

Abstract
We have previously demonstrated that continuous i.c.v. infusion of amyloid β-peptide (Aβ), the major constituent of senile plaques in the brains of patients with Alzheimer's disease, results in learning and memory deficits in rats.
In the present study, we investigated the effects of nefiracetam [N-(2,6-dimethylphenyl)-2-(2-oxo-1-pyrrolidinyl) acetamide, DM-9384] on Aβ-(1-42)-induced learning and memory deficits in rats.
In the Aβ-(1-42)-infused rats, spontaneous alternation behaviour in a Y-maze task, spatial reference and working memory in a water maze task, and retention of passive avoidance learning were significantly impaired as compared with Aβ-(40-1)-infused control rats.
Nefiracetam, at a dose range of 1–10 mg kg−1, improved learning and memory deficits in the Aβ-(1-42)-infused rats when it was administered p.o. 1 h before the behavioural tests.
Nefiracetam at a dose of 3 mg kg−1 p.o. increased the activity of choline acetyltransferase in the hippocampus of Aβ-(1-42)-infused rats.
Nefiracetam increased dopamine turnover in the cerebral cortex and striatum of Aβ-(1-42)-infused rats, but failed to affect the noradrenaline, serotonin and 5-hydroxyindoleacetic acid content.
These results suggest that nefiracetam may be useful for the treatment of patients with Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid β-peptide, nefiracetam (DM-9384), reference memory, voltage-sensitive calcium channels, working memory
Introduction
Alzheimer's disease (AD) is the most common cause of progressive decline of cognitive function in aged humans. The disease is characterized neuropathologically by the presence of numerous senile plaques and neurofibrillary tangles accompanied by neuronal loss. The senile plaques are composed of amyloid β-peptide (Aβ), a 40–42 amino acid peptide fragment of the β-amyloid precursor protein (APP) (Kang et al., 1987). Transgenic mice, which overexpress human APP containing the mutations associated with familial AD, develop many of the pathological characterizations associated with AD (Games et al., 1995; Johnson-Wood et al., 1997; Sturchler-Pierrat et al., 1997). Furthermore, Aβ is cytotoxic to neurons (Yankner et al., 1990) and renders neurons vulnerable to various insults including excitotoxicity (Koh et al., 1990; Mattson et al., 1992). These previous findings suggest that Aβ has a central role in the pathogenesis of AD.
We have previously demonstrated that continuous infusion of Aβ-(1-40) into the cerebral ventricle of rats results in learning and memory deficits, suggesting that accumulation of Aβ in the brain is related to cognitive impairments in AD (Nitta et al., 1994; Nabeshima & Nitta, 1994). Continuous Aβ-(1-40) infusion causes a decrease in choline acetyltransferase (ChAT) activity in the cerebral cortex and hippocampus (Nitta et al., 1994), changes in ciliary neurotrophic factor levels (Yamada et al., 1995a), activation of glial cells as shown by an increase in glial fibrillary acidic protein immunoreactivity (Nitta et al., 1997), and a deficiency of long-term potentiation in the CA1 field of the hippocampus (Nabeshima & Itoh, 1997). Repeated administration of propentofylline, which stimulates nerve growth factor synthesis/secretion in quiescent astroglial cells (Shinoda et al., 1990), improves Aβ-(1-40)-induced learning and memory deficits (Yamada et al., 1998). We have also demonstrated that NC-1900, an active fragment analogue of arginine vasopressin, improves cognitive dysfunction in this non-transgenic animal model of AD (Tanaka et al., 1998). Although the mechanisms of the brain dysfunction caused by accumulation of Aβ in the brain are obscure, dysfunction of cholinergic and dopaminergic neuronal systems may be responsible, since in vivo brain microdialysis revealed a marked reduction of nicotine- and/or KCl-induced stimulation of acetylcholine (ACh) release in the hippocampus/cerebral cortex and 3,4-dihydroxyphenylethylamine (dopamine) release occurred in the striatum without affecting the basal levels of these neurotransmitters (Itoh et al., 1996).
Nefiracetam, a pyrrolidone derivative, is a novel compound developed for drug therapy against cognitive disorders in aged humans (for review see Nabeshima, 1994; Yamada & Nabeshima, 1996). This compound shows remarkable effects in animals on cognitive impairments caused by various drug treatments (Nabeshima et al., 1990; 1991b; 1994), basal forebrain lesion (Nabeshima et al., 1991a), cerebral ischemia (Tanaka et al., 1992), and carbon monoxide exposure (Hiramatsu et al., 1992). Electrophysiological studies have demonstrated that nefiracetam modulates the currents through L-type voltage-sensitive calcium channels (VSCC) (Yoshii & Watabe, 1994), γ-aminobutyric acid (GABA)A receptors (Huang et al., 1996), and nicotinic ACh receptors (Nishizaki et al., 1998), by interacting with a cyclic AMP-dependent protein kinase and/or a Ca2+-dependent protein kinase C pathway. Since the ameliorating effect of nefiracetam on scopolamine-induced memory impairments is antagonized by treatment of VSCC antagonists, it is suggested that activation of VSCC is involved in the improving effect of nefiracetam on cognitive deficits (Yamada et al., 1994).
In the present study, we investigated the effects of nefiracetam on Aβ-induced impairments of spontaneous alternation behaviour in a Y-maze task, spatial reference and working memory in a water maze task, and retention of passive avoidance learning. Furthermore, the effect of this compound on ChAT activity and monoamine metabolism were examined in the Aβ-