

Abstract
We have investigated the antagonist properties of 6 α-substituted phenylglycine analogues based on the structure of 4-carboxyphenylglycine (4-CPG) for group I metabotropic glutamate receptors (mGlu1α and mGlu5a) permanently expressed in CHO cells.
(S)-4-CPG and (S)-MCPG were the most selective mGlu1α receptor antagonists. Longer chain α-carbon substitutions resulted in a progressive loss of antagonist affinity at mGlu1α receptors but not at mGlu5a receptors. Thus mGlu1α receptor antagonists require small aliphatic groups at the α-position. α-cyclopropyl-4-CPG showed a tendency towards mGlu5a selectivity, suggesting that bulky groups at this position may favour mGlu5a receptor antagonism.
We demonstrate that the mGlu5a receptor displays agonist-dependent antagonism. L-glutamate-induced Ca2+ release in mGlu5a receptor expressing cells was more susceptible to antagonism by cyclic α-carbon derivatives than (S)-3,5-dihydroxyphenylglycine (DHPG)-induced Ca2+ release in the same cell line.
The data presented suggests that mGlu1α and mGlu5a receptors have different steric and/or conformational requirements for the binding of antagonists and different amino acids which could interact with agonists.
These phenylglycine analogues could provide leads for the development of subtype selective antagonists.
Keywords: Metabotropic glutamate receptor, mGluR, phenylglycine, excitory amino acid, CHO
Introduction
The mGlu family of glutamate receptors are G-protein-linked receptors currently comprising eight members. These are divided into three groups based on structural homology, pharmacology and signal transduction mechanisms when expressed in clonal cell lines (for review, see Pin & Duvoisin, 1995). Group I mGlu receptors (mGlu1 and mGlu5) are linked to phosphoinositide turnover and thus diacylglycerol production and Ca2+ mobilization (Tanabe et al., 1992; Abe et al., 1992). These receptors are specifically activated by (S)-3,5-dihydroxyphenylglycine ((S)-3,5-DHPG, (Ito et al., 1992; Schoepp et al., 1994)) and may play a role in long-term potentiation and long-term depression (e.g. Bashir et al., 1993; Bashir & Collingridge, 1994; Cohen & Abraham, 1996; Davis & Laroche, 1996; Fukuda et al., 1997; Manahan-Vaughan, 1997). (S)-3,5-DHPG has also been shown to potentiate NMDA induced depolarizations in the CA1 region of the hippocampus (Harvey & Collingridge, 1993; Fitzjohn et al., 1996) and to induce a novel form of long term depression in the same region (Palmer et al., 1997).
A number of pharmacological tools are available with which to distinguish groups of mGlu receptors and much effort has gone into the synthesis and analysis of phenylglycine derivatives as potential selective agonists and antagonists (Watkins & Collingridge, 1994; Roberts, 1995). (S)-4-carboxyphenylglycine ((S)-4-CPG) and many of it's derivatives display antagonist activity at group I mGlu receptors while (S)-α-methyl-4-carboxyphenylglycine ((S)-MCPG) is a well known antagonist at all groups of metabotropic glutamate receptors (Kemp et al., 1994; Bedingfield et al., 1995; Thomsen et al., 1994; Sekiyama et al., 1996). However, only a few compounds have been synthesized which discriminate between individual subtypes within a group (S)-4-CPG and (S)-MCPG have been shown to be more potent antagonists at mGlu1 than mGlu5 receptors cloned from both rat and human cDNA libraries (Brabet et al., 1995; Kingston et al., 1995) and (+)-2-methyl-4-carboxyphenylglycine was recently shown to be an antagonist at mGlu1 receptors (Clark et al., 1997). Such work has also lead to the discovery of (S)-4-carboxy-3-hydroxyphenylglycine ((S)-4C3HPG), a partial agonist selective for mGlu5 and an antagonist of mGlu1 receptors (Brabet et al., 1995) as well as (RS)-2-chloro-5-hydroxyphenylglycine ((RS)-CHPG), an mGlu5 selective agonist (Doherty et al., 1997).
