

Abstract
The cardiac depressant actions of TNF were investigated in the isolated perfused rat heart under constant flow (10 ml min−1) and constant pressure (70 mmHg) conditions, using a recirculating (50 ml) mode of perfusion.
Under constant flow conditions TNF (20 ng ml−1) caused an early (<25 min) decrease in left ventricular developed pressure (LVDP), which was maintained for 90 min (LVDP after 90 min: control vs TNF; 110±4 vs 82±10 mmHg, P<0.01).
The depression in cardiac function seen with TNF under constant flow conditions, was blocked by the ceramidase inhibitor N-oleoylethanolamine (NOE), 1 μM, (LVDP after 90 min: TNF vs TNF with NOE; 82±10 vs 111±5 mmHg, P<0.05).
In hearts perfused at constant pressure, TNF caused a decrease in coronary flow rate (change in flow 20 min after TNF: control vs TNF; −3.0±0.9 vs −8.7±1.2 ml min−1, P<0.01). This was paralleled by a negative inotropic effect (change in LVDP 20 min after TNF: control vs TNF; −17±7 vs −46±6 mmHg, P<0.01). The decline in function was more rapid and more severe than that seen under conditions of constant flow.
These data indicate that cardiac function can be disrupted by TNF on two levels, firstly via a direct, ceramidase dependant negative inotropic effect, and secondly via an indirect coronary vasoconstriction.
Keywords: TNFα, cardiac contractility, N-oleoylethanolamine, sphingosine, sphingomyelinase, coronary circulation, Starling curve
Introduction
Tumour necrosis factor-α (TNF) is an essential mediator of the profound cardiovascular changes observed during the shocked-state associated with bacterial sepsis, and its experimental counterpart, endotoxic shock (Tracey et al., 1986; Mathison et al., 1988). Indeed, immunization of mice against TNF can protect against a lethal injection of endotoxin (Beutler et al., 1985). The negative inotropic action observed during septic shock is mediated by circulating substances capable of depressing myocyte performance in vitro (Parrillo et al., 1985). Monoclonal TNF antibodies can protect against both the hypotension and the decline in cardiac output observed upon live Escherichia coli administration to baboons (Tracey et al., 1987), and TNF administration to dogs has been shown to have negative inotropic actions (Pagani et al., 1992; Murray & Freeman, 1996). High circulating levels of TNF have also been associated with a variety of cardiac diseased states which are independent of bacterial infections (Levine et al., 1990; Latini et al., 1994; Vaddi et al., 1994; Arbustini et al., 1991; Smith & Allen, 1992) while overexpression of TNF in the murine heart leads to dilated cardiomyopathy associated with ventricular hypertrophy (Kubota et al., 1997).
TNF-induced cardiac depression has been shown to have at least two phases. There appears to be an immediate depression of contractility as well as a later phase of contractile dysfunction. Cytokines, either alone or in combination, have the ability to cause upregulation of inducible nitric oxide synthase (iNOS) in cardiac myocytes (Schulz et al., 1992; Balligand et al., 1994; Pinsky et al., 1995), and nitric oxide from iNOS can subsequently adversely affect cardiac function. This altered cardiac function has been observed both as a direct decrease in cardiac performance (Schulz et al., 1995), and as decreased responsiveness of cardiac myocytes to β-adrenergic stimulation (Balligand et al., 1993). By definition iNOS is not constituatively expressed, and the protein synthesis required for its expression can take several hours. Another delayed action of TNF in the heart may involve the release of endothelins, whereby prior treatment of rats with lipopolysaccharide or TNF has been shown to cause the release of endothelin which results in a subsequent coronary constriction (Hohlfeld et al., 1995; Klemm et al., 1995).
In contrast to the delayed cardiac depressant effects of TNF described above, an immediate negative inotropic action has been observed within minutes of treatment, this has proven more difficult to characterize. The acute negative inotropic action of TNF has been shown to be sensitive to nitric oxide synthase inhibitors (Finkel et al., 1992). Another report presented evidence to suggest that the immediate depression of function seen with TNF was via a nitric oxide-mediated decrease in myofilament calcium responsiveness (Goldhaber et al., 1996). However, other authors have shown TNF-induced alterations in myocyte calcium handling and depressed myocyte calcium transients which are independent of nitric oxide (Yokoyama et al., 1993). The same group have recently shown the potential involvement of sphingosine in TNF-induced changes in calcium homeostasis, and subsequent negative inotropic action (Oral et al., 1997). Another recent report from the same group shows that the involvement of sphingosine in TNF-mediated cardiac depression is also evident in vivo, and is accompanied by a ventricular remodeling characterized as left ventricular dilation (Bozkurt et al., 1998). In addition to this, short term TNF treatment has been shown to inhibit L-type calcium currents in cardiac myocytes, this was also accompanied by a decrease in calcium transients (Krown et al., 1995). Using a Langendorff perfused heart preparation under constant flow conditions we have recently observed another early action of TNF in the heart. This is an immediate coronary vasoconstrictor action which could also contribute to the cardiac depressant action of TNF (Edmunds & Woodward, 1998).
The sphingomyelinase (SMase) pathway has been recognized as a major signalling pathway of TNF (Kim et al., 1991; Schutze et al., 1992; Dressler et al., 1992; Yang et al., 1993), and this pathway has recently been demonstrated in cardiac myocytes (Oral et al., 1997). Sphingomyelinase can be activated by TNF to cause the breakdown of the membrane bound phospholipid, sphingomyelin to its metabolite ceramide, which can be subsequently converted to sphingosine by ceramidase. Both ceramide and sphingosine have potential second messenger functions (Kolesnick, 1991; Hannun, 1994; Merrill et al., 1997). Sphingosine can depress cardiac function by decreasing calcium-induced calcium release from the sarcoplasmic reticulum (Webster et al., 1994), as well as by depressing the L-type calcium current (McDonough et al., 1994). N-oleoylethanolamine (NOE) is a relatively specific inhibitor of the ceramidase enzyme (Sugita et al., 1975), and is a compound which we have made use of in our studies.
In a previous report we have described a coronary constrictor action of TNF in the isolated perfused rat heart (Edmunds & Woodward, 1998). This appears to be mediated through an interaction between sphingosine and thromboxane A2. In the present paper we provide evidence to show that the immediate negative inotropic action of TNF, observed in the isolated perfused rat heart also involves the sphingosine pathway. We also show that in a model where coronary flow is allowed to vary with changes in coronary tone, the vasoconstriction observed in our previous study can act in synergy with the direct negative inotropic actions of TNF to cause a further decrease in cardiac function.
Methods
Throughout this study male Wistar rats, 280–310 g, have been used. Animals were anaesthetized by an interperitoneal injection of sodium pentabarbitone (200 mg kg−1) and then killed by cervical dislocation. The heart of each animal was isolated and perfused, via the aorta, according to the Langendorff technique under both constant flow and constant pressure conditions, with prefiltered, oxygenated (02 95%, CO2 5%) Krebs-Henseleit solution of the following composition (mM): NaCl 118, NaHCO3 25, KCl 4.7, KH2PO4 1.2, MgSO4.7H20 1.2, CaCl2.6H20 1.2 and D-glucose 11.6, pH 7.4 and a temperature of 37°C. A fluid filled clingfilm balloon was placed in the left ventricle, and left ventricular developed pressure (LVDP) was used as a measure of cardiac contractility. Left ventricular end diastolic pressure (LVEDP) was set between 5 and 10 mmHg, with the use of a 1 ml syringe connected to a micrometer head, allowing fine adjustment of the intraventricular balloon volume. Hearts were allowed to beat spontaneously throughout the experimental procedure. All hearts were perfused in a non-recirculating system for 30 min, before being switched to a recirculating system, with a total volume of 50 ml, for the remainder of the experiment, which lasted 90 min. The reason for recirculating the Krebs buffer was to reduce the amount of TNF used during each experiment, as well as to allow the build up of possible metabolites released by TNF. The recirculating perfusate was constantly gassed with 95% O2, 5% CO2. Starling curves were performed after 15, 45 and 75 min, whereby, the intraventricular balloon was emptied then slowly filled, in 0.03 ml aliquots every 30 s, to see if TNF altered the Frank-Starling response. Hearts were rejected from this study if LVDP failed to reach 100 mmHg.
For constant flow studies, hearts were perfused with a flow rate of 10 ml min−1. Coronary perfusion pressure (CPP) was recorded by a pressure transducer, and used as an index of coronary tone. In another set of experiments hearts were initially perfused under constant flow conditions (10 ml min−1) before being switched to a constant head of pressure of 70 mmHg. In this situation coronary flow was measured using a T 206 Transonic Systems Inc. flow probe attached to the aortic canula and represented the volume of Krebs buffer perfused into each heart with time.
Recombinant human TNF, 20 ng ml−1 (stock solution: 100 μg ml−1 in phosphate buffered saline with 0.1% bovine serum albumin) was added to the perfusate at 25 min, 5 min prior to recirculation. In preliminary experiments a range of concentrations, up to 100 ng ml−1, were investigated, and 20 ng ml−1 found to be optimum for cardiac depression (results not shown), this was a similar concentration to that used in other in vitro studies showing TNF-induced cardiac depression (Schulz et al., 1995). The enzyme sphingomyelinase, (SMase, from Bacillus cereus) was added exogenously in one set of experiments to cause the breakdown of the phospholipid sphingomyelin. When added to the perfuate at 25 min, i.e. 5 min before recirculation, it showed a concentration dependant (concentrations ranging from 0.001–0.01 u l−1 negative inotropic action (results not shown). In subsequent experiments a submaximal concentration of SMase, 0.003 u l−1, was added 5 min prior to recirculation.
All antagonists were added directly to the Krebs buffer after 5 min, to allow 20 min for equilibration before TNF addition. The ceramidase inhibitor NOE (1 μM, dissolved in ethanol) was used to block sphingosine production from the SMase pathway (Oral et al., 1997). The non-specific nitric oxide synthase inhibitor, nitro-L-arginine (100 μM, dissolved in saline) was used to block any nitric oxide release. The cylo-oxygenase inhibitor, indomethacin (10 μM, stock solution dissolved in equimolar Na2CO3) was added to block prostanoid accumulation from TNF-induced AA release. Bosentan (3 μM, dissolved in saline) a combined endothelin A and B receptor antagonist (Clozel et al., 1994) was used to block the actions of any TNF-induced endothelin release. Control experiments were conducted with all of the above agents in the absence of TNF. Control experiments were also conducted with vehicles for each of the drugs used, none of these altered cardiac function (data not shown).
A series of experiments was conducted to observe the actions of endothelin-1, 3 pM-3 nM, in hearts perfused under a constant head of pressure, in order to mimic the coronary constriction previously seen by us under constant flow conditions with TNF (Edmunds & Woodward, 1998), with the objective of demonstrating the dependency of LVDP on coronary flow.
In some of these constant pressure experiments, and despite having the heart surrounded by a heated water jacket, coronary flow decreased to such an extent that there was a fall in cardiac temperature, as measured by a temperature probe inserted into the right ventricle. For this reason some ex-periments were performed in order to investigate the effect of drop in temperature from 37–33°C on cardiac contractility.
Statistics
All results are expressed as mean±standard error of the mean. Statistical analysis was carried out, where appropriate, using one-way ANOVA coupled to either Dunett's test or Tukey's multiple comparison test. For non-parametric data a Mann-Whitney U test was performed. Analysis was completed by the use of a computer programme (Minitab 10 for Windows).
Materials
Bosentan was also a gift from Dr A.G. Roach (Rhone-Poulenc Rorer, Dagenham, U.K.). rhTNFα was a kind gift from Bayer (Slough, U.K.). All of the other drugs used in this study were purchased from Sigma Chemical Co. (Poole, Dorset, U.K.).
Results
Actions of TNF in the isolated rat heart perfused under constant flow conditions
It can be seen from the typical experimental trace shown in Figure 1a that in control hearts left ventricular developed pressure (LVDP) decreased only slightly throughout the 90 min perfusion period. Recirculation after 30 min had no direct effect on LVDP (LVDP before recirculation vs after recirculation: 127±3 mmHg vs 120±5 mmHg, P=NS, n=13). Addition of TNF, 20 ng ml−1, into the perfusate resulted in a depression in cardiac function which was evident within a few minutes of TNF addition (Figures 1b, 2 and 6). This depression became significant 25 min after TNF addition, and was evident for the remainder of the protocol (LVDP after 90 min: control vs TNF; 110±4 mmHg vs 82±10 mmHg, P<0.05, n=10 for TNF). TNF had no chronotropic effect, (heart rate after 90 min: control vs TNF; 285±11 beats per minute (b.p.m.) vs 288±12 b.p.m. P=NS).
Figure 1.
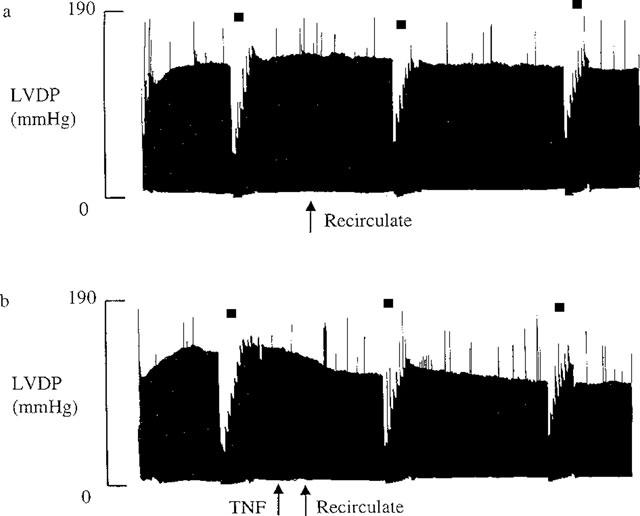
Experimental traces showing LVDP throughout the experiment. These traces are typical for (a) n=13 control hearts and (b) n=10 TNF, 20 ng ml−1 treated hearts. Starling curves were performed as indicated by *.
Figure 2.
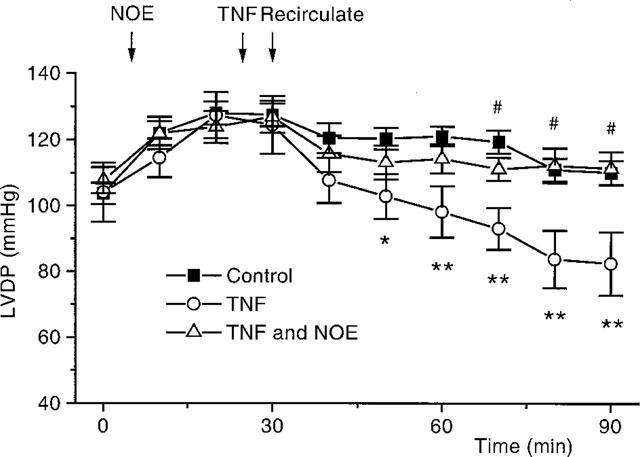
A graph to show changes in LVDP, in hearts, which occur with time, for control (n=13) TNF treated, 20 ng ml−1 (n=10) and TNF in the presence of NOE, 1 μM (n=9). *P<0.05, **P<0.01, TNF vs control. #P<0.05, TNF with NOE vs TNF alone.
Figure 6.
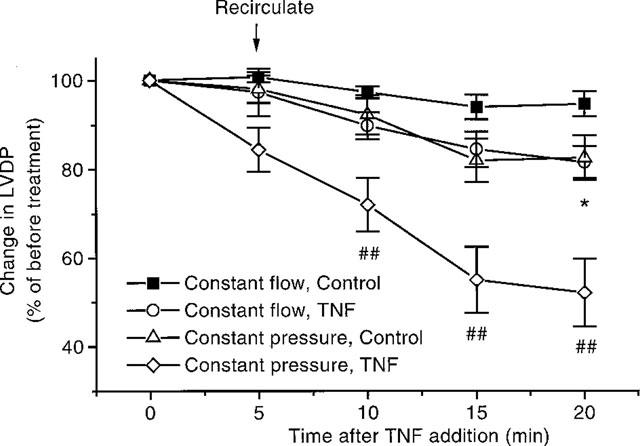
Effects of TNF, 20 ng ml−1, in hearts perfused under constant pressure and constant flow conditions (n=6–7). *P<0.05, TNF vs control when perfused under constant flow conditions. *P<0.05, TNF vs control when perfused under constant flow conditions. P<0.01. TNF under constant flow conditions vs TNF under constant pressure conditions. Where initial LVDP at 0 min: 122±4 mmHg; 126±6 mmHg; 101±6 mmHg and 106±3 mmHg for constant flow control and TNF treated heart and constant pressure control and TNF treated hearts respectively.
Actions of SMase in the isolated rat heart perfused under constant flow conditions
When added 5 min prior to recirculation SMase caused a negative inotropic action (Figure 3). This depression in function was seen immediately upon addition of SMase and became significant after 15 min. SMase also caused coronary constriction (increase in CPP 20 min after SMase: control, n=7, vs SMase, n=8; 13±5 mmHg vs 75±20 mmHg, P<0.01). SMase did not alter heart rate (heart rate after 90 min: control vs SMase; 285±11 b.p.m. vs 273±25 b.p.m., P=NS).
Figure 3.
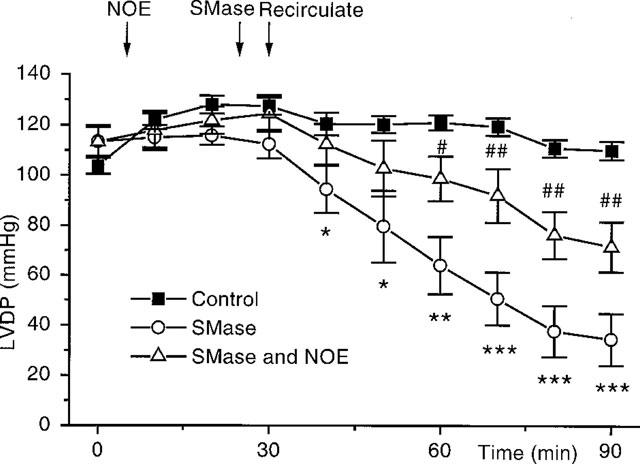
A graph to show the action of sphingomyelinase (SMase), alone and in combination with NOE, on the contractile parameters of the perfused heart. Control hearts (n=13), SMase, 0.003 u l−1, (n=7). SMase, 0.003 u l−1 and NOE, 1 μM, (n=8). *P<0.05, **P<0.01, ***P<0.001 for SMase vs control. #P<0.05, ##P<0.01, for SMase with NOE vs SMase alone.
Effects of antagonists and inhibitors on changes in contractility caused by TNF and SMase under constant flow conditions
N-oleoylethanolamine (NOE)
Inclusion of the ceramidase inhibitor NOE, 1 μM, into the perfusate 20 min before addition of TNF completely blocked the negative inotropic actions of the cytokine (Figure 2), without altering basal contractility (LVDP before NOE vs after NOE: 110±4 mmHg vs 109±4 mmHg, P=NS) or heart rate (heart rate before NOE vs after NOE: 267±10 b.p.m. vs 263±9 b.p.m., P=NS). NOE also caused a significant attenuation of the depression in function seen upon addition of SMase (Figure 3).
Nitro-L-arginine
The nitric oxide synthase inhibitor, nitro-L-arginine, 100 μM, caused a rapid and sustained increase in coronary perfusion pressure (CPP before nitro-L-arginine vs 20 min after nitro-L-arginine: 56±2 mmHg vs 126±9 mmHg, P<0.001). However, Figure 4 shows that nitric oxide synthase inhibition did not ameliorate the decreased contractility seen with TNF. Heart rate was not altered by nitro-L-arginine (heart rate before nitro-L-arginine vs after nitro-L-arginine: 289±12 b.p.m. vs 290±12 b.p.m., P=NS).
Figure 4.
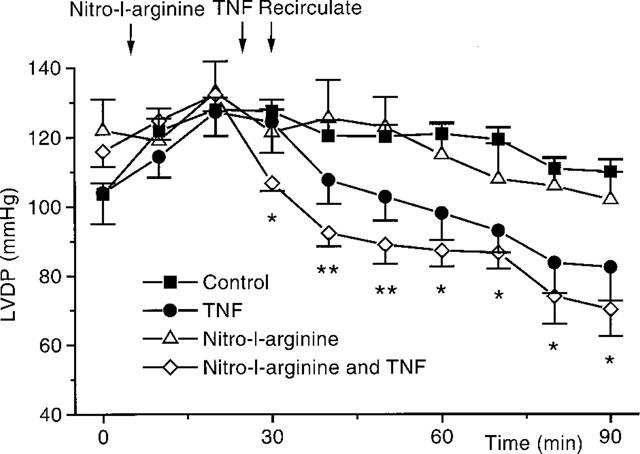
This graph shows the actions of nitro-L-arginine, 100 μM, on the depression in LVDP seen with TNF (n=8). Control hearts (n=13), TNF (n=10), and control hearts in the presence of nitro-L-arginine (n=4) are all shown. *P<0.05, **P<0.01, for TNF with nitro-L-arginine vs nitro-L-arginine alone.
Indomethacin
We have already reported that indomethacin was able to block the increase in CPP seen with TNF (Edmunds & Woodward, 1998). However, indomethacin did not alter the cardiac depression seen with TNF (LVDP after 90 min: indomethacin vs indomethacin and TNF; 101±6 mmHg vs 66±6 mmHg, P<0.01). Indomethacin had no effect on basal parameters of LVDP (LVDP before indomethacin vs after indomethacin: 101±4 mmHg vs 105±4 mmHg, P=NS) or heart rate (heart rate before indomethacin vs after indomethacin: 263±4 b.p.m. vs 278±13 b.p.m., P=NS).
Bosentan
Addition of the ETA-ETB-receptor antagonist bosentan (3 μM) to the perfusate did not block the TNF-induced alterations in cardiac function (LVDP after 90 min: bosentan vs bosentan and TNF; 103±6 mmHg vs 73±6 mmHg, P=0.01). Bosentan also did not alter the basal LVDP (LVDP before bosentan vs after bosentan: 104±2 mmHg vs 106±2 mmHg, P=NS), heart rate (heart rate before bosentan vs after bosentan: 271±9 b.p.m. vs 271±6 b.p.m., P=NS) or CPP (CPP before bosentan vs after bosentan: 50±2 mmHg vs 54±3 mmHg, P=NS).
Actions of TNF in the isolated rat heart perfused under a constant head of pressure
We have reported earlier that TNF causes coronary constriction under constant flow conditions (Edmunds & Woodward, 1998). Under constant pressure conditions this constriction would be expected to adversely affect cardiac contractility. A set of experiments were completed, where hearts were perfused under a constant head of pressure. Upon recirculation in control hearts there was a slight decrease in coronary flow, which continued throughout the experiment (Figure 5). Addition of TNF caused a more pronounced decrease in coronary flow (Figure 5). The LVDP in hearts perfused under a constant head of pressure follow the same pattern observed in hearts perfused under constant flow conditions. Namely there was a decline in LVDP after recirculation (Figure 6). However from Figure 6 it is clear that TNF caused a more pronounced decline in cardiac function under constant pressure conditions than with constant flow perfusion. Prior addition of NOE blocked both the decrease in coronary flow (Figure 5), as well as inhibiting the depression in cardiac function observed with TNF (Figure 7).
Figure 5.
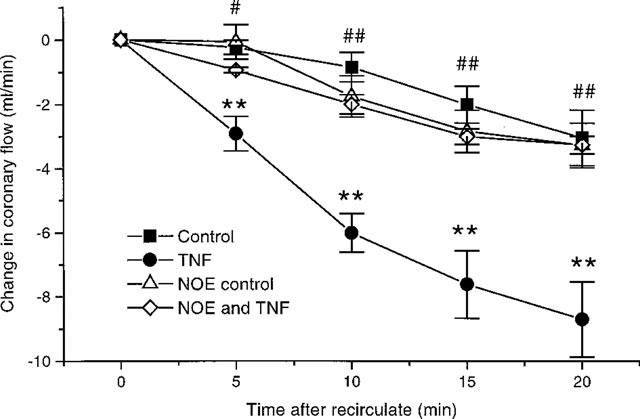
Effects of TNF (20 ng ml−1) and NOE (1 μM), alone and in combination on coronary flow in hearts perfused at constant pressure (n=6–7). **P<0.01 TNF vs control, #P<0.05, ##P<0.01 NOE alone vs TNF with NOE. Initial coronary flow at 0 min: 12.4±0.6 ml min−1; 11±1.2 ml min−1; 8.9±1 ml min−1 and 10.3±2.4 ml min−1 for control hearts, TNF treated, NOE treated and NOE with TNF treated hearts respectively.
Figure 7.
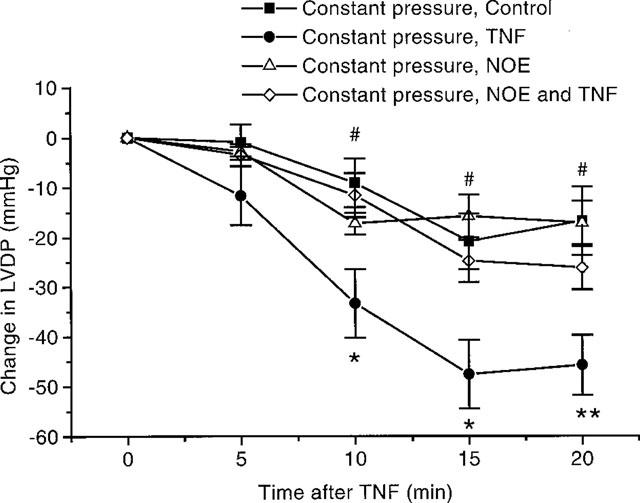
Effects of NOE on the depression in cardiac function seen with TNF, 20 ng ml−1 under constant pressure conditions. Shown as a change in LVDP after TNF administration (n=6–7). *P<0.05, **P<0.01 TNF vs control. #P<0.05 NOE vs TNF with NOE. Where LVDP at 0 min for NOE treated controls and NOE with TNF treated animals: 107±6 mmHg and 100±5 mmHg respectively.
Actions of endothelin-1 in hearts perfused under a constant head of pressure
Endothelin-1, when perfused through hearts under a constant head of pressure, showed a concentration dependant decrease in coronary flow, indicative of coronary constriction. This decrease in coronary flow was accompanied by a concentration dependant depression in cardiac function. At 1 nM, endothelin-1 caused a decrease in coronary flow from 9.5±0.5 ml min−1 to 2.5±0.7 ml min−1 (P<0.001, n=5). At the same concentration of endothelin-1 LVDP was depressed from 114±9 mmHg to 56±14 mmHg (P<0.01, n=5).
Effects of decreased temperature on cardiac function
A drop in temperature of the Krebs buffer from 37–33°C caused a decrease in heart rate, from 286±9 b.p.m. to 201±9 b.p.m. (n=4, P<0.05) and a concomitant increase in LVDP, from 123±2–133±5 mmHg (n=4, P<0.05).
Starling curves
Figure 8 shows a Starling curve at 75 min (50 min after TNF addition), where LVDP increases with balloon volume. It can be seen that there is a tendency for TNF to decrease LVDP for a given balloon volume, although this did not prove to be a significant effect. However in hearts perfused under a constant head of pressure there was a significant depression in the hearts ability to perform Starling curves when in the presence of TNF (Figure 9). Under these conditions, prior addition of NOE was able to block these actions of TNF (Figure 9).
Figure 8.
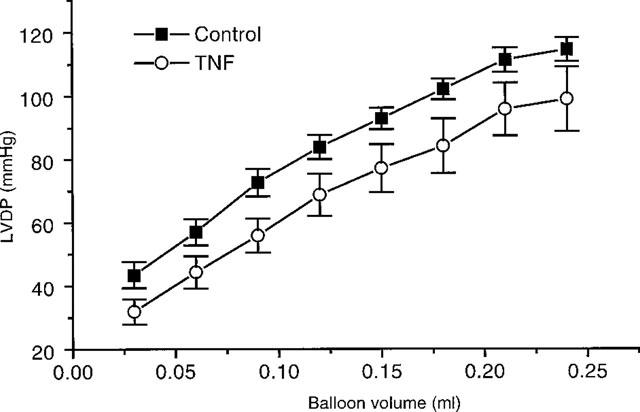
Starling curves for hearts perfused under constant flow conditions, 50 min after TNF addition. Control hearts (n=13), and TNF, 20 ng ml−1 (n=10).
Figure 9.
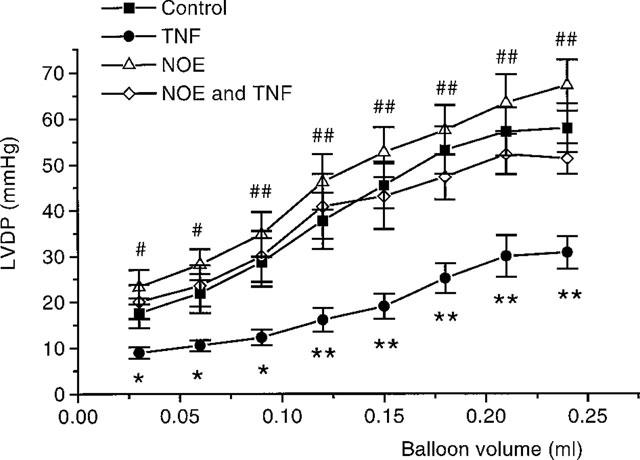
Starling curves in hearts perfused under a constant head of pressure, 50 min after TNF addition. Control hearts (n=7), TNF, 20 ng.ml−1, alone (n=7), NOE, 1 μM, alone (n=6) and TNF with NOE. *P<0.05, **P<0.01, TNF vs control. #P<0.05, ##P<0.01, NOE alone vs TNF with NOE.
Discussion
The aim of this study was to examine the mechanisms underlying the early cardiac depressant actions of TNF in the isolated perfused rat heart. We have shown an early negative inotropic action which was exacerbated by the coronary constrictor actions of TNF. The fact that these effects of TNF can be attenuated by the ceramidase inhibitor NOE implicates sphingosine in these actions. The findings of the study presented herein can be summarized as follows: isolated hearts perfused under constant flow conditions with a pathophysiologically relevant concentration of TNF (Redl et al., 1993), showed a depression in ventricular contractility, as indicated by the decrease in LVDP which was evident within minutes of TNF treatment and was sustained for the remainder of the experiment. There was also a tendency for TNF to cause a depression in the Starling curves under constant flow conditions, although this was not shown to be statistically significant. The direct depression in LVDP, under constant flow conditions, was not blocked by nitro-L-arginine, bosentan or indomethacin, which inhibit nitric oxide release, endothelin actions and arachidonic acid metabolism respectively (Gross et al., 1990; Clozel et al., 1994; Vane, 1971). Thus NO, endothelins and arachidonic acid metabolites are unlikely to be involved in the TNF-induced decrease in LVDP. The inability of indomethacin to block the direct depression in cardiac function is interesting as indomethacin did block the increase in coronary tone previously observed by us (Edmunds & Woodward, 1998). However, the negative inotropic actions of TNF were completely blocked by the ceramidase inhibitor NOE. This suggests that these actions of TNF are, at least in part, dependant on ceramidase converting ceramide to sphin-gosine, and is in agreement with the recent data of Oral et al. (1997) and Bozkurt et al. (1998). The fact that NOE also attenuated the negative inotropic actions of SMase provides indirect evidence that activation of the sphingomyelinase path-way can depress cardiac contractility in the whole heart, and that this effect can be blocked with an inhibitor of the enzyme responsible for the conversion of ceramide to sphingosine.
We have previously reported that TNF causes coronary constriction in the rat heart (Edmunds & Woodward, 1998). The fact that TNF depressed cardiac contractility under constant flow conditions indicates that in this situation TNF is having a direct cardiac depressant action independant of changes in coronary flow. However, it is possible for the coronary circulation to alter cardiac function under constant flow condition via the ‘garden hose' effect (Katz, 1992), which is something which we can not rule out. However, nitro-L-arginine also caused a vasoconstriction under constant flow conditions, but this was not accompanied by cardiac depression. This suggests that the garden hose effect is not important under these conditions and it provides evidence for a dissociation between cardiac depression and coronary constriction in this situation.
When hearts were perfused under a constant head of pressure, and coronary flow was allowed to vary, it was shown that TNF produced a greater, and earlier, depression of LVDP and of the Starling curves. This greater effect on contractility was coupled with a decrease in coronary flow to below 5 ml min−1. Therefore under these constant pressure conditions, the TNF-induced coronary constriction appears to be contributing to its enhanced depressant actions. The TNF-induced depression in LVDP could also be blocked by prior addition of NOE. These data extend our previous observations showing a role for the coronary constriction in the cardiac depressant effects of TNF. Both depressant actions, coronary (indirect) and cardiac (direct), appear to involve the sphingomyelin pathway, but the vasoconstriction was also dependant thromboxane A2 (Edmunds & Woodward, 1998), however the direct action of TNF on cardiac function is independent of arachidonic acid metabolism as it was not inhibited by indomethacin. Therefore there appears to be a link between the sphingosine and cyclo-oxygenase pathways in the TNF-mediated coronary constriction, but the direct cardiac depressant actions of TNF are dependant on the conversion of ceramide of sphingosine, and do not involve the cyclo-oxygenase pathway.
The TNF-induced decrease in coronary flow observed under a constant head of pressure could be completely blocked by NOE. This constriction followed a very similar time course to that seen when hearts were perfused at a constant flow, the majority of the effect being within 10 min of TNF administration. It can be seen when comparing the graphs shown in Figures 6 and 7 that the earlier depression in LVDP seen in hearts perfused under a constant head of pressure paralleled this decrease in coronary flow.
The studies utilizing the vasoconstrictor peptide endothelin-1, also show that coronary constriction under constant pressure conditions has the ability to depress contractile function. This depression in cardiac contractility induced by endothelin-1 in the whole heart under constant pressure conditions is clearly outweighing the direct positive inotropic actions that endothelin-1 has on rat cardiac myocytes (Kramer et al., 1991), adding further weight to the argument that the TNF-induced coronary constriction can contribute to depressed cardiac function.
Whilst activation of the sphingomyelinase pathway by TNF has been known for some time (Dressler et al., 1992; Schutze et al., 1992), this has only recently been shown in cardiac tissue (Oral et al., 1997). These authors demonstrated that TNF-induced sphingosine accumulation was responsible for a negative inotropic action of TNF in isolated feline cardiac myocytes, our data supports this work and indicates that similar mechanisms exist in the rat heart.
The early depression in function was not dependent on nitric oxide because it was not altered by the nitric oxide synthase inhibitor nitro-L-arginine. This is in contrast to the observations of Finkel et al., 1992, but is in agreement with other reports showing that the early direct decrease in cardiac function produced by TNF was not prevented by nitric oxide synthase inhibition (Yokoyama et al., 1993; Oral et al., 1997; Bozkurt et al., 1998). Schulz et al. (1995) showed that TNF-induced and IL-1β-induced synthesis of iNOS was responsible for a late depression (>2 h) in function in the isolated perfused rat heart. In contrast to our experiments, these authors did not observe an early depression in cardiac contractility, in fact a small increase in cardiac work was seen, we have no explanation for this apparent discrepancy. To our knowledge, alterations in cardiac function involving iNOS take place over a longer time course than used in our experiments (Schulz et al., 1992; Szabo et al., 1993; Shindo et al., 1994; Balligand et al., 1994; Pinsky et al., 1995). Our results also show that the release of endothelin, which occurs after treatment of animals with lipopolysaccharide or TNF (Hohlfeld et al., 1995; Klemm et al., 1995), does not play a role in the early negative inotropic action of TNF. This is evidenced by the fact that the ETA/B receptor antagonist, bosentan, did not alter the TNF-induced decrease in LVDP.
The ceramidase inhibitor, NOE, which attenuated the depressant actions of TNF under constant flow and constant pressure conditions, disrupts signalling through sphingomyelin breakdown by inhibiting the conversion of ceramide to sphingosine (Sugita et al., 1975), an action which appears to be specific for the SMase pathway (Coroneos et al., 1995). This suggests that the depressant effect of TNF is due to increased sphingosine production. Sphingosine has been shown to have the ability to disrupt cardiac myocyte calcium handling, both by a decrease in calcium release from the sarcoplasmic reticulum (Sabbadini et al., 1992, Dettbarn et al., 1994; Webster et al., 1994), and also by a depression in the L-type calcium current (McDonough et al., 1994). We have shown that the addition of SMase also caused a decrease in cardiac function, however the site of action of this exogenous SMase was not known. The sphingomyelinase enzyme used has a molecular size of 25 kDa (Matsuyama et al., 1992), so it is likely that it will have access to the cardiac myocytes from the coronary circulation. A potential limitation of this study is that the site of action of this enzyme was not defined and it may have been on the endothelial cells or the smooth muscle cells of the coronary circulation and not directly on the cardiac myocytes. However, the cardiac depression seen with SMase was still attenuated by NOE, suggesting activation of the SMase pathway, and release of sphingosine is responsible for its cardiac depressant action. However, it should be noted that the source of the SMase used in this study was bacterial in origin, and hence it is possible that it contained trace amounts of endotoxin. Contamination with endotoxin, and hence subsequent activation of constitutive nitric oxide synthase by endotoxin, could possibly explain why NOE only partially blocked the SMase induced depression in cardiac function.
When perfusing under a constant head of pressure, the coronary constriction caused the flow rate to decrease, and despite the fact that the heart was surrounded by a water jacket at 37°C, a fall in temperature of the heart occurred. However in control experiment where hearts were perfused at 33°C, a temperature comparable to that seen when coronary flow was maximally decreased by TNF, heart rate decreased and LVDP actually increased. Therefore if this was to occur upon perfusion with TNF one would expect slight protection against the decrease in myocardial contractility, and this may have masked an even more severe depressant action of TNF.
In summary, we add further evidence in confirmation of the study by Oral et al. (1997), showing that a direct and early negative inotropic action of TNF in the isolated perfused rat heart is likely to be mediated via activation of SMase. Further to this, we have shown that the direct negative inotropic action of TNF can be accentuated by its coronary vasoconstrictor action.
Acknowledgments
This work has been funded by the British Heart Foundation, grant numbers: FS/95044 and PG/96173.
Abbreviations
- bpm
beats per minute
- iNOS
inducible nitric oxide synthase
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end-diastolic pressure
- NOE
N-oleoylethanolamine
- SMase
sphingomyelinase
- TNF
tumour necrosis factor-α
References
- ARBUSTINI E., GRASSO M., DIEGOLI M., BRAMERIO M., FOGLIEM A.S., ALBERTARIO M., MARTINELLI L., GARAZZI A., GOGGI G., CAMPANA C., VIGARO M. Expression of tumor necrosis factor in human acute cardiac rejection- An immunohistochemical and immunoblotting study. Am. J. Path. 1991;139:709–715. [PMC free article] [PubMed] [Google Scholar]
- BALLIGAND J.L., UNGUREANU D., KELLY R.A., KOBZIK L., PIMENTAL D., MICHEL T., SMITH T.W. Abnormal contractile function due to induction of nitric oxide synthesis in rat cardiac myocytes follows exposure to activated macrophage-conditioned medium. J. Clin. Invest. 1993;91:2314–2319. doi: 10.1172/JCI116461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALLIGAND J.L., UNGUREANU D., SIMMONS W.W., PIMENTAL D., MALINSKI T.A., KAPTURCZAK M., TAHA Z., LOWENSTEIN C.J., DAVIDOFF A.J., KELLY R.A., SMITH T.W., MICHEL T. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. J. Biol. Chem. 1994;44:27580–27588. [PubMed] [Google Scholar]
- BEUTLER B., MILSARK J., CERAMI A. Passive immunisation against cachectin / tumour necrosis factor from lethal effect of endotoxin. Science. 1985;229:869–871. [Google Scholar]
- BOZKURT B., KRIBBS S.B., CLUBB F.J., MICHAEL L.H., DIDENKO V.V., HORNSBY P.J., SETA Y., ORAL H., SPINALE F.G., MANN D.L. Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- CLOZEL M., BREU V., GRAY G.A., KALINA B., LOFFER B.M., BURRI K., CASSAL J.M., HIRTH G., MULLER M., NEIDHART W., RAMUZ H. Pharmacological characterisation of bosentan, a new potent orally active non-peptide endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 1994;270:228–235. [PubMed] [Google Scholar]
- CORONEOS E., MARTINEZ M., MCKENNA S., KESTER M. Differential regulation of sphingomyelinase and ceramidase activities by growth factors and cytokines. J. Biol. Chem. 1995;270:23305–23309. doi: 10.1074/jbc.270.40.23305. [DOI] [PubMed] [Google Scholar]
- DETTBARN C.A., BETTO R., SALVIATI G., PALADE P., JENKINS G.M., SABBADINI R.A. Modulation of cardiac sarcoplasmic reticulum ryanodine receptor by sphingosine. J. Mol. Cell Cardiol. 1994;26:229–242. doi: 10.1006/jmcc.1994.1026. [DOI] [PubMed] [Google Scholar]
- DRESSLER K.A., MATHIAS S., KOLESNICK R.N. Tumor Necrosis Factor-α activates the sphingomyelin signal transduction pathway in a cell free system. Science. 1992;255:1715–1718. doi: 10.1126/science.1313189. [DOI] [PubMed] [Google Scholar]
- EDMUNDS N.J., WOODWARD B. Effects of tumour necrosis factor-α on the coronary circulation of the rat isolated perfused heart: a potential role for thromboxane A2 and sphingosine. Br. J. Pharmacol. 1998;124:493–498. doi: 10.1038/sj.bjp.0701863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINKEL M.S., ODDIS C.V., JACOB T.D., WATKINS S.C., HATTLER B.G., SIMMONS R.L. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–389. doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- GOLDHABER J.I., KIM K.H., NATTERSON P.D., LAWRENCE T., YANG P., WEISS J.N. Effects of TNF-α on [Ca2+]I and contractility in isolated adult rabbit ventricular myocytes. Am. J. Physiol. 1996;271:H1449–H1455. doi: 10.1152/ajpheart.1996.271.4.H1449. [DOI] [PubMed] [Google Scholar]
- GROSS S.S., STUEHR D.J., AISAHA K., JAFFE E.A., LEVI R., GRIFFITH O.W. Macropharge and endothelial cell nitric oxide synthesis–cell type selective inhibition by NG–Aminoarginine, NG–nitroarginine and NG-met hylarginine. Biochem. Biophys. Res. Comm. 1990;170:96–103. doi: 10.1016/0006-291x(90)91245-n. [DOI] [PubMed] [Google Scholar]
- HANNUN Y.A. The sphingomyelin cycle and the second messenger function of ceramide. J. Biol. Chem. 1994;269:3125–3128. [PubMed] [Google Scholar]
- HOHLFELD T., KLEMM P., THIEMERMANN C., WARNER T.D., SCHROR K., VANE J.R. The contribution of tumour necrosis factor-α and endothelin-1 to the increase of coronary resistance in hearts from rats treated with endotoxin. Br. J. Pharmacol. 1995;116:3309–3315. doi: 10.1111/j.1476-5381.1995.tb15140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATZ A.M.Structure of the heart and cardiac muscle Physiology of the heart 1992New York: Raven Press; ed. Katz, A.M. pp. 613 [Google Scholar]
- KIM M.Y., LINARDIC C., OBEID L., HANNUN Y. Identification of sphingomyelin turnover as an effector mechanism for the actions of tumor necrosis factor α and γ-interferon. J. Biol. Chem. 1991;266:484–489. [PubMed] [Google Scholar]
- KLEMM P., WARNER T.D., HOHLFELD T., CORDER R., VANE J.R. Endothelin 1 mediates ex vivo coronary vasoconstriction caused by exogenous and endogenous cytokines. Proc. Natl. Acad. Sci. USA. 1995;92:2691–2695. doi: 10.1073/pnas.92.7.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLESNICK R.N. Sphingomyelin and derivatives as cellular signals. Prog. Lipid Res. 1991;30:1–38. doi: 10.1016/0163-7827(91)90005-p. [DOI] [PubMed] [Google Scholar]
- KRAMER B.K., SMITH T.W., KELLY R.A. Endothelin and increased contractility in adult-rat ventricular myocytes–role of intracellular alkalosis induced by activation of the protein kinase-C-dependant Na+ / H+ exchanger. Circ. Res. 1991;68:269–279. doi: 10.1161/01.res.68.1.269. [DOI] [PubMed] [Google Scholar]
- KROWN K.A., YASUI K., BROOKER M.J., DUBIN A.E., NGUYEN C., HARRIS G.L., MCDONOUGH P.M., GLEMBOTSKI C.C., PALADE P.T., SABBADINI R.A. TNFα receptor expression in rat cardiac myocytes: TNFα inhibition of L-type Ca2+ current and Ca2+ transients. FEBS Lett. 1995;376:24–30. doi: 10.1016/0014-5793(95)01238-5. [DOI] [PubMed] [Google Scholar]
- KUBOTA T., MCTIERNAN C.F., FRYE C.S., SLAWSON S.E., LEMSTER B.H., KORETSKY A.P., DEMETRIS A.J., FELDMAN A.M. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- LATINI R., BIANCHI M., CORREALE E., DINARELLO C.A., FRESCO C., MAGGIONI A.P., MENGOZZI M., ROMANO S., SHAPIRO L., SIRONI M., TOGNONI G., TURATO R., GHEZZI P. Cytokines in acute myocardial infarction: selective increase in circulating tumor necrosis factor, its soluble receptor, and interleukin-1 receptor antagonist. J. Cardiovasc. Pharmacol. 1994;23:1–6. [PubMed] [Google Scholar]
- LEVINE B., KALMAN J., MAYER L., FILLIT H.M., PACKER M. Elevated circulating levels of tumor necrosis factor in severe chronic heart disease. New England Journal of Medicine. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- MATHISON J.C., WOLFSON E., ULEVITCH R.J. Participation of tumor necrosis factor in the mediation of gram negative bacterial lipipolysaccharide-induced injury in rabbits. J. Clin. Invest. 1988;81:1925–1937. doi: 10.1172/JCI113540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUYAMA H., TOMITA M., TAGUCHI R., IKEZAWA H. Changes in the enzymatic-adsorbing activities of sphingomyelinase from bacillus-cereus by modification with a polyethylene-glycol derivative. Chemical and Pharmaceutical Bulletin. 1992;40:2478–2482. doi: 10.1248/cpb.40.2478. [DOI] [PubMed] [Google Scholar]
- MCDONOUGH P.M., YASUI K., BETTO R., SALVIATI G., GLEMBOTSKI C.C., PALADEM P.T., SABBADINI R.A. Coronary cardiac Ca2+ levels. Inhibitory actions of sphingosine on Ca2+ transients and L-type Ca2+ channel conductance. Circ. Res. 1994;75:981–989. doi: 10.1161/01.res.75.6.981. [DOI] [PubMed] [Google Scholar]
- MERRILL A.H., SCHMELZ E.-M., DILLEHAY D.L., SPIEGEL S., SHAYMAN J.A., SCHROEDER, RILEY R.T., VOSS K.A., WANG E. Sphingolipids-the enigmatic lipid class: biochemistry, physioloy, and pathophysiology. Toxicol. Appl. Pharmacol. 1997;142:208–225. doi: 10.1006/taap.1996.8029. [DOI] [PubMed] [Google Scholar]
- MURRAY D.R., FREEMAN G.L. Tumor necrosis factor-α induces a biphasic effect on myocardial contractility in concious dogs. Circ. Res. 1996;78:154–160. doi: 10.1161/01.res.78.1.154. [DOI] [PubMed] [Google Scholar]
- ORAL H., DORN G.W., III, MANN D.L. Sphingosine mediates the immediates negative inotropic effects of tumour necrosis factor-α in the adult mammalian cardiac myocyte. J. Biol. Chem. 1997;272:4836–4842. doi: 10.1074/jbc.272.8.4836. [DOI] [PubMed] [Google Scholar]
- PAGANI F.D., BAKER L.S., HIS C., KNOX M., FINK M.P., VISNER M.S. Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor-α in conscious dogs. J. Clin. Invest. 1992;90:389–398. doi: 10.1172/JCI115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARRILLO J.E., BURCH C., SHELHAMER J.H., PARKER M.M., NATANSON C., SCHUETTE W. A circulating myocardial depressant substance in humans with septic shock. J. Clin. Invest. 1985;76:1539–1553. doi: 10.1172/JCI112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINSKY D.J., CAI B., YANG X., RODRIGUEZ C., SCIACCA R.R., CANNON P.J. The lethal effects of cytokine-induced nitrc oxide on cardiac myocytes are blocked by nitric oxide synthase antagonism or transforming growth factor β. J. Clin. Invest. 1995;95:677–685. doi: 10.1172/JCI117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REDL H., SCHLAG G., BAHRAMI S., DAVIES J., WAAGE A., CESKA M., BUURMAN W.A., ADOLF G.The cytokine network in trauma and sepsis I: TNF and IL-8 Pathophysiology of shock, sepsis and organ failure 1993Berlin Heidelberg: Springer-Verlag; 468–490.ed. Schlag, G. & Redl, H. pp [Google Scholar]
- SABBADINI R.A., BETTO R., TERESI A., FACHECHI-CASSANO G., SALVIATI G. The effects of sphingosine on sarcoplasmic reticulum membrane calcium release. J. Biol. Chem. 1992;267:15475–15484. [PubMed] [Google Scholar]
- SCHULZ R., NAVA E., MONCADA S. Induction and potential biological relevence of a Ca2+-independant nitric oxide synthase in myocardium. Br. J. Pharmacol. 1992;105:575–580. doi: 10.1111/j.1476-5381.1992.tb09021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHULZ R., PANAS D.L., CATENA R., MONCADA S., OLLEY P.M., LOPASCHUK G.D. The role of nitric oxide in cardiodepression induced by interleukin-1β and tumour necrosis factor-α. Br. J. Pharmacol. 1995;114:27–34. doi: 10.1111/j.1476-5381.1995.tb14901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHUTZE S., POTTHOFF K., MACHLEIDT T., BERKOVIC D., WIEGMANN K., KRONKE M. TNF acitvates NF-κB by phosphatidylcholine-specific phospholipase C-induced ‘acidic' sphingomyelin breakdown. Cell. 1992;71:765–776. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- SHINDO T., IKEDA U., OHKAWA F., TAKAHASHI M., FUNAYAMA H., NISHINAGA M., KAWAHARA Y., YOKOYAMA M., KASAHARA T., SHIMADA K. Nitric oxide synthesis in rat cardiac myocytes and fibroblasts. Life Sciences. 1994;55:1101–1108. doi: 10.1016/0024-3205(94)00238-x. [DOI] [PubMed] [Google Scholar]
- SMITH S.C., ALLEN P.M. Neutralization of endogenous tumor necrosis factor ameliorates the severity of myosin-induced myocarditis. Circ. Res. 1992;70:856–863. doi: 10.1161/01.res.70.4.856. [DOI] [PubMed] [Google Scholar]
- SUGITA M., WILLIAMS M., DULANEY J.T., MOSER H.W. Ceramidase and ceramide synthesis in human kidney and cerebellum. Biochem. Biophys. Acta. 1975;398:125–131. doi: 10.1016/0005-2760(75)90176-9. [DOI] [PubMed] [Google Scholar]
- SZABO C., WU C.C., MITCHELL J.A., GROSS S.S., THIEMERMANN C., VANE J.R. Platelet-activating factor contributes to the induction of nitric oxide synthase by bacterial lipopolysaccharide. Circ. Res. 1993;73:991–999. doi: 10.1161/01.res.73.6.991. [DOI] [PubMed] [Google Scholar]
- TRACEY K.J., BEUTLER B., LOWRY S.F., MERRYWEATHER J., WOLPE S., MILSARK I.W., HARIRI R.J., FAHEY T.J., III, ZENTELLA A., ALBERT J.D., SHIRES G.T., CERAMI A. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- TRACEY K.J., FONG Y., HESSE D.G., MANOGUE K.R., LEE A.T., KUO G.C., LOWRY S.F., CERAMI A. Anti-cachectin / TNF monoclonal antibodies prevent septic shock during lathal bacteraemia. Nature. 1987;330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- VADDI K., NICOLINI F.A., MEHTA P., MEHTA J.L. Increased secretion of tumor necrosis factor-α and interferon-γ by mononuclear leukocytes in patients with ischaemic heart disease. Circ. 1994;90:694–699. doi: 10.1161/01.cir.90.2.694. [DOI] [PubMed] [Google Scholar]
- VANE J.R. Inhibition of prostaglandin synthesis as a mechanism of action for asprin-like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- WEBSTER R.J., SABBADINI R.A., DETBARN C.A., PAOLINI P.J. Sphingosine effects on the contractily behavior of skinned cardiac myocytes. J. Mol. Cell Cardiol. 1994;26:1273–1290. doi: 10.1006/jmcc.1994.1147. [DOI] [PubMed] [Google Scholar]
- YANG Z., COSTANZO M., GOLDE D.W., KOLESNICK R.N. Tumor necrosis factor activation of the sphingomyelin pathway signals nuclear factor κB translocation in intact HL-60 cells. J. Biol. Chem. 1993;268:20520–20523. [PubMed] [Google Scholar]
- YOKOYAMA T., VACA L., ROSSEN R.D., DURANTE W., HAZARIKA P., MANN D.L. Cellular basis for the negative inotropic effects of tumor necrosis factor-α in the adult mammalian heart. J. Clin. Invest. 1993;92:2303–2012. doi: 10.1172/JCI116834. [DOI] [PMC free article] [PubMed] [Google Scholar]