT2A and 5-HT2C receptor sites (Bard et al., 1993; Hoyer et al., 1994; Roth et al., 1994). Furthermore, sumatriptan was found to have micromolar affinity in this study whilst it has only millimolar affinity at 5-HT2A and 5-HT2C receptors (Hoyer et al., 1994).
The high affinity of 5-CT and 5-MeOT also ruled out the involvement of 5-HT3 receptors, where the agonists are inactive (Bard et al., 1993, Hoyer et al., 1994 Sleight et al., 1995b).
The involvement of 5-HT4, 5-ht5 and 5-ht6 receptors is excluded due to the use of a nanomolar concentration of [3H]-mesulergine in displacement studies. Mesulergine has only micromolar affinity at 5-ht5A, 5-ht5B and 5-ht6 receptors (Plassat et al., 1993; Erlander et al., 1993; Monsma et al., 1993) and is inactive at 5-HT4 receptors (Bard et al., 1993). Further evidence against 5-ht5B and 5-ht6 receptor involvement is the low affinity of 5-CT (pKi=7.4 and 6.6 respectively) for these receptors (Hoyer et al., 1994).
While the 5-HT orphan receptor reported by Castro et al. (1997) has a high affinity for 5-CT, it cannot be the receptor in question since mesulergine has only a micromolar affinity at the orphan receptor (Castro et al., 1997). In addition the 5-HT orphan receptor has a similar affinity for both 5-CT and 5-HT (Castro et al., 1997), a characteristic not displayed by the receptor in the present study.
Functional significance
5-HT7 sites have been found to exist in high densities in the limbic system, hypothalamus and thalamocortical regions, hence leading to the suggestion that they may have a role in affective behaviours (Plassat et al., 1993; Eglen et al., 1994; Gustafson et al., 1996).
This suggestion is further supported by the high affinity of atypical antipsychotics such as clozapine and risperidone for the 5-HT7 receptor (Roth et al., 1994). Moreover, the 5-HT7 receptor has also been implicated in other affective disorders such as depression, where it was found that a down-regulation of the receptor occurs after chronic antidepressant treatment (Sleight et al., 1995a). In addition 5-HT7 receptors may have a role in the regulation of mammalian circadian rhythms (Lovenberg et al., 1993).
The high affinity binding of LSD in rat brain in the present study adds further support to a 5-HT7 site where a pKi value similar to that reported in guinea-pig brain by To et al. (1995) was obtained (pKi 8.2 compared with 7.8). This raises the possibility that the hallucinogenic action of LSD may be mediated in part via the 5-HT7 receptor.
Peripheral tissue studies
The results of the binding studies in guinea-pig ileum lend further support to the functional findings of Feniuk et al. (1984), Kalkman et al. (1986) and Carter et al. (1995) where a postjunctional 5-HT site was seen to induce relaxation of pre-contracted guinea-pig ileum. The binding studies performed in this investigation are concordant with those of Kalkman et al. (1986), where [125I]-LSD was used to characterize a postjunctional 5-HT site in guinea-pig ileum. In the study by Kalkman et al. (1986), cinanserin (300 nM) was used to mask 5-HT2 receptors, a concentration which the authors claim theoretically inhibits 98% of 5-HT2 receptors. Unfortunately, this concentration of cinanserin was also found to occupy close to 50% of the 5-HT7-like site in a displacement study in the same investigation. This may have led to an underestimation of the KD value of [125I]-LSD at the 5-HT7 site. Nevertheless, the antagonist order of affinity reported (iodo-LSD>metergoline = mesulergine>spiperone> >haloperidol>propranolol) correlates well with a 5-HT7 receptor.
Despite the lack of binding of [3H]-mesulergine in the rat jejunum, the functional experiments performed in the present study suggest the presence of a 5-HT7-like receptor in this tissue, in agreement with the findings of McLean & Coupar (1996). The low affinity of pindolol and lack of antagonist affinity of yohimbine, cinanserin, RS 102221 and GR 113808 rule out a 5-HT1A, α2, 5-HT2A, 5-HT2C and 5-HT4 receptor site, in contrast to the findings of Javid & Naylor (1997), who reported the involvement of 5-HT2 and 5-HT4 receptors in mediating the contractile response of 5-HT in the proximal region of the rat small intestine. The use of ondansetron (1 μM) and the relatively high potency of 5-CT rules out 5-HT3 receptors. The presence of 5-ht5 and 5-ht6 receptors can also be excluded by the potency of 5-CT at this site. The relatively low potency of 5-CT compared to its high affinity in binding studies is characteristic of the 5-HT7 receptor and was also reported by Carter et al. (1995), and Martin & Wilson (1995). The correlation between the pKB and apparent pKB values obtained in the rat jejunum and recombinant 5-HT7 receptors from rat or mouse expressed in transfected cells (Plassat et al., 1993; Ruat et al., 1993; Shen et al., 1993; Roth et al., 1994) was very high (see Results).
It is interesting to note that the affinity values of mesulergine (7.3) and ritanserin (7.3) were lower in the present study with the rat isolated jejunum than reported in a similar study by McLean & Coupar (1996), where mesulergine and ritanserin were reported to have pA2 values of 8.1 and 8.0 respectively. Affinity values for LSD, mesulergine and ritanserin reported in the guinea-pig ileum, receptors expressed from 5-HT7 receptor genes (Ruat et al., 1993; Carter et al., 1995) and binding studies in the rat brain and guinea-pig ileum in this investigation were also slightly higher than those obtained in the rat jejunum. However, binding studies performed in the guinea-pig brain (To et al., 1994), have shown a pKD value of 7.8 for LSD, which agrees with the pKB value obtained in the present study. In addition, risperidone, ritanserin and LSD resulted in a non-competitive antagonism of the serotonin response in the presence of ondansetron (1 μM). At higher concentrations, risperidone (10–100 nM) and ritanserin (100 nM–1 μM) caused a depression of the maximum response to 5-HT without further shifting the control curve. A comparison of the functional characteristics of LSD and risperidone at the 5-HT7 receptor can not be made, as the effect of these compounds has not been investigated in other studies. However, at a concentration range of 10–100 nM, ritanserin was reported to act as a competitive inhibitor of the 5-HT7-like response in the rat jejunum, (McLean & Coupar, 1996). The observation that an increased concentration of ritanserin and risperidone did not further shift the mean curve to 5-HT may indicate that the interaction of the antagonists with the 5-HT7 receptor is of a non-competitive nature such as that seen with allosteric modulation. Negative allosteric modulators are known to produce parallel shifts of agonist concentration-response curves up to a limiting value (Ehlert, 1988; Lanzafame et al., 1996). Often, as seen with ritanserin, they may appear to be competitive at lower concentration ranges. Further studies are required to fully define the mechanism by which ritanserin and risperidone interact with the 5-HT7 site.
The lack of [3H]-mesulergine binding in the rat jejunum in this study may be explained by a low 5-HT7 receptor density. This is supported by the observation that a response to 5-HT in the rat jejunum after blockade of 5-HT3 receptors with ondansetron was not always present; (see Results). The response observed in the rat jejunum differs from other smooth muscles where activation of the 5-HT7 receptor results in relaxation or various preparations in different species, such as porcine vena cava and myometrium (Sumner et al., 1989; Kitazawa et al., 1998), canine coronary artery (Cushing & Cohen, 1992), marmoset aorta (Dyer et al., 1994) guinea-pig ileum (Feniuk et al., 1984; Kalkman et al., 1986; Carter et al., 1995) and cat saphenous vein (Hoyer et al., 1994). In addition, the contractile response is not consistent with the coupling mechanism of the 5-HT7 receptor, which to date has been shown to only involve activation of adenylate cyclase, (Shenker et al., 1987; Eglen et al., 1997).
5-HT7 receptor subtypes?
The possibility of 5-HT7 receptor subtypes cannot be excluded; Ruat et al. (1993) isolated a 448 amino acid cDNA in the rat (5-HT7(a)), while Lovenberg et al. (1993) isolated a 435 amino acid cDNA (5-HT7(b)). The difference between the two cDNAs was only in the carboxy-terminal of the seven transmembrane structure of the expressed receptor and was said to result from alternative splicing (Boess & Martin, 1994). The generation of two receptor variants differing in the carboxy regions, can result in differential coupling to G-proteins (Lucas & Hen, 1995) and possibly a different physiological response as has been reported with the prostaglandin EP3 (Namba et al., 1993) and somatostatin SST2 receptors (Vanetti et al., 1993). In fact, the neuronal 5-HT7(a) isoform has been reported to increase intracellular calcium resulting in activation of calmodulin-stimulated, adenylate cyclase isoforms AC1 and AC8, independent of phosphoinositide and protein kinase C (Baker et al., 1998). This unique mechanism of coupling for any of the serotonin receptors was reported to be Gs-independent in studies with whole cells (see Wayman et al., 1994; Sunahara et al., 1996). It is possible some similar mechanism may exist in the rat jejunum, where a contraction instead of a relaxation to a 5-HT7-like receptor was observed.
Furthermore, an additional rat 5-HT7 isoform, a 470 amino acid cDNA, (5-HT7(c)) resulting from a retained exon cassette, has also been reported (Heidmann et al., 1997). However, unlike the other two isoforms, the rat 5-HT7(c) was reported not to be present in human tissue, which suggests a species difference for the presence of various 5-HT7 isoforms (Heidmann et al., 1997). This again raises the possibility of a difference in the physiological response produced by 5-HT7 receptors and may explain why an ‘atypical' response was obtained in the rat jejunum.
In conclusion binding studies performed with [3H]-mesulergine were able to detect 5-HT7 sites in rat brain and guinea-pig ileum, but not rat jejunum, where a functional 5-HT7-like site was present.
Acknowledgments
We would like to thank Dr Arthur Christopoulos for assistance with data analysis and Dr David Taylor, Shaunagh Darroch and Alfred Lanzafame for helpful discussions. The authors are grateful to Glaxo for the generous gifts of ondansetron, sumatriptan and GR 113808.
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKER L.P., NIELSEN M.D., IMPEY S., METCALF M.A., POSER S.W., CHAN G., OBRIETAN K., HAMBLIN M.W., STORM D.R. Stimulation of type I and type 8 Ca2+ calmodulin-sensitive adenylyl cyclases by the GS-coupled 5-hydroxytryptamine subtype 5-HT7A receptor. J. Biol. Chem. 1998;273:17469–17476. doi: 10.1074/jbc.273.28.17469. [DOI] [PubMed] [Google Scholar]
- BARD J.A., ZGOMBICK J., ADHAM N., VAYSSE P., BRANCHEK T.A., WEINSHANK R.L. Cloning of a novel serotonin receptor (5-HT7) positively linked to adenylate cyclase. J. Biol. Chem. 1993;31:23422–23426. [PubMed] [Google Scholar]
- BOESS F.G., MARTIN I.L. Review: molecular biology of 5-HT receptors. Neuropharmacol. 1994;33:275–317. doi: 10.1016/0028-3908(94)90059-0. [DOI] [PubMed] [Google Scholar]
- BONHAUS D.W., WEINHARDT K.K., TAYLOR M., DESOUZA A., MCNEELEY P.M., SZCZEPANSKI K., FONTANA D.J., TRINH J., ROCHA C.L., DAWSON M.W., FLIPPIN L.A., EGLEN R.M. RS-102221: a novel high affinity and selective, 5-HT2C receptor antagonist. Neuropharmacol. 1997;36:621–629. doi: 10.1016/s0028-3908(97)00049-x. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CARTER D., CHAMPNEY M., HWANG B., EGLEN R.M. Characterization of post-junctional 5-hydroxytryptamine (5-HT) receptor mediating relaxation of guinea-pig isolated ileum. Eur. J. Pharmacol. 1995;280:243–250. doi: 10.1016/0014-2999(95)00195-q. [DOI] [PubMed] [Google Scholar]
- CASTRO E.M., ROMÓN T., CASTILLO M.J., DEL OLMO E., PAZOS A., DE ARCO C. Identification and characterisation of a new serotonergic recognition site with high affinity for 5-carboxamidotryptamine in mammalian brain. J. Neuropharmacol. 1997;69:2123–2131. doi: 10.1046/j.1471-4159.1997.69052123.x. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibitor constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CLOSSE A. [3H]-Mesulergine, a selective ligand for serotonin-2 receptors. Life Sci. 1983;32:2485–2495. doi: 10.1016/0024-3205(83)90375-2. [DOI] [PubMed] [Google Scholar]
- CUSHING D.J., COHEN M.L. Sertonin-induced relaxation in canine coronary artery smooth muscle. J. Pharmacol. Exp. Ther. 1992;263:123–129. [PubMed] [Google Scholar]
- DYER S.M., BEXIS S., MANE M.T., DE LA LANDE I.S., FREWIN D.B., HEAD R.J. Interactions between 5-hydroxytryptamine, noradrenaline and the thromboxane-A(2) mimetic U44069 in the marmoset isolated aorta. Clin. Exp. Pharmacol. Physiol. 1994;21:201–206. doi: 10.1111/j.1440-1681.1994.tb02496.x. [DOI] [PubMed] [Google Scholar]
- EGLEN R.M., JAKEMAN L., ALVAREZ R.A. The 5-hydroxytryptamine7 receptor. Expert Opin. Invest. Drugs. 1994;3:175–177. [Google Scholar]
- EGLEN R.M., JASPER J.R., CHANG D.J., MARTIN G.R. The 5-HT7 receptor: orphan found. Trends in Pharmacol. Sci. 1997;18:104–107. doi: 10.1016/s0165-6147(97)01043-2. [DOI] [PubMed] [Google Scholar]
- EHLERT F.J. Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol. Pharmacol. 1988;33:187–194. [PubMed] [Google Scholar]
- ERLANDER M.G., LOVENBERG T.W., BARON B.M., DELECEA L., DANIELSON P.E., RACKE M., SLONE A.L., SIEGEL B.W., FOYE P.E., CANNON K., BRUNS J.E., SUTCLIFFE J.G. Two members of a distinct subfamily of 5-hydroxytryptamine receptors differentially expressed in rat brain. Proc. Natl. Acad. Sci. USA. 1993;90:3452–3457. doi: 10.1073/pnas.90.8.3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENIUK W., HUMPHREY P.P.A., WATTS A.D. 5-hydroxytryptamine-induced relaxation of isolated mammalian smooth muscle. Eur. J. Pharmacol. 1984;96:71–78. doi: 10.1016/0014-2999(83)90530-7. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F.The classification of adrenoceptors (adrenergic receptors). An evaluation from the standpoint of receptor theory Handbook of experimental pharmacology, catecholamines 1972Vol 33New York: SpringerVerlag; 285–335.(eds.) Blaschko, H. & Muscholl E. pp [Google Scholar]
- GUSTAFSON E.L., DURKIN M.M., BARD J.A., ZGOMBICK J., BRANCHEK T.A. A receptor autoradiographic and in situ hybridization analysis of the distribution of the 5-ht7 receptor in rat brain. Br. J. Pharmacol. 1996;117:657–666. doi: 10.1111/j.1476-5381.1996.tb15241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEIDMANN D.E.A., METCALF M.A., KOHEN R., HAMBLIN M.W. Four 5-hydroxytryptamine7 (5-HT7) receptor isoforms in human and rat produced by alternative splicing: species differences due to altered intron–exon organisation. J. Neurochem. 1997;68:1372–1381. doi: 10.1046/j.1471-4159.1997.68041372.x. [DOI] [PubMed] [Google Scholar]
- HOYER D., CLARKE D.E., FOZARD J.R., HARTIG P.R., MARTIN G.R., MYLECHARANE E.J., SAXENA P.R., HUMPHREY P.P.A. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol. Rev. 1994;46:157–204. [PubMed] [Google Scholar]
- JAVID F.A., NAYLOR R.J. Characterisation of the 5-HT receptors mediating the contractile effect of 5-HT in the terminal region of the rat small intestine. Br. J. Pharmacol. 1997;112:311P. [Google Scholar]
- KALKMAN H.O., ENGEL G., HOYER D. Inhibition of 5-carboxamidotryptamine-induced relaxation of guinea-pig ileum correlates with [125l]LSD binding. Eur. J. Pharmacol. 1986;129:139–145. doi: 10.1016/0014-2999(86)90345-6. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. The classification of drugs and drug receptors in isolated tissues. Pharmacol. Rev. 1984;36:165–222. [PubMed] [Google Scholar]
- KITAZAWA T., KUBO O., SATOH M., TANEIKE T. Involvement of 5-hydroxytryptamine7 receptors in inhibition of porcine myometrial contractility by 5-hydroxytryptamine. Br. J. Pharmacol. 1998;123:173–182. doi: 10.1038/sj.bjp.0701583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANZAFAME A., CHRISTOPOULOS A., MITCHELSON F. Interactions of agonists with an allosteric antagonist at muscarinic acetylcholine M2 receptors. Eur. J. Pharmacol. 1996;316:27–32. doi: 10.1016/s0014-2999(96)00639-5. [DOI] [PubMed] [Google Scholar]
- LOVENBERG T.W., BARON B., DE LECEA L., MILLER J.D., PROSSER R.A., REA M.A., FOYE P.E., RACKE M., SLONE A.L., SIEGEL B.W., DANIELSON P.E., SUTCLIFFE J.G., ERLANDER M.G. A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms. Neuron. 1993;11:449–458. doi: 10.1016/0896-6273(93)90149-l. [DOI] [PubMed] [Google Scholar]
- LUCAS J.J., HEN R. New players in the 5-HT receptor field: genes and knockouts. Trends in Pharmacol. Sci. 1995;16:246–252. doi: 10.1016/s0165-6147(00)89034-3. [DOI] [PubMed] [Google Scholar]
- MARTIN G.R., WILSON R.J. Operational characteristics of a 5-HT receptor mediating direct vascular relaxation: identity with the 5-HT7 receptor. Br. J. Pharmacol. 1995;114:383P. [Google Scholar]
- McLEAN P., COUPAR I.M. Characterisation of a postjunctional 5-ht7-like and a prejunctional 5-HT3 receptor mediating contraction of rat isolated jejunum. Eur. J. Pharmacol. 1996;312:215–225. doi: 10.1016/0014-2999(96)00456-6. [DOI] [PubMed] [Google Scholar]
- MONSMA F.J., SHEN Y., WARD R.P., HAMBLIN M.W., SIBLEY D.R. Cloning and expression of a novel serotonin receptor with high affinity for tricyclin psychotropic drugs. Mol. Pharmacol. 1993;43:320–327. [PubMed] [Google Scholar]
- NAMBA T., SUGIMOTO Y., NEGISHI M., IRIE A., USHIKUBI F., KAZIZUKA A., ITO I., ICHIKAWA A., NARUMIYA S. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- PAZOS A., HOYER D., PALACIOS J.M. Mesulergine, a selective serotonin-2 ligand in the rat cortex, does not label these receptors in porcine and human cortex: evidence for species differences in brain serotonin-2 receptors. Eur. J. Pharmacol. 1985;106:531–538. doi: 10.1016/0014-2999(84)90056-6. [DOI] [PubMed] [Google Scholar]
- PINKUS L.M., SARBIN N.S., GORDON J.C., MUNSON H.R., JR Antagonism of [3H]zacopride binding to 5-HT3 recognition sites by its (R) and (S) enantiomers. Eur. J. Pharmacol. 1990;179:231–235. doi: 10.1016/0014-2999(90)90425-6. [DOI] [PubMed] [Google Scholar]
- PLASSAT J.L., AMLAIKY N., HEN R. Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase. Mol. Pharmacol. 1993;44:229–236. [PubMed] [Google Scholar]
- RINNE U.K.New ergot derivatives in the treatment of Parkinson's disease Lisuride and other dopamine agonists 1983New York: Raven Press; 431–442.(eds.). Caine, D.B. et al. pp [Google Scholar]
- ROTH B.L., CRAIGO S.C., CHOUDHARY M.S., ULUER A., MONSMA F.J., SHEN Y., MELTZER H.Y., SIBLEY D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994;268:1403–1410. [PubMed] [Google Scholar]
- RUAT M., TRAIFFORT E., LEURS R., TARDIVEL-LACOMBE J., DIAZ J., ARRANG J.-M., SCHWARTZ J.-C. Molecular cloning, characterization, and localization of a high affinity serotonin receptor (5-HT7) activating cyclic AMP formation. Proc. Natl. Acad. Sci. USA. 1993;90:8547–8551. doi: 10.1073/pnas.90.18.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN Y., MONSMA F.J., Jr, METCALF M.A., JOSE P.A., HAMBLIN M.W., SIBLEY D.R. Molecular cloning and expression of a 5-hydroxytryptamine7 serotonin receptor subtype. J. Biol. Chem. 1993;268:18200–18204. [PubMed] [Google Scholar]
- SHENKER A., MAAYANI S., WEINSTEIN H., GREEN J.P. Pharmacological characterization of two 5-hydroxytryptamine receptors coupled to adenylate cyclase in guinea-pig hippocampal membranes. Mol. Pharmacol. 1987;31:357–367. [PubMed] [Google Scholar]
- SLEIGHT A.J., BOESS F.G., BOURSON A., SIBLEY D.R., MONSMA F.J., JR 5-HT6 and 5-HT7 serotonin receptors: Molecular biology and pharmacology. Neurotransmissions. 1995b;11:1–5. [Google Scholar]
- SLEIGHT A.J., CAROLO C., PETIT N., ZWINGELSTEIN C., BOURSON A. Identification of 5-Hydroxytryptamine7 receptor binding sites in rat hypothalamus: sensitivity of chronic antidepressant treatment. Mol. Pharmacol. 1995a;47:99–103. [PubMed] [Google Scholar]
- SUMNER M.J., FENIUK W., HUMPHREY P.P.A. Further characterisation of the 5-HT receptor mediating vascular relaxation and elevation of cyclic AMP in porcine isolated vena cava. Br. J. Pharmacol. 1989;97:292–300. doi: 10.1111/j.1476-5381.1989.tb11953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUNAHARA R.K., DESSAUER C.W., GILMAN A.G. Complexity and diversity of mammalian adenylyl cyclases. Ann. Rev. Pharmacol. Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- TO Z.P., BONHAUS D.W., EGLEN R.M., JAKEMAN L.B. Characterization and distribution of putative 5-ht7 receptors in guinea-pig brain. Br. J. Pharmacol. 1995;115:107–116. doi: 10.1111/j.1476-5381.1995.tb16327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSOU A.P., KOSAKA A., BACH C., ZUPPAN P., YEE C., TOM L., ALVAREZ R., RAMSEY S., BONHAUS D.W., STEFANICH E., JAKEMAN L., EGLEN R.M., CHAN H.W. Cloning and expression of a 5-hydroxytryptamine7 receptor positively coupled to adenylyl cyclase. J. Neurochem. 1994;63:456–464. doi: 10.1046/j.1471-4159.1994.63020456.x. [DOI] [PubMed] [Google Scholar]
- VANETTI M., VOGT G., HOLLT V. The two isoforms of the mouse somatostatin receptor (mSSTR2A and mSSTR2B) differ in coupling efficiency to adenylate cyclase and in agonist-induced receptor desensitisation. FEBS Lett. 1993;331:260–266. doi: 10.1016/0014-5793(93)80349-y. [DOI] [PubMed] [Google Scholar]
- WAYMAN G.A., IMPEY S., WU Z., KINDSVOGEL W., PRICHARD L., STORM D.R. Synergistic action of the type 1 adenylyl cyclase by Ca2+ and Gs-coupled receptors in vivo. J. Biol. Chem. 1994;269:25400–25405. [PubMed] [Google Scholar]