Most of the phenylglycine derivatives described to date which have activity on the group I mGlu receptors have either hydroxyl or carboxyl substitutes on the phenyl ring. Relatively little work, however, has been carried out using α-substituted analogues (Bedingfield et al., 1995; Sekiyama et al., 1996). These studies have suggested that alkylation of the α-carbon with a methyl or an ethyl group increases the affinity of (S)-4-CPG for mGlu1 receptors. However, with the exception of (S)-MCPG, no studies to date have been reported showing any discrimination between mGlu1 and mGlu5 receptors by such compounds. We have therefore tested a variety of α-substituted derivatives of 4-CPG for their antagonist effects on recombinant mGlu1α and mGlu5a receptors permanently expressed in CHO cells (Figure 1). We report that short-chain alkyl substituents maintain the mGlu1 receptor selectivity of (S)-4-CPG, but that this selectivity is lost when bulkier α-substituents are used. In addition, we report that, in common with the rat mGlu1α receptor (Brabet et al., 1995), the mGlu5a receptor displays agonist-dependent antagonism by some α-substituted phenylglycine analogues.
Figure 1.
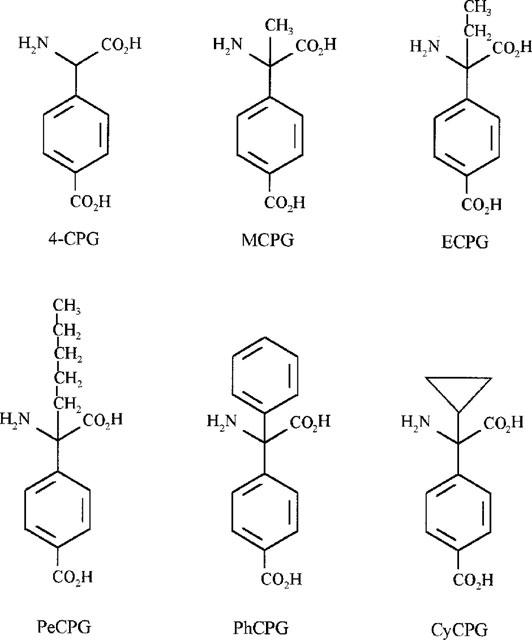
Structures of α-substituted phenylglycine derivatives. Analogues are 4-carboxyphenylglycine (4-CPG); α-methyl-4-CPG (MCPG); α-ethyl-4-CPG (ECPG); α-pentyl-4-CPG (PeCPG); α-phenyl-4-CPG (PhCPG), and α-cyclopropyl-4-CPG (CyCPG).
Methods
Synthesis of phenylglycine analogues
(RS)-α-Ethyl-4-carboxyphenylglycine ((RS)-ECPG) was synthesized by the previously reported method (Bedingfield et al., 1995). The α-cyclopropyl-, α-pentyl- and α-phenyl-analogues of 4-carboxyphenylglycine were synthesized in a similar manner. The new phenylglycine analogues had 1H and 13C n.m.r. spectra and elemental analytical data consistent with the proposed structure.
Cell culture
CHO cells stably expressing mGlu1α and mGlu5a receptors were kindly provided by Professor S. Nakanishi and were cultured in Dulbecco's Modified Eagle's Medium (DMEM, 4.5 g l−1 D-glucose) supplemented with 10% dialysed foetal bovine serum (FBS, 10,000 Mr cut-off, Sigma), glutamine (2 mM), 1% L-proline, penicillin (100 U ml−1) and streptomycin (100 μg ml−1). Cells were maintained at 37°C in a 5% CO2 humidified atmosphere. For imaging experiments, cells were plated onto glass coverslips (22 mm diameter, 1 thickness, 1–2×105 cells coverslip−1) and used within 48 h.
Ca2+ imaging
Cells were washed three times in Krebs buffer (in mM, NaCl 124, KCl 2.9, NaHCO3 25, NaH2PO4.H2O 1.4, MgSO4.7H2O 1, to which is added 4.5 g l−1 D-glucose and CaCl2 (2 mM) and loaded with 5 μM of the membrane permeable Ca2+ indicator Fluo3-AM made up in 1 mg ml−1 bovine serum albumin/Krebs at 37°C for 20 min. Cells were then washed three times in Krebs buffer and incubated for 30 min in a 5% CO2 atmosphere at 22°C in order to allow for de-esterification of the fluorophore. Cells were then viewed ona Bio-Rad MRC600 confocal microscope equipped with an argon ion laser using standard green filter sets and perfused continuously with Krebs buffer at a rate of approximately 2 ml min−1. Kalman integrations of five individual images were obtained every 10 s. Agonists (2 ml) were bath applied while antagonists were pre-incubated for 1 min prior to co-application with agonist. The fluorescence of between five and ten individual cells in each preparation was measured on a Macintosh computer using the public domain NIH Image program (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nihimage/) and expressed relative to baseline. The mean peak fluorescence was taken as a measure of mGlu receptor activation.
Data analysis
Dose response profiles were analysed by non-linear least-squares analysis and EC50 values derived using SigmaPlot (Jandel Scientific). KB values were calculated using the equation KB=L/r−1 where L is the antagonist concentration and r is the ratio of EC50 values in the presence and absence of antagonist. Unless stated otherwise, results are given as means±s.e.mean (n). Statistical analysis was by unpaired t-test.
Materials
(S)-4-CPG and (S)-MCPG were obtained from Tocris-Cookson (Bristol, U.K.), DMEM and penicillin/streptomycin solution from GibcoBRL (Paisley, U.K.), and dialysed FBS, L-proline, L-glutamine (cell culture grade) and Fluo3-AM from Sigma Chemical Co. (Poole, U.K.). All other chemicals used were of analytical grade.
Results
Effect of a single concentration of antagonist on L-glutamate-mediated Ca2+ release
The ability of 1 mM of each phenylglycine analogue to antagonize L-glutamate (10 μM)-evoked Ca2+ release in cells expressing mGlu1α and mGlu5a receptors was analysed. In each experiment, control responses to L-glutamate at 1, 10 and 100 μM were carried out to ensure that the concentration of agonist used was within the linear phase of the dose response profile. The EC50 for L-glutamate-evoked Ca2+ release in both these cell lines using this methodology has previously been shown to be 8 μM (Doherty et al., 1997). The results are shown in Figure 2a,b. (S)-4-CPG was the most selective compound tested, depressing responses to 0.9±0.4 (5) and 73±10% (4) of control (P<0.01) in mGlu1α and mGlu5a receptor-expressing cells followed by (RS)-ECPG, which reduced responses to 2±1 (4) and 34±8% (3) of control, respectively (P<0.01). (S)-MCPG was an effective antagonist on both cell lines, completely blocking the responses in mGlu1α receptor-expressing cells in all experiments (n=4) and reducing responses to 6±3% (4) of control in cells expressing the mGlu5a receptor. Conversely, (RS)-CyCPG was a more effective antagonist in cells expressing mGlu5a receptors than in mGlu1α receptor-expressing cells, depressing responses to 7±4 (3) and 20±7% (3) of control, respectively, although this did not reach statistical significance.
Figure 2 .
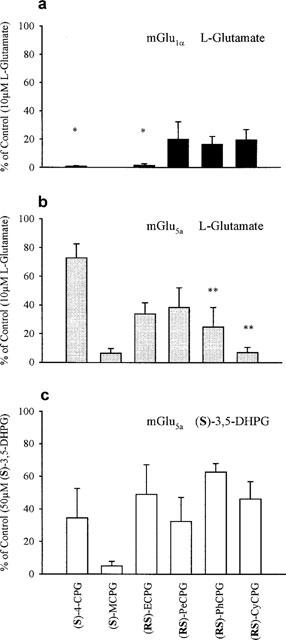
Effects of α-substituted phenylglycine derivatives on agonist-induced Ca2+ release in CHO cells expressing either mGlu1α receptors (a) or mGlu5a receptors (b,c). Results are displayed as percentage of control responses (10 μM glutamate (panels a,b) or 50 μM (S)-3,5-DHPG (panel c)) following incubation with 1 mM of each antagonist. *Denotes significant difference between mGlu1α and mGlu5a receptor-expressing cells, P<0.01. **Denotes significant difference between effects on L-glutamate and (S)-3,5-DHPG-induced Ca2+ release, P<0.05.
Effect of a single concentration of antagonist on (S)-3,5-DHPG-mediated Ca2+ release in mGlu5a receptor expressing cells
It has previously been reported that the group I mGlu receptors display agonist-dependent antagonism (Brabet et al., 1995). It is therefore of great interest to determine if the antagonist characteristics of these phenylglycine analogues is retained when the commonly used group I mGlu selective agonist (S)-3,5-DHPG is used to elicit the functional response. As with the experiments using L-glutamate as the agonist, a single concentration of antagonist (1 mM) was used to block mGlu5a receptor function mediated by an agonist concentration (50 μM) near it's EC50 value (40±8 μM (4)). The results are shown in Figure 2c. The ability of (RS)-PhCPG and (RS)-CyCPG to antagonize mGlu5a receptor-mediated Ca2+ release was significantly reduced when using (S)-3,5-DHPG as the agonist compared to that evoked by L-glutamate. Responses were reduced to 63±5 (3) and 46±11% (3) of control respective (P<0.05). In contrast, (S)-4-CPG appeared to a more effective antagonist when (S)-3,5-DHPG rather than L-glutamate was used as agonist, reducing the maximal Ca2+ release to 35±18% (3) of control, although this did not reach statistical significance. No difference was detected in the ability of any other analogue to antagonize mGlu5a receptor function evoked by either agonist.
KB value determination
The experiments carried out above suggest that modification of the α-carbon alters the ability of the phenylglycine derivative to antagonize mGlu1α and mGlu5a receptor function. In order to investigate this in more detail, the aliphatic series of compounds (S)-4-CPG, (S)-MCPG, (RS)-ECPG and (RS)-PeCPG were chosen for further study. Increasing concentrations of L-glutamate were used to evoke Ca2+ release in the absence and presence of 1 mM of each compound. Agonist applications were made in a random fashion. EC50 values derived from the resulting dose response profiles were then used to calculate a KB value as described in Methods. The results from a representative experiment are shown in Figure 3. (RS)-PeCPG caused a parallel shift in the dose response profile of L-glutamate in cells expressing the mGlu5a receptor with no loss of maximal activity, a response consistent with competitive antagonism. Similar parallel shifts were shown by all the compounds which showed antagonism. The EC50 values derived from the dose response curves in Figure 3 were 5±2 and 27±11 μM in the absence and presence of (RS)-PeCPG, respectively giving a KB value of 312 μM.
Figure 3 .
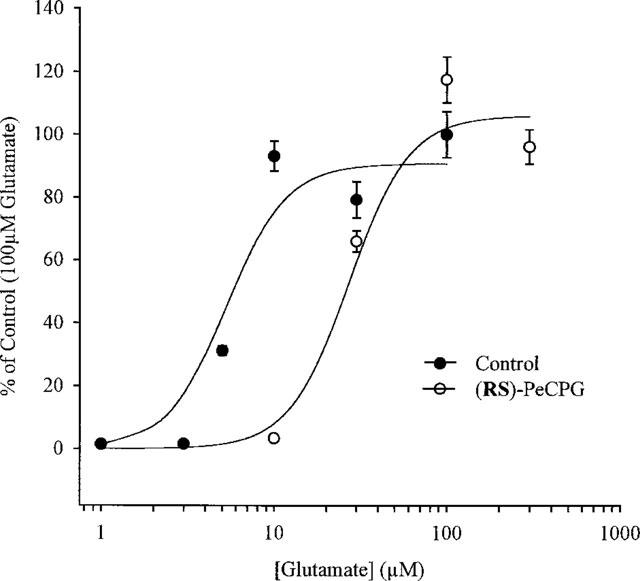
Effect of (RS)-PeCPG on L-glutamate-evoked Ca2+ release in CHO cells expressing the mGlu5a receptor. Representative experiment demonstrating the method for determining the KB value for a single phenylglycine derivative. Increasing concentrations of L-glutamate used to evoke a Ca2+ release in the absence and presence of 1 mM (RS)-PeCPG. EC50 values derived using non-linear least squares analysis were 5±2 and 21±11 μM, respectively giving a KB value of 312 μM.
Similar experiments were carried out for each phenylglycine analogue at least three times with cells expressing either mGlu1α or mGlu5a receptors. The results from these experiments are shown in Figure 4. (The KB values quoted are those for the single active enantiomer of each compound; ECPG and PeCPG were only available as racemic mixtures in a 50 : 50 ratio). In common with the results for using a single concentration of antagonist, (S)-4-CPG demonstrated selectively for mGlu1α-receptor expressing cells (P<0.01), with a KB value of 163±43 μM. In mGlu5a receptor-expressing cells, no shift in the L-glutamate dose response was detected in two out of three preparations. Methylation of (S)-4-CPG to (S)-MCPG induced antagonist activity at mGlu5a receptor-expressing cells (KB=316±43 μM) while maintaining the selectivity for mGlu1α receptors, (KB value of 50±12 μM, P<0.05). Increasing the chain length of the α-carbon substituent had no further effect on the ability of the phenylglycine analogues to antagonize mGlu5a receptor function. However, in mGlu1α receptor-expressing cells, increasing the chain length of the substituent group to two and then five carbon atoms results in a progressive lowering of antagonist affinity when used such that (S)-PeCPG has a 6 fold higher KB value than (S)-MCPG.
Figure 4.
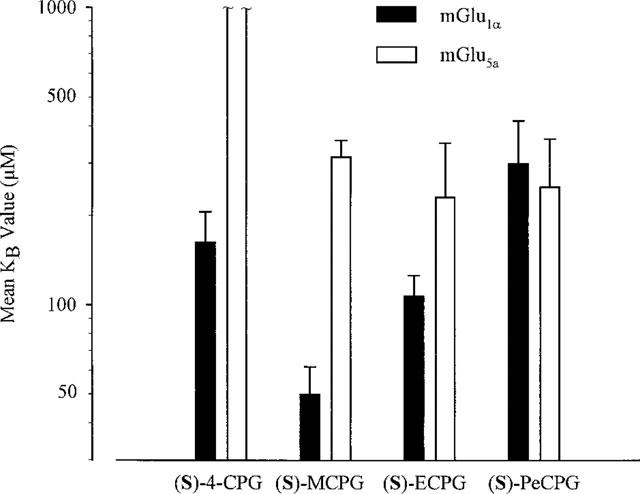
Graphical representation showing KB values derived for the aliphatic series of α-substituted phenylglycine analogues when used with mGlu1α receptor- and mGlu5a receptor-expressing cells. KB values were derived from EC50 values calculated from dose responses carried out in the absence and presence of 1 mM of each antagonist. Results are means±s.e.mean from three independent experiments carried out on each cell line.
Discussion
Selectivity of α-substituted phenylglycine analogues on mGlu1α and mGlu5a receptors
The aim of this study was to investigate the effects of substitution on the α-carbon of 4-CPG on the ability of this compound to discriminate between mGlu1α and mGlu5a receptors. The results show that, in common with previous studies (Kingston et al., 1995; Brabet et al., 1995; Bedingfield et al., 1995), both (S)-4-CPG and (S)-MCPG selectively antagonize mGlu1α receptors over mGlu5a receptors and that (RS)-ECPG is a good antagonist of the mGlu1α receptor. In addition, we have demonstrated that this mGlu1α receptor selectivity is maintained in (RS)-ECPG (Figures 2 and 4).
It has previously been reported that (S)-MCPG is a poor antagonist of L-glutamate-induced rat mGlu5a receptor function (Brabet et al., 1995). In contrast, we have found that although (S)-MCPG displays selectivity for mGlu1α receptors, it is also an effective antagonist of mGlu5a receptors expressed in CHO cells. The reasons for this discrepancy may well be due to the methodologies used in these two studies. Here, we have used cell lines permanently expressing the appropriate receptor, whereas Brabet et al. (1995) used receptors transiently expressed in LLC-PK1 cells, thus potentially giving much higher expression levels of receptor. The KB for (S)-MCPG antagonism of L-glutamate-evoked mGlu1α receptor activation derived in that study was 10 fold higher than we find here. In addition, only 500 μM (S)-MCPG was used to antagonize the functional response. Thus, under those conditions, any effect of (S)-MCPG on the mGlu5a receptor would probably be insignificant although a small shift is discernible in their data. Interestingly, Kingston et al. (1995) found that (S)-MCPG acted as an antagonist at the human mGlu5 receptor, with an affinity similar to that derived in this study, when using quisqualate as the agonist.
As well as confirming the mGlu1α receptor selective nature of (S)-4-CPG and (S)-MCPG, this study has extended the analysis to analogues substituted at the α-position with longer chain aliphatic, cyclopropyl or phenyl groups. The data presented here demonstrates that the ability of α-substituted derivatives of 4-CPG to antagonize mGlu receptor activity is dependent on the nature of the substituent group and on the particular receptor subtype involved. Short chain aliphatic substitutions of the α-carbon of 4-CPG produce effective antagonists of mGlu1α receptors, while bulky cyclic substituent groups lead to analogues with greater mGlu5a selectivity (Figure 2). Thus, mGlu1α and mGlu5a receptors have different steric and/or conformational requirements for the binding of these analogues and it should therefore be possible to design selective antagonists for each of these subtypes.
A question remains regarding the effects of these compounds on the other subgroups of mGlu receptors. Many phenylglycine derivates have both agonist and antagonist actions when used with different mGlu receptor subgroups. For example, (S)-4-CPG and (S)-4-carboxy-3-hydroxyphenylglycine ((S)-4C3HPG) are antagonists of group I mGlu receptors but agonists of group II receptors (Hayashi et al., 1994). Previous work has indicated that, in addition to the effects on group I mGlu receptors, (RS)-ECPG is able to antagonize mGlu2 but not mGlu6 receptors when expressed in CHO cells (Sekiyama et al., 1996). However, the ability of (RS)-ECPG to antagonize mGlu2 receptors was reduced compared to mGlu1α receptors, unlike (S)-MCPG which has a similar affinity for both receptors (Hayashi et al., 1994) and is also known to antagonize group III mGlu receptors (Kemp et al., 1994; Bushell et al., 1996). This may make (RS)-ECPG a more useful antagonist for mGlu1α receptors than (S)-MCPG. Little data is available regarding the effects of the other compounds tested here on individual mGlu receptor subtypes, although all the α-substituted compounds used in this study are able to antagonize group II and group III receptors in spinal cord (unpublished observations).
Agonist dependent antagonism
A further important question regarding the ability of compounds to antagonize the group I mGlu receptors is the phenomenon of agonist-dependent antagonism. It has previously been reported that (S)-4-CPG, (S)-MCPG and (S)-4C3HPG were found to display higher affinities for the antagonism of (1S,3R)-ACPD-mediated phosphoinositide turnover in mGlu1α-expressing cells than the same functional response mediated by L-glutamate (Brabet et al., 1995). It was therefore of great interest to determine if the same phenomenon was displayed by the mGlu5a receptor. The antagonist effects of the phenylglycine analogues on (S)-3,5-DHPG-mediated Ca2+ release were measured (Figure 2c) and compared to the effects on L-glutamate-mediated Ca2+ release (Figure 2b). The results indicate that indeed the ability of phenylglycines to antagonize mGlu5a receptor function is also dependent on the agonist used to elicit the functional response. (S)-4-CPG appeared to be a better antagonist of (S)-3,5-DHPG-evoked Ca2+ release in this cell line than of L-glutamate-mediated release (65 and 28% inhibition of control responses, respectively). Conversely, (RS)-CyCPG showed a greater ability to antagonize L-glutamate-evoked Ca2+ release than that mediated by (S)-3,5-DHPG (93 and 46% inhibition of control responses, respectively) as did (RS)-PhCPG. This effect is unlikely to be simply a matter of a bulky substituent on the α-carbon as (RS)-PeCPG showed no tendency towards agonist dependency. Thus the cyclic α-substituents may block the interaction of L-glutamate with amino-acids which are critical for binding, but which are less important for the binding of (S)-3,5-DHPG. It may well be, therefore, that the ligand binding domain of subgroup I mGlu receptors contains a number of sites within it capable of interacting with different agonists, thus allowing different modes of agonist binding and giving rise to agonist-dependent antagonism.
These data demonstrate the complexity of the pharmacology of the group I mGlu receptors which may provide greater opportunities for the development of subtype selective antagonists. It is also clear that the phenomenon of agonist-dependent antagonism must be taken into consideration in studies using phenylglycine analogues.
Effects of α-alkylation on phenylglycine antagonism of the mGlu1α receptor
In order to investigate the effects of α-alkylation in more detail, experiments were undertaken to measure the affinity of the alkyl series of analogues at both mGlu1α and mGlu5a receptors. The affinities of (S)-4-CPG, (S)-MCPG and (RS)-ECPG for the mGlu1α receptor measured here are in line with previous work using different methodologies (Thomsen et al., 1994; Bedingfield et al., 1995; Kingston et al., 1995). KB values for the α-alkylphenylglycines showed that alkylation of the α-carbon of 4-CPG with a methyl group increases it's affinity at both mGlu1α and mGlu5a receptors, but substitution with longer alkyl chains results in a chain length-dependent loss of affinity at the mGlu1α receptor (Figure 4). In contrast, at the mGlu5a receptor, no significant effect of either ethyl- or pentyl-alkylation could be detected over that produced by methylation. These results show that the effectiveness of phenylglycine analogues as antagonists of group I mGlu receptors is considerably influenced by α-carbon substitution. In this light, it is interesting that the phenyl- and cyclopropyl-derivatives of 4-CPG also showed a reduced ability to antagonize mGlu1α receptor function (Figure 2a) while showing greater effectiveness against the mGlu5a receptor. Thus the loss of antagonist affinity at mGlu1α receptors may be due to steric hindrance caused by bulky α-substituents or may be due to changes to the preferred conformation of the phenylglycines induced by such α-substituents while such changes may increase mGlu5a selectivity.
The data presented in this paper suggest that the mode of binding of the 4-CPG moiety and thus the ligand binding domains of the mGlu1α and mGlu5a receptors may be very different. The mode of binding and antagonist potency of α-substituted 4-carboxyphenylglycine analogues is dependent on the particular modification made, the agonist chosen to mediate the functional response and the receptor subtype used. In general, short chain aliphatic substitutions retain the mGlu1α receptor selectivity of 4-CPG itself. But this is lost when long chain aliphatic groups or bulky cyclic groups are used. Indeed it may well be that these latter compounds will provide lead compounds from which mGlu5 selective antagonists may be developed.
Acknowledgments
This work was funded by BIOTEC (grant no. BIO4 CT96 0049) and the M.R.C.
Abbreviations
- (S)-4-CPG
(S)-4-carboxyphenylglycine
- (S)-4C3HPG
(S)-4-carboxy-3-hydroxyphenylglycine
- (RS)-CHPG
(RS)-2-chloro-5-hydroxyphenylglycine
- (RS)-CyCPG
(RS)-α-cyclopropyl-4-carboxyphenylglycine
- (S)-3,5-DHPG
(S)-3,5-dihydroxyphenylglycine
- DMEM
Dulbecco's Modified Eagle's Medium
- (RS)-ECPG
(RS)-α-ethyl-4-carboxyphenylglycine
- FBS
foetal bovine serum
- (S)-MCPG
(S)-α-methyl-4-carboxyphenylglycine
- mGlu receptor
metabotropic glutamate receptor
- NMDA
N-methyl-D-aspartate
- (RS)-PeCPG
(RS)-α-pentyl-4-carboxyphenylglycine
- (RS)-PhCPG
(RS)-α-phenyl-4-carboxyphenylglycine
References
- ABE T., SUGIHARA H., NAWA H., SHIGEMOTO R., MIZUNO N., NAKANISHI S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J. Biol. Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- BASHIR Z.I., COLLINGRIDGE G.L. An investigation of depotentiation of long-term potentiation in the CA1 region of the hippocampus. Exp. Brain Res. 1994;100:437–443. doi: 10.1007/BF02738403. [DOI] [PubMed] [Google Scholar]
- BASHIR Z.I., JANE D.E., SUNTER D.C., WATKINS J.C., COLLINGRIDGE G.L. Metabotropic glutamate receptors contribute to the induction of long-term depression in the CA1 region of the hippocampus. Eur. J. Pharmacol. 1993;239:265–266. doi: 10.1016/0014-2999(93)91009-c. [DOI] [PubMed] [Google Scholar]
- BEDINGFIELD J.S., KEMP M.C., JANE D.E., TSE H.W., ROBERTS P.J., WATKINS J.C. Structure-activity relationships for a series of phenylglycine derivatives acting at metabotropic glutamate receptors (mGluRs) Br. J. Pharmacol. 1995;116:3323–3329. doi: 10.1111/j.1476-5381.1995.tb15142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRABET I., MARY S., BOCKAERT J., PIN J.-P. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharmacol. 1995;34:895–903. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- BUSHELL T.J., JANE D.E., TSE H.-W., WATKINS J.C., GARTHWAITE J., COLLINGRIDGE G.L. Pharmacological antagonism of the actions of group II and III mGluR agonists in the lateral perforant path of rat hippocampal slices. Br. J. Pharmacol. 1996;117:1457–1462. doi: 10.1111/j.1476-5381.1996.tb15306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK B.P., BAKER S.R., GOLDSWORTHY J., HARRIS J.R., KINGSTON A.E. (+)-2-methyl-4-carboxyphenylglycine ( LY367385) selectively antagonises metabotropic glutamate mGluR1 receptors. Bioorg. Med. Chem. Lett. 1997;7:2777–2780. [Google Scholar]
- COHEN A.S., ABRAHAM W.C. Facilitation of long-term potentiation by prior activation of metabotropic glutamate receptors. J. Neurophysiol. 1996;76:953–962. doi: 10.1152/jn.1996.76.2.953. [DOI] [PubMed] [Google Scholar]
- DAVIS S., LAROCHE S. Activation of metabotropic glutamate receptors induce differential effects on synaptic transmission in the dentate gyrus and CA1 of the hippocampus in the anaesthetized rat. Neuropharmacol. 1996;35:337–346. doi: 10.1016/0028-3908(95)00185-9. [DOI] [PubMed] [Google Scholar]
- DOHERTY A.J., PALMER M.J., HENLEY J.M., COLLINGRIDGE G.L., JANE D.E. (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5 but not mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacol. 1997;36:265–267. doi: 10.1016/s0028-3908(97)00001-4. [DOI] [PubMed] [Google Scholar]
- FITZJOHN S.M., IRVING A.J., PALMER M.J., HARVEY J., LODGE D., COLLINGRIDGE G.L. Activation of group I mGluRs potentiates NMDA responses in rat hippocampal slices. Neurosci. Lett. 1996;203:211–213. doi: 10.1016/0304-3940(96)12301-6. [DOI] [PubMed] [Google Scholar]
- FUKUDA T., HEIZMANN C.W., KOSAKA T. Quantitative analysis of GAD65 and GAD67 immunoreactivities in somata of GABAergic neurons in the mouse hippocampus proper (CA1 and CA3 regions), with special reference to parvalbumin-containing neurons. Brain Res. 1997;764:237–243. doi: 10.1016/s0006-8993(97)00683-5. [DOI] [PubMed] [Google Scholar]
- HARVEY J., COLLINGRIDGE G.L. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S,3R-ACPD in rat hippocampal slices. Br. J. Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAYASHI Y., SEKIYAMA N., NAKANISHI S., JANE D.E., SUNTER D.C., BIRSE E.F., UDVARHELYI P.M., WATKINS J.C. Analysis of agonist and antagonist activitys of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J. Neurosci. 1994;14:3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO I., KOHDA A., TANABE S., HIROSE E., HAYASHI N., MITSUNAGA S., SUGIYAMA H. 3,5-Dihydroxyphenyl-glycine. A potent agonist of metabotropic glutamate receptors. NeuroReport. 1992;3:1013–1016. [PubMed] [Google Scholar]
- KEMP M.C., ROBERTS P.J., POOK P.C.-K., JANE D.E., JONES A.W., JONES P.L. ST. J., SUNTER D.C., UDVARHELYI P.M., WATKINS J.C. Antagonism of presynaptically mediated depressant responses and cyclic AMP-coupled metabotropic glutamate receptors. Eur. J. Pharmacol. Mol. Pharmacol. 1994;266:187–192. doi: 10.1016/0922-4106(94)90109-0. [DOI] [PubMed] [Google Scholar]
- KINGSTON A.E., BURNETT J.P., MAYNE N.G., LODGE D. Pharmacological analysis of 4-carboxyphenylglycine derivatives: comparison of effects on mGluR1α and mGluR5a subtypes. Neuropharmacol. 1995;34:887–894. doi: 10.1016/0028-3908(95)00069-i. [DOI] [PubMed] [Google Scholar]
- MANAHAN-VAUGHAN D. Group 1 and 2 metabotropic glutamate receptors play differential roles in hippocampal long-term depression and long-term potentiation in freely moving rats. J. Neurosci. 1997;17:3303–3311. doi: 10.1523/JNEUROSCI.17-09-03303.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALMER M.J., IRVING A.J., SEABROOK G.R., JANE D.E., COLLINGRIDGE G.L. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacol. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- PIN J.-P., DUVOISIN R. The metabotropic glutamate receptors: Structure and functions. Neuropharmacol. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- ROBERTS P.J. Pharmacological tools for the investigation of metabotropic glutamate receptors (mGluRs): phenylglycine derivatives and other selective antagonists–an update. Neuropharmacol. 1995;34:813–819. doi: 10.1016/0028-3908(95)00094-m. [DOI] [PubMed] [Google Scholar]
- SCHOEPP D.D., GOLDSWORTHY J., JOHNSON B.G., SALHOFF C.R., BAKER S.R. 3,5-dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J. Neurochem. 1997;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- SEKIYAMA N., HAYASHI Y., NAKANISHI S., JANE D., TSE H.W., BIRSE E.F., WATKINS J.C. Structure-activity relationships of new agonists and antagonists of different metabotropic glutamate receptor subtypes. Br. J. Pharmacol. 1996;117:1493–1503. doi: 10.1111/j.1476-5381.1996.tb15312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANABE Y., MASU M., ISHII T., SHIGEMOTO R., NAKANISHI S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- THOMSEN C., BOEL E., SUZDAK P.D. Actions of phenylglycine analogs at subtypes of the metabotropic glutamate receptor family. Eur. J. Pharmacol. Mol. Pharmacol. 1994;267:77–84. doi: 10.1016/0922-4106(94)90227-5. [DOI] [PubMed] [Google Scholar]
- WATKINS J.C., COLLINGRIDGE G.L. Phenylglycine derivatives as antagonists of metabotropic glutamate receptors. Trends Pharmac. Sci. 1994;15:333–342. doi: 10.1016/0165-6147(94)90028-0. [DOI] [PubMed] [Google Scholar]