

Abstract
Isometric tension was recorded in isolated rings of aorta, carotid, coronary and mesenteric arteries taken from endothelial nitric oxide synthase knockout mice (eNOS(−/−) mice) and the corresponding wild-type strain (eNOS(+/+) mice). The membrane potential of smooth muscle cells was measured in coronary arteries with intracellular microelectrodes.
In the isolated aorta, carotid and coronary arteries from the eNOS(+/+) mice, acetylcholine induced an endothelium-dependent relaxation which was inhibited by Nω-L-nitro-arginine. In contrast, in the mesenteric arteries, the inhibition of the cholinergic relaxation required the combination of Nω-L-nitro-arginine and indomethacin.
The isolated aorta, carotid and coronary arteries from the eNOS(−/−) mice did not relax in response to acetylcholine. However, acetylcholine produced an indomethacin-sensitive relaxation in the mesenteric artery from eNOS(−/−) mice.
The resting membrane potential of smooth muscle cells from isolated coronary arteries was significantly less negative in the eNOS(−/−) mice (−64.8±1.8 mV, n=20 and −58.4±1.9 mV, n=17, for eNOS(+/+) and eNOS(−/−) mice, respectively). In both strains, acetylcholine, bradykinin and substance P did not induce endothelium-dependent hyperpolarizations whereas cromakalim consistently produced hyperpolarizations (−7.9±1.1 mV, n=8 and −13.8±2.6 mV, n=4, for eNOS(+/+) and eNOS(−/−) mice, respectively).
These findings demonstrate that in the blood vessels studied: (1) in the eNOS(+/+) mice, the endothelium-dependent relaxations to acetylcholine involve either NO or the combination of NO plus a product of cyclo-oxygenase but not EDHF; (2) in the eNOS(−/−) mice, NO-dependent responses and EDHF-like responses were not observed. In the mesenteric arteries acetylcholine releases a cyclo-oxygenase derivative.
Keywords: Cyclo-oxygenase products, EDHF, endothelium, eNOS knockout mice, nitric oxide, smooth muscle
Introduction
The production by the endothelial cells of vasoactive substances, such as nitric oxide (NO; Furchgott & Zawadzki, 1980; Palmer et al., 1987), prostacyclin (Moncada & Vane, 1979) and a still unidentified molecule, endothelium-derived hyperpolarizing factor (EDHF; Chen et al., 1988; Félétou & Vanhoutte, 1988; Taylor & Weston, 1988), contributes to the local regulation of vascular tone. The relative importance of these factors for endothelium-dependent vasodilation depends on the size of the blood vessels and the vascular bed investigated. In resistance arteries, the contribution of EDHF to endothelium-dependent vasodilation may be major (Nagao et al., 1992; Bauersachs et al., 1996; Garland & Plane, 1996; Shimokawa et al., 1996).
Chronic inhibition of nitric oxide synthase (NOS) has increased vascular resistance in all species studied. This was associated with an increase in blood pressure in rats and pigs but no such changes in rabbits and dogs (Oliveira et al., 1992; Cayatte et al., 1994; Ito et al., 1995; Puybasset et al., 1995). However, since inhibitors of NO production are not selective, and the three isoforms of NOS are inhibited during chronic treatment with those drugs, the cardiovascular changes observed are not necessarily linked to the inhibition of the endothelial isoform. In contrast, disruption of the gene encoding the eNOS in mice produces a specific suppression of endothelial NO production and an increase in systemic blood pressure (Huang et al., 1995).
EDHF may act as a back-up mechanism when the endothelial production of NO is impaired (Kilpatrick & Cocks, 1994; Corriu et al., 1998). Conversely, NO may regulate the production and the effect of EDHF (Olmos et al., 1995; Mombouli et al., 1996a; Popp et al., 1996; Kristof et al., 1997). The purpose of the present work was to address these two possibilities by studying endothelium-dependent relaxations and hyperpolarizations in isolated arteries of mutant eNOS(−/−) mice and the corresponding control wild-type strain (eNOS(+/+) mice).
Methods
C57BL6 mice (wild-type strain; eNOS(+/+) mice) and homozygotous mutant mice lacking the gene for endothelial NOS (eNOS(−/−), Iffa Credo, L'Arbresle, France), 8–10 weeks of age, were used for the study. The generation of eNOS deficient mice has been described elsewhere (Huang et al., 1995). The mice were maintained under pathogen-free conditions in filter-topped cages in an air-conditioned room at 21±1°C, fed a standard laboratory diet and given water ad libitum.
Contraction
eNOS (+/+) and eNOS(−/−) mice were killed by CO2 inhalation. The aorta and the carotid, left anterior coronary and mesenteric (first order) arteries were removed and dissected free of adherent connective tissues. Aortic rings (4 mm in length) were suspended in organ chambers and segments of the other vessels (2 mm in length for carotid and mesenteric arteries, 1.5–3 mm for the coronary artery depending on the branching pattern) were mounted in a microvessel myographs for isometric tension recording (Mulvany & Halpern, 1977). Blood vessels were immersed in Krebs' Ringer bicarbonate solution (37°C, aerated with a 95% O2/5% CO2 gas mixture, pH 7.4) of the following composition (in mM): NaCl 118.3; KCl 4.7; CaCl2 2.5; MgSO4 1.2; KH2PO4 1.2; NaHCO3 25; calcium-disodium EDTA 0.026 and glucose 11.1 (control solution). Segments were stretched step by step and contracted repeatedly with KCl (60 mM) until maximal and reproducible contractions were obtained (Félétou and Teissere 1990). A passive tension of approximately 1000 mg was exerted on the aortas and 100–200 mg on the other arteries. Under these conditions, the imposed diameter of the carotid, mesenteric and coronary arteries was 250, 120 and 70 μm, respectively. After a 45 min equilibration period, endothelium-dependent relaxations to acetylcholine were studied. Aortic rings were contracted with noradrenaline (1–10 μM) and carotid arteries with U 46619 (10–30 nM), in order to achieve a level of tension representing 50–80% of the reference contraction to KCl (60 mM). Cumulative concentration-response curves (1 nM or 10 nM to 100 μM) were performed in these two arteries. Successive concentrations of acetylcholine were applied after stabilization of the relaxation or every 5 min. Murine mesenteric and coronary arteries were contracted with noradrenaline (3 μM) and U 46619 (100 nM), respectively. In preliminary experiments, complete acetylcholine-relaxation curve in mesenteric and coronary arteries were tentatively performed. A sustained level of tension, in all the murine arteries studied, was difficult to obtain. In contrast to the aorta and the carotid artery, in which acetylcholine-induced relaxation could easily be differentiated from a spontaneous decrease in tone, in the mesenteric and coronary arteries the maximal amplitude of acetylcholine-induced relaxation represented only 30–50% of the precontracted level. By using a single, supramaximal concentration of acetylcholine (10 μM), it was easier to evaluate the effects of Nω-nitro-L-arginine (L-NA) or/and indomethacin. Indeed, the effect of this single concentration of acetylcholine produced fast and sharp relaxation which could not be mistaken for a spontaneous decrease in tone. The incubation time with the various inhibitors studied (L-NA and/or indomethacin) was at least 30 min. At the end of the experiment, maximal relaxation was obtained with sodium nitroprusside (1 mM). In some arterial rings the endothelium was mechanically removed (aorta: with jeweller forceps; other arteries: by inserting a human hair into the lumen) to verify the endothelium-dependency of the relaxation induced by acetylcholine (10 μM).
Electrophysiology
Segments of left anterior descending coronary artery (luminal diameter: 70–90 μm) were pinned down to the bottom of an organ chamber (0.5 ml in volume) and superfused at a constant flow (2.5 ml min−1) with modified Krebs' Ringer bicarbonate solution. Care was taken to preserve the integrity of the endothelium. In preliminary experiments, the preservation of the endothelial lining was verified histologically in preparations fixed in Bouin solution and stained with either haematein and eosin or Masson trichrome at the end of the electrophysiological study (data not shown). Smooth muscle cell impalements were performed from the adventitial side of the vessels. The transmembrane potential was recorded with glass capillary microelectrodes (tip resistance of 30–90 MΩ) filled with KCl (3 M) connected to the headstage of a recording amplifier (Intra 767, World Precision Instruments Ltd, New Haven, CT, U.S.A.) equipped with capacitance neutralization; an Ag/AgCl pellet, in contact with the bath solution and directly connected to the amplifier, served as the reference electrode. The electrophysiological signal was continuously monitored on an oscilloscope (3091 Nicolet, Madison, WI, U.S.A.) and simultaneously recorded on paper (Gould, Valley View, OH, U.S.A.) and on a video recorder (TEAC XR310, Tokyo, Japan). Successful impalements were signalled by a sudden negative drop in potential from the baseline (zero potential reference) followed by a stable negative potential for at least 3 min.
Drugs
The drugs used were: noradrenaline hydrochloride, acetylcholine chloride, bradykinin, indomethacin, Nω-nitro-L-arginine (L-NA), sodium nitroprusside (Sigma, La Verpillère, France); substance P (Bachem, Voisins-Le-Bretonneux, France); U 46619 (9,11-dideoxy-9α, 11α-methanoepoxyprostaglandin F2α, Cayman Chemical Company, Ann Arbor, MI, U.S.A.). Cromakalim and SIN-1 were synthesized in the chemistry department of the Servier Research Center (Suresnes, France). Indomethacin was dissolved in deionized water with an equimolar concentration of Na2CO3. Cromakalim was dissolved in equivalent volumes of deionized water and propylene glycol. SIN-1 was prepared in methanol. U 46619 was solubilized in ethanol. Final concentrations of propylene glycol, methanol and ethanol in the bath were respectively 6.8, 25 and 0.2 mM. The other drugs were dissolved in distilled water.
Statistics
Data are shown as mean±s.e.mean. n indicates the number of animals from which arteries were taken. Statistical analysis was performed using Student's t-test for paired or unpaired observations. Differences were considered to be statistically significant when P was less than 0.05.
Results
Contraction
The contractions induced by KCl (60 mM) were not significantly different in arteries from eNOS(+/+) and eNOS(−/−) mice (contraction, in arteries from eNOS(+/+) and eNOS(−/−) mice, respectively: aorta: 910±110 mg, n=13 and 890±210 mg, n=11; carotid arteries: 52±13 mg, n=5 and 77±18 mg, n=6; coronary arteries: 20±3 mg, n=17 and 21±3 mg, n=17; mesenteric arteries: 54±5 mg, n=18 and 54±5 mg, n=20).
Isolated rings of aorta and carotid arteries from both strains were studied in control solution or in the presence of L-NA (100 μM), indomethacin (5 μM), or the combination of both inhibitors. Aortic rings were contracted with noradrenaline (1–10 μM) and carotid arteries with U 46619 (10–30 nM), in order to achieve a level of tension representing 50–80% of the reference contraction to KCl (60 mM). Acetylcholine (10 nM to 100 μM) induced endothelium-dependent relaxations in aortas and carotid arteries from the eNOS(+/+) mice but not in those from eNOS(−/−). In the eNOS(+/+), these relaxations were unaffected by indomethacin but were blocked by L-NA or by the combination of the two inhibitors (Figure 1 and 2). In the eNOS(−/−), the decrease in tension was not significantly different from that observed in time-matched controls (data not shown).
Figure 1.
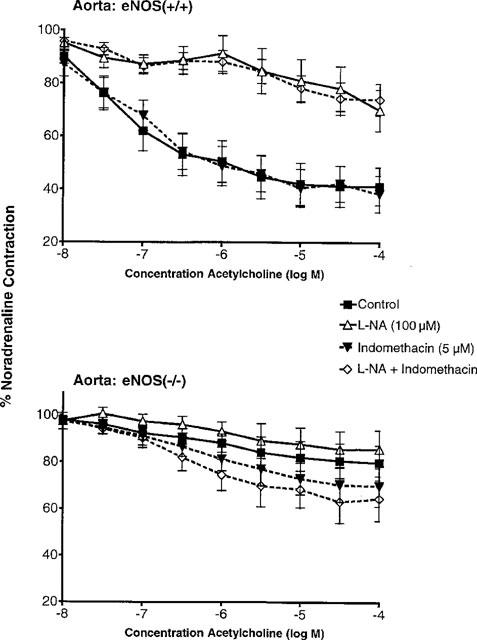
Concentration-relaxation curves to acetylcholine in aortas with endothelium from eNOS(+/+) (upper panel; n=13) and eNOS(−/−) mice (lower panel; n=11) in the absence and presence of indomethacin (5 μM) and/or Nω-nitro-L-arginine (L-NA, 100 μM). Data are shown as means±s.e.mean.
Figure 2.
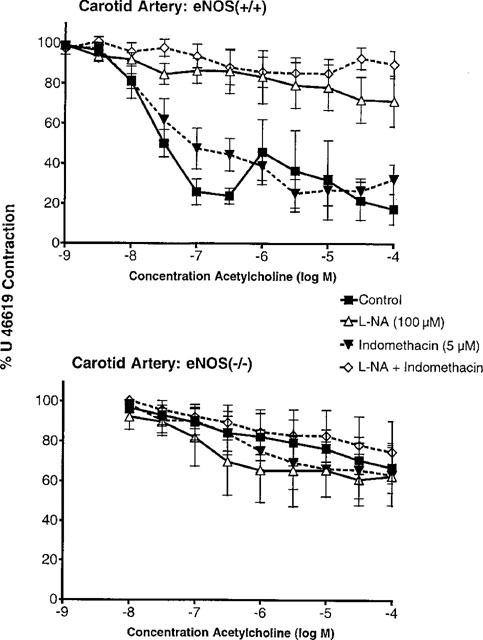
Concentration-relaxation curves to acetylcholine in carotid arteries with endothelium from eNOS(+/+) (upper panel; n=5–6) and eNOS(−/−) mice (lower panel; n=4–5) in the absence and presence of indomethacin (5 μM) and/or Nω-nitro-L-arginine (L-NA, 100 μM). Data are shown as means±s.e.mean.
Mesenteric arteries were contracted with noradrenaline (3 μM). The contractile response was significantly larger in mesenteric arteries from the eNOS(−/−) mice (48±4 mg, n=33 and 67±7 mg, n=31, in eNOS(+/+) and eNOS(−/−) mice, respectively, P<0.05). The addition of L-NA (100 μM) induced a further increase in tension in mesenteric arteries from eNOS(+/+) but not in eNOS(−/−) mice (+14±7 mg, n=7 and −8±6 mg, n=8 in eNOS(+/+) and eNOS(−/−) mice, respectively). Indomethacin (5 μM) decreased the contractile response to noradrenaline in eNOS(+/+) mice but did not influence the level of tension in the eNOS(−/−) mice. The combination of L-NA and indomethacin provoked an increase in tension in arteries in eNOS(+/+) mice but not in eNOS(−/−) mice (+13±4 mg, n=10 and −3±11 mg, n=7 in eNOS(+/+) and eNOS(−/−) mice, respectively). Acetylcholine (10 μM) induced relaxation in the mesenteric arteries from both strains. In the wild-type strain the relaxation to acetylcholine was not significantly affected by indomethacin or L-NA but was abolished by the combination of the two inhibitors. In mesenteric arteries from eNOS(−/−) mice, acetylcholine-induced relaxation was not affected by L-NA but was significantly inhibited either by indomethacin or the combination of both inhibitors (Figure 3).
Figure 3.
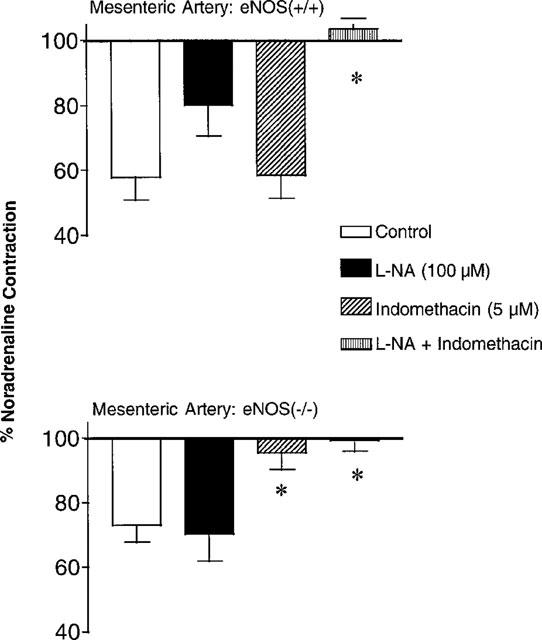
Acetylcholine (10 μM)-induced relaxations in mesenteric arteries with endothelium from eNOS(+/+) (upper panel; n=8–10) and eNOS(−/−) mice (lower panel; n=8–9), in the absence and presence of indomethacin (5 μM) and/or Nω-nitro-L-arginine (L-NA, 100 μM). Data are shown as means±s.e.mean. Asterisks indicate a statistically significant difference with the corresponding control value.
Coronary arteries were contracted with U 46619 (100 nM). The contractile response was significantly larger in coronary arteries from the eNOS(−/−) mice (27±3 mg, n=25 and 45±7 mg, n=28, in eNOS(+/+) and eNOS(−/−) mice, respectively). The addition of L-NA (100 μM) induced a further increase in tension in coronary arteries in eNOS(+/+) but not in eNOS(−/−) mice (+17±4 mg, n=5 and +2±1 mg, n=6 in eNOS(+/+) and eNOS(−/−) mice, respectively). Indomethacin (5 μM) did not significantly affect the contractile response to U 46619 in either strain. The combination of L-NA and indomethacin provoked an increase in tension in eNOS(+/+) mice but not in eNOS(−/−) mice (+8±1 mg, n=7 and −6±2 mg, n=6 in eNOS(+/+) and eNOS(−/−) mice, respectively). Acetylcholine (10 μM) induced relaxation in the coronary artery from eNOS(+/+) mice. This relaxation was unaffected by indomethacin but was inhibited by L-NA or by the combination of both inhibitors. In coronary arteries from eNOS(−/−) mice, acetylcholine did not produce any significant relaxations either in control or in presence of inhibitors (Figure 4).
Figure 4.
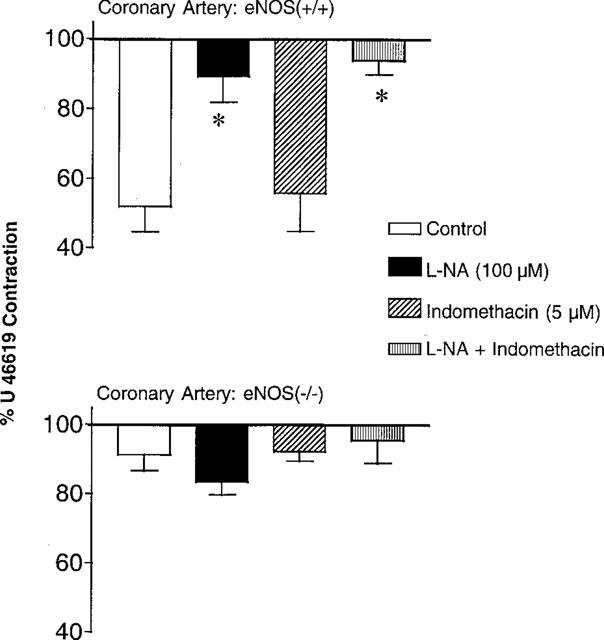
Acetylcholine (10 μM)-induced relaxations in coronary arteries with endothelium from eNOS(+/+) (upper panel; n=6–8) and eNOS(−/−) mice (lower panel; n=6–8), in presence or not of indomethacin (5 μM) and/or Nω-nitro-L-arginine (L-NA, 100 μM). Data are shown as means±s.e.mean. Asterisks indicate a statistically significant difference with the corresponding control value.
Sodium nitroprusside (1 mM) induced complete relaxation in all the arteries studied from both strains (data not shown).
Electrophysiology
Under control conditions, the resting membrane potential of eNOS(+/+) mice coronary arterial smooth muscle cells was not altered significantly by the presence of L-NA and/or indomethacin (Table 1). In eNOS(−/−) mice, under control conditions, the resting membrane potential of the coronary arterial smooth muscle cells was significantly less negative. This value was not altered significantly by the presence of L-NA and/or indomethacin (Table 1).
Table 1.
Resting membrane potential (mV) in coronary artery smooth muscle cells from eNOS(+/+) and eNOS(−/−) mice

In coronary smooth muscle cells from eNOS(+/+) mice, acetylcholine (1 μM) did not modify the resting membrane potential in the absence or the presence of L-NA and/or indomethacin. At a higher concentration (10 μM), acetylcholine induced a depolarization of the smooth muscle cells in approximately 50% of the cells (five depolarizations in nine blood vessels studied) in control solution and in the presence of L-NA (Table 2) which was in some cases accompanied by a rhythmic activity (Figure 5). The depolarization was observed in only one artery treated with indomethacin or L-NA plus indomethacin (one depolarization in seven blood vessels studied; Table 2). Substance P (100 nM) and bradykinin (100 nM) did not influence the resting membrane potential. In two smooth muscle cells out of twelve, SIN-1 (10 μM) induced a marked hyperpolarization (−5 and −9 mV) but no significant changes in the other cells (data not shown). In contrast, cromakalim (10 μM) induced a significant hyperpolarization under the various experimental conditions (Figure 5, Table 2).
Table 2.
Changes in membrane potential induced by acetylcholine SIN-1 and cromakalin in coronary artery smooth muscle cells from eNOS (+/+) and eNOS (−/−) mice

Figure 5.
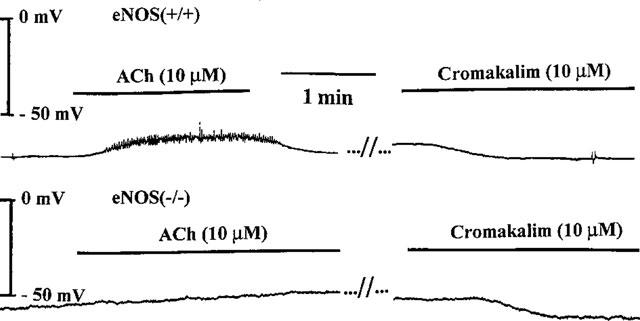
Original traces showing the effects of acetylcholine (ACh, 10 μM) and cromakalim (10 μM) on the resting membrane potential of smooth muscle cell from isolated coronary artery with endothelium taken from eNOS(+/+) (upper panel) or eNOS (−/−) mice (lower panel) mice.
In coronary smooth muscle cells from eNOS(−/−) mice, the effects of acetylcholine (1 μM) bradykinin (100 nM), substance P (100 nM), SIN-1 (10 μM) and cromakalim (10 μM) were qualitatively and quantitatively similar to those observed in eNOS(+/+) mice (Figure 5, Table 2). Acetylcholine (10 μM) induced a depolarization of the smooth muscle cells in six out of seven blood vessels studied, either under control conditions or in the presence of L-NA (Table 2), which was in some cases accompanied by a rhythmic activity. The depolarization was inhibited significantly by indomethacin.
Discussion
The disruption of the eNOS gene did not alter the intrinsic contractile properties of the smooth muscle cells, since isolated rings of aorta, carotid, mesenteric and coronary arteries from eNOS(−/−) mice developed a similar level of tension in response to KCl (60 mM) to that of corresponding arterial rings from eNOS(+/+) mice. The absolute value of the resting membrane potential of the coronary artery smooth muscle cells from eNOS(−/−) mice was less negative than that observed in eNOS(+/+) mice. The modest depolarization observed in smooth muscle cells from eNOS(−/−) mice is unlikely to be attributed to the disappearance of endothelial NO per se. Indeed, in both eNOS(−/−) and eNOS(+/+) mice as well as in various arteries from different species, treatment of the isolated artery rings with either L-NA or indomethacin or their combination did not affect the resting membrane potential. Furthermore, in other species, endothelial denudation does not affect vascular smooth muscle membrane potential indicating that basal production of NO or/and cyclo-oxygenase products does not exert any modulatory role on the membrane potential of vascular smooth muscle cells (Parkington et al., 1993; 1995; Zygmunt et al., 1994; Corriu et al., 1996). eNOS(−/−) mice have systemic arterial and moderate pulmonary hypertension (Huang et al., 1995; 1996; Steudel et al., 1997). Depolarization of the arterial smooth muscle cells has been associated with various experimental models of hypertension and may be linked to the physical strain exerted by high blood pressure on the blood vessel wall (Tomobe et al., 1991; Fujii et al., 1992; Morel et al., 1996; Vanheel & Van De Voorde, 1996).
In mesenteric and coronary arteries from eNOS(+/+) mice, the addition of L-NA after stabilization of the contractile response produced either by noradrenaline or U 46619, the analogue of thromboxane A2, resulted in a further increase in tension indicating a basal endothelial release of NO and possibly the release of NO by activation of endothelial α2-adrenoceptor (Cocks & Angus 1983; Thorin et al., 1998). In the corresponding arteries from eNOS(−/−) mice, the addition of L-NA did not produce a contraction, indicating the absence of endothelial NO production in these blood vessels. However, when compared with eNOS(+/+) mice, the contractile responses to noradrenaline and U 46619 observed in the mesenteric and the coronary arteries from eNOS(−/−) mice were significantly larger. The difference observed was abolished after L-NA treatment demonstrating the role of endothelial NO release in modulating agonist-induced contraction of vascular smooth muscle.
The endothelium-dependent relaxations induced by acetylcholine in the aorta, carotid and left anterior descending coronary arteries from eNOS(+/+) mice were abolished by acute inhibition of NOS indicating that, in these blood vessels, NO is the sole endogenous endothelium-derived vasodilator (Ku et al., 1996). This conclusion is stengthened by the disappearance of these relaxations in eNOS(−/−) mice, in agreement with previous studies (Huang et al., 1995; Faraci et al., 1998). In the isolated perfused murine heart, the vasodilatation produced by acetylcholine involved both NO and a cyclo-oxygenase derivative in eNOS(+/+) mice and only the latter in eNOS(−/−) mice (Godecke et al., 1998) which may indicate that the cyclo-oxygenase component is more proeminent in distal coronary arteries than in the left anterior decending coronary artery studied in the present work. Acetylcholine, bradykinin and substance P did not hyperpolarize the smooth muscle cells of the murine coronary artery, which, in conjunction with the absence of relaxations resistant to inhibitors of NOS and cyclo-oxygenase, indicates that EDHF does not contribute to endothelium-dependent relaxations in the various murine blood vessels studied. NO could suppress or regulate the expression of EDHF (Olmos et al., 1995; Mombouli et al., 1996a; Popp et al., 1996; Kristof et al., 1997) which has been described in various species as a back-up mechanism in case of dysfunction of the endothelial NO pathway (Kilpatrick & Cocks, 1994; Corriu et al., 1998). However, studies performed in eNOS(+/+) mice in presence of an inhibitor of NOS and eNOS(−/−) mice did not unmask an EDHF-like response. To date, the mouse, appears to be the only mammal studied so far that appears to lack an endothelium-dependent hyperpolarizing mechanism in isolated coronary arteries (for review: Félétou & Vanhoutte, 1996). Compensatory mechanisms have been described in isolated perfused heart from eNOS(−/−) mice, in response to reactive hyperaemia, but the origin of these mechanisms is not known at present (Godecke et al., 1998).
Although endothelium-dependent hyperpolarization could not be observed in murine coronary smooth muscle cells, cromakalim induced hyperpolarization of the coronary arteries of both murine strains, under all the experimental conditions tested. However, cromakalim activates ATP-sensitive potassium channels while EDHF in most blood vessels, taken from other species, activates a different but yet undetermined potassium conductance (Corriu et al., 1996). Therefore, this observation does not allow to determine whether the absence of EDHF-mediated responses lies in the absence of EDHF-synthesis/release by the endothelial cells or in the absence of the target for EDHF such as a specific population of potassium channel on the vascular smooth muscle cells.
Exogenous NO released by SIN-1 produced a hyperpolarization in depolarized smooth muscle cells. Repolarization of the smooth muscle cells by endogenous release of NO or by the addition of nitrovasodilators in the absence of any hyperpolarizing effect has been described in other species (Tare et al., 1990; Vanheel et al., 1994). Sodium nitroprusside, another nitrovasodilator, consistently induced maximal relaxations in all the vessels studied from both murine strains. This is in agreement with previous studies demonstrating that the relaxing effect of exogenous NO, which can be attributed to activation of soluble guanylate cyclase, was not altered (and even enhanced) in eNOS(−/−) mice (Huang et al., 1995; Faraci et al., 1998).
The endothelium-dependent relaxation induced by acetylcholine in the mesenteric arteries from eNOS(+/+) mice was partially blocked by the NOS inhibitor, as previously shown in the murine perfused mesenteric bed (Berthiaume et al., 1997). However, in contrast with the aorta and carotid arteries, a combined exposure to L-NA plus indomethacin was necessary to inhibit acetylcholine-induced relaxation fully. The cyclo-oxygenase component is unmasked only after NOS inhibition and the effects of the two endothelial mediators are not additive. Similar observations have been previously reported concerning the relative participation of NO and prostacyclin in the endothelium-dependent relaxations and hyperpolarizations of rat and guinea-pig arteries (Corriu et al., 1996; Zygmunt & Högestätt, 1996). In the mesenteric arteries from eNOS(−/−) mice, acetylcholine induced a relaxation which was insensitive to L-NA but abolished by indomethacin indicating that nitric oxide is not involved in this relaxation and that the products of cyclo-oxygenase present in mesenteric arteries from eNOS(+/+) mice can still be produced in eNOS(−/−) mice. An upregulation of the endothelial cyclo-oxygenases has been shown in dogs (COX-1) and rats (COX-2) chronically treated with inhibitors of NOS (Puybasset et al., 1996; Beverelli et al., 1997; Henrion et al., 1997). The present study was not designed to determine whether or not cyclo-oxygenase is upregulated in eNOS(−/−) mice. However, intravenous indomethacin injection does not modify systemic haemodynamic parameters either in eNOS(+/+) or in eNOS(−/−) mice (Steudel et al., 1997). In isolated mesenteric artery from both eNOS(+/+) and eNOS(−/−) mice, Thorin et al. (1998) have recently shown that an α2-adrenergic agonist could elicit NO-sensitive, endothelium-dependent relaxations which are resistant to cyclo-oxygenase inhibition. These experiments suggest that an endothelium-derived hyperpolarizing factor, whose release and/or production is reduced by NO, can be produced by the murine mesenteric endothelial cells. Whether or not this relaxation is due to the hyperpolarization of the underlying vascular smooth muscle cells remains to be determined. The specificity of this response upon α2-adrenergic stimulation and the unusual occurrence of an endothelium-dependent relaxation markedly potentiated by a NOS inhibitor has to be further explored.
Neuronal NO may compensate for the absence of endothelial NO during acetylcholine-induced dilatation of cerebral arterioles of eNOS(−/−) mice (Meng et al., 1996, 1998) and contribute to the increase in regional cerebral blood flow in response to hypercapnia (Ma et al., 1996). The present study experiments did not reveal a L-NA sensitive component in the various peripheral arteries from the eNOS(−/−) mice. Thus the compensatory role of neuronal NOS might be restricted to the cerebral circulation.
Acetylcholine-induced depolarizations were observed in coronary smooth muscle cells from both eNOS(−/−) and eNOS(+/+) mice. The depolarization to acetylcholine could be due to a direct effect of acetylcholine on the smooth muscle cells. Alternatively, acetylcholine may release endothelial-derived depolarizing factor (Corriu et al., 1996; Mombouli et al., 1996b), or vasoconstricting endothelial products of cyclo-oxygenase observed in several models of hypertension (Lüscher & Vanhoutte, 1986; Zanchi et al., 1995). The depolarization was not affected by L-NA but was inhibited by indomethacin suggesting the latter possibility.
In conclusion, in the murine eNOS(+/+) blood vessels studied, endothelium-dependent relaxations involve either NO or the combination of NO and a product of cyclo-oxygenase, but EDHF is not involved. In the eNOS(−/−) mice, NO-dependent responses and EDHF-like responses were not observed suggesting that the latter pathway is not upregulated by eNOS gene disruption. The only back-up mechanism which was observed in mesenteric arteries from the eNOS(−/−) mice was a cyclo-oxygenase-dependent response.
Abbreviations
- EDHF
endothelium-derived hyperpolarizing factor
- eNOS
endothelial nitric oxide synthase
- eNOS(−/−) mice
endothelial nitric oxide synthase knock-out mice
- eNOS(+/+) mice
wild-type strain mice
- L-NA
Nω-nitro-L-arginine
References
- BAUERSACHS J., LISCHKE V., BUSSE R., HECKER M.Endothelium-dependent hyperpolarization in the coronary microcirculation Endothelium-derived hyperpolarizing factor 1996Amsterdam, The Netherlands: Harwood Academic Publishers; 211–216.ed. Vanhoutte P.M., pp [Google Scholar]
- BERTHIAUME N., HESS F., CHEN A., REGOLI D., D'ORLEANS-JUSTE P. Pharmacology of kinins in the arterial and venous mesenteric bed of normal and B2 knockout transgenic mice. Eur. J. Pharmacol. 1997;333:55–61. doi: 10.1016/s0014-2999(97)01096-0. [DOI] [PubMed] [Google Scholar]
- BEVERELLI F., BEA M.L., PUYBASSET L., GIUDICELLI J.F., BERDEAUX A. Chronic inhibition of NOS enhances the production of prostacyclin in coronary arteries through upregulation of the cyclo-oxygenase type 1 isoform. Fundam. Clin. Pharmacol. 1997;11:252–259. doi: 10.1111/j.1472-8206.1997.tb00193.x. [DOI] [PubMed] [Google Scholar]
- CAYATTE A.J., PALACINO J.J., HORTEN K., COHEN R.A. Chronic inhibition of nitric oxide production accelerates neointima formation and impairs endothelial function in hypercholesterolemic rabbits. Arterioscler. Thromb. 1994;14:753–759. doi: 10.1161/01.atv.14.5.753. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COCKS T.M., ANGUS J.A. Endothelium-dependent relaxation of coronary arteries by noradrenaline and serotonin. Nature. 1983;305:627–630. doi: 10.1038/305627a0. [DOI] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., CANET E., VANHOUTTE P.M. Endothelium-derived factors and hyperpolarizations of the isolated carotid artery of the guinea-pig. Br. J. Pharmacol. 1996;119:959–964. doi: 10.1111/j.1476-5381.1996.tb15765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORRIU C., FELETOU M., PUYBASSET L., BEA M.-L., BERDEAUX A., VANHOUTTE P. M.Endothelium-dependent hyperpolarization in isolated arteries from animal treated with NO-synthase inhibitors J. Cardiovasc. Pharmacol. 199832in press [DOI] [PubMed] [Google Scholar]
- FARACI F.M., SIGMUND C.D., SHESELY E.G., MAEDA N., HEISTAD D.D. Responses of carotid artery in mice deficient in expression of the gene for endothelial NOS. Am. J. Physiol. 1998;274:H564–H570. doi: 10.1152/ajpheart.1998.274.2.H564. [DOI] [PubMed] [Google Scholar]
- FELETOU M., TEISSEIRE B. Converting enzyme inhibition in isolated porcine resistance artery potentiates bradykinin relaxation. Eur. J. Pharmacol. 1990;190:159–166. doi: 10.1016/0014-2999(90)94122-e. [DOI] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988;93:515–524. doi: 10.1111/j.1476-5381.1988.tb10306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELETOU M., VANHOUTTE P.M. Endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 1996;23:1082–1090. doi: 10.1111/j.1440-1681.1996.tb01174.x. [DOI] [PubMed] [Google Scholar]
- FUJII K., TOMINAGA M., OHMORI S., KOBAYASHI K., KOGA T., TAKATA Y., FUJISHIMA M. Decreased endothelium-dependent hyperpolarization to acetylcholine in smooth muscle of the mesenteric artery of spontaneously hypertensive rats. Circ. Res. 1992;70:660–669. doi: 10.1161/01.res.70.4.660. [DOI] [PubMed] [Google Scholar]
- FURCHGOTT R.F., ZAWADZKI J.V. The obligatory role of the endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F.Relative importance of endothelium-derived hyperpolarizing factor for the relaxation of vascular smooth muscle in different arterial beds Endothelium-derived hyperpolarizing factor 1996Amsterdam, The Netherlands: Harwood Academic Publishers; 173–179.ed. Vanhoutte P.M., pp [Google Scholar]
- GODECKE A., DECKING U.K.M., DING Z., HIRCHENHAIN J., BIDMON H.J., GODECKE S., SCHRADER J. Coronary hemodynamics in endothelial NOS knockout mice. Circ. Res. 1998;82:186–194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- HENRION D., DECHAUX E., DOWELL F.J., MACLOUF J., SAMUEL J-L, LEVY B.I., MICHEL J.-B. Alteration of flow-induced dilatation in mesenteric resistance arteries of L-NAME treated rats and its partial association with induction of cyclo-oxygenase-2. Br. J. Pharmacol. 1997;121:83–90. doi: 10.1038/sj.bjp.0701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG P.L., HUANG Z., MASHIMO H., BLOCH K.D., MOSKOWITZ M.A., BEVAN J.A., FISHMAN M.C. Hypertension in mice lacking the gene for endothelial NOS. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- HUANG Z., HUANG P.L., MA J., MENG W., AYATA C., FISHMAN M.C., MOSKOWITZ M.A. Enlarged infarcts in endothelial NOS knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- ITO A., EGASHIRA K., KADOKAMI T., FUKUMOTO Y., TAKAYANAGI T., NAKAIKE R., KUGA T., SUEISHI K., SHIMOKAWA H., TAKESHITA A. Chronic inhibition of endothelium-derived nitric oxide synthesis causes coronary microvascular structural changes and hyperreactivity to serotonin in pigs. Circulation. 1995;92:2636–2644. doi: 10.1161/01.cir.92.9.2636. [DOI] [PubMed] [Google Scholar]
- KILPATRICK E.V., COCKS T.M. Evidence for differential roles of nitric oxide (NO) and hyperpolarization in endothelium-dependent relaxation of pig isolated coronary artery. Br. J. Pharmacol. 1994;112:557–565. doi: 10.1111/j.1476-5381.1994.tb13110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRISTOF A.S., NOORHOSSEINI H., HUSSAIN N.A.S. Attenuation of endothelium-dependent hyperpolarizing factor by bacterial lipopolysaccharides. Eur. J. Pharmacol. 1997;325:69–73. doi: 10.1016/s0014-2999(97)83030-0. [DOI] [PubMed] [Google Scholar]
- KU D., GUO L., DAI J., ACUFF C.G., STEINHELPER M.E. Coronary vascular and endothelial reactivity changes in transgenic mice overexpressing atrial natriuretic factor. Am. J. Physiol. 1996;271:H2368–H2376. doi: 10.1152/ajpheart.1996.271.6.H2368. [DOI] [PubMed] [Google Scholar]
- LÜSCHER T.F., VANHOUTTE P.M. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension. 1986;8:344–348. doi: 10.1161/01.hyp.8.4.344. [DOI] [PubMed] [Google Scholar]
- MA J., MENG W., AYATA C., HUANG P.L., FISHMAN M.C., MOSKOWITZ M.A. L-NNA-sensitive regional cerebral blood flow augmentation during hypercapnia in type III NOS mutant mice. Am. J. Physiol. 1996;271:H1717–H1719. doi: 10.1152/ajpheart.1996.271.4.H1717. [DOI] [PubMed] [Google Scholar]
- MENG W., AYATA C., WAEBER C., HUANG P.L., FISHMAN M.C., MOSKOWITZ M.A. Neuronal NOSc-GMP-dependent ACh-induced relaxation in pial arterioles of endothelial NOS knockout mice. Am. J. Physiol. 1998;274:H411–H494. doi: 10.1152/ajpheart.1998.274.2.H411. [DOI] [PubMed] [Google Scholar]
- MENG W., MA J., AYATA C., HARA H., HUANG P.L., FISHMAN M.C., MOSKOWITZ M.A. ACh dilates pial arterioles in endothelial and neuronal NOS knockout mice by NO-dependent mechanisms. Am. J. Physiol. 1996;271:H1145–H1150. doi: 10.1152/ajpheart.1996.271.3.H1145. [DOI] [PubMed] [Google Scholar]
- MOMBOULI J.V, , BISSIRIOU I., AGBOTON V.D., ALVEAR J., CHELLY F., KILBOURN R., VANHOUTTE P.M.Endotoxemia reduces the contribution of endothelium-derived hyperpolarizing factor, but augments that of prostanoids in canine coronary arteries Endothelium-derived hyperpolarizing factor 1996aAmsterdam The Netherlands: Harwood Academic Publishers; 271–278.ed. Vanhoutte, P.M., pp [Google Scholar]
- MOMBOULI J.V., BISSIRIOU I., VANHOUTTE P.M.Biossay of Endothelium-derived hyperpolarizing factor: is endothelium-derived depolarizing factor a confounding element Endothelium-derived hyperpolarizing factor 1996bAmsterdam, The Netherlands: Harwood Academic Publishers; 51–56.ed. Vanhoutte, P.M., pp [Google Scholar]
- MONCADA S., VANE J.R. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2 and prostacyclin. Pharmacol. Rev. 1979;30:293–331. [PubMed] [Google Scholar]
- MOREL N., GHISDAL P., GODFRAIND T.Hyperpolarization to acetylcholine in the mesenteric artery from NaCl-loaded hypertensive rats Endothelium-derived hyperpolarizing factor 1996Amsterdam, The Netherlands: Harwood Academic Publishers; 255–261.ed. Vanhoutte, P.M., pp [Google Scholar]
- MULVANY M.J., HALPERN W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ. Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- NAGAO T., ILLIANO S.C., VANHOUTTE P.M. Heterogeneous distribution of endothelium-dependent relaxations resistant to NG-nitro-L-arginine in rats. Am. J. Physiol. 1992;263:H1090–H1094. doi: 10.1152/ajpheart.1992.263.4.H1090. [DOI] [PubMed] [Google Scholar]
- OLIVEIRA M., ANTUNES E., DE NUCCI G., LOVISOLO S.M., ZATZ R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertens. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- OLMOS L., MOMBOULI J.V., ILLIANO S., VANHOUTTE P.M. cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. Am. J. Physiol. 1995;37:H865–H870. doi: 10.1152/ajpheart.1995.268.2.H865. [DOI] [PubMed] [Google Scholar]
- PALMER R.M.J., FERRIDGE A.G., MONCADA S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- PARKINGTON H.C., TARE M., TONTA M., COLEMAN H.A. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKINGTON H.C., TONTA M., COLEMAN H.A., TARE M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. J. Physiol. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPP R., BAUERSACHS J., SAUER E., HECKER M., FLEMING I., BUSSE R.The cytochrome P450 monooxygenase pathway and nitric oxide-independent relaxations Endothelium-derived hyperpolarizing factor 1996Amsterdam, The Netherlands: Harwood Academic Publishers; 65–72.ed. Vanhoutte, P.M., pp [Google Scholar]
- PUYBASSET L., BEA M.L., GHALEH B., GIUDICELLI J.F., BERDEAUX A. Coronary and systemic hemodynamic effects of sustained inhibition of nitric oxide synthesis in conscious dogs. Circ. Res. 1996;79:343–357. doi: 10.1161/01.res.79.2.343. [DOI] [PubMed] [Google Scholar]
- PUYBASSET L., BEA M.L., SIMON L., GHALEH B., GIUDICELLI J.F., BERDEAUX A. Hemodynamic effects of sub-chronic NOS inhibition in conscious dogs. Role of EDRF. Arch. Mal. Coeur Vaisseaux. 1995;88:1217–1221. [PubMed] [Google Scholar]
- SHIMOKAWA H., YASUTAKE H., FUJII K., OWADA M.K., NAKAIKE R., FUKUMOTO Y., TAKAYANAGI T., NAGAO T., EGASHIRA K., FUJISHIMA M., TAKESHITA A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J. Cardiovasc. Pharmacol. 1996;28:703–711. doi: 10.1097/00005344-199611000-00014. [DOI] [PubMed] [Google Scholar]
- STEUDEL W., ICHINOSE F., HUANG P.L., HURFORD W.E., JONES R.C., BEVAN J.A., FISHMAN M.C., ZAPOL W.M. Pulmonary vasoconstriction and hypertension in mice with targeted disruption of the endothelial NOS (NOS 3) gene. Circ. Res. 1997;81:34–40. doi: 10.1161/01.res.81.1.34. [DOI] [PubMed] [Google Scholar]
- TARE M., PARKINGTON H.C., COLEMAN M., NEILD T.O., DUSTING G.J. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- TAYLOR S.G., WESTON A.H. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends Pharmacol. Sci. 1988;9:272–274. doi: 10.1016/0165-6147(88)90003-x. [DOI] [PubMed] [Google Scholar]
- THORIN E., HUANG P.L., FISHMAN M.C., BEVAN J.A. Nitric oxide inhibits α2-adrenoceptor-mediated endothelium-dependent vasodilatation. Circ. Res. 1998;82:1323–1329. doi: 10.1161/01.res.82.12.1323. [DOI] [PubMed] [Google Scholar]
- TOMOBE Y., ISHIKAWA T., YANAGISAWA M., KIMURA S., MASAKI T., GOTO K. Mechanisms of altered sensitivity to endothelin-1 between aortic smooth muscles of spontaneously hypertensive and Wystar-Kyoto rats. J. Pharmacol. Exp. Ther. 1991;257:555–561. [PubMed] [Google Scholar]
- VANHEEL B., VAN DE VOORDE J.Impaired endothelium-dependent hyperpolarization and relaxation in the aorta from the renal hypertensive rat Endothelium-derived hyperpolarizing factor 1996Amsterdam, The Netherlands: Harwood Academic Publishers; 235–245.ed. Vanhoutte, P.M., pp [Google Scholar]
- VANHEEL B., VAN DE VOORDE J., LEUSEN I. Contribution of nitric oxide to the endothelium-dependent hyperpolarization in rat aorta. J. Physiol. 1994;475:277–284. doi: 10.1113/jphysiol.1994.sp020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZANCHI A., AUBERT J.F., BRUNNER H.R., WAEBER B. Vascular acetylcholine response during chronic NOS inhibition: in vivo versus in vitro. Cardiovasc. Res. 1995;30:122–129. [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTÄTT E.D.Endothelium-dependent hyperpolarization and relaxation in the hepatic artery of the rat Endothelium-derived hyperpolarizing factor 1996Amsterdam, The Netherlands: Harwood Academic Publishers; 191–201.ed. Vanhoutte, P.M., pp [Google Scholar]
- ZYGMUNT P.M., WALDECK K., HÖGESTÄTT E.D. The endothelium-mediates a nitric-oxide independent hyperpolarization and relaxation in the rat hepatic artery. Acta Physiol. Scand. 1994;152:375–384. doi: 10.1111/j.1748-1716.1994.tb09819.x. [DOI] [PubMed] [Google Scholar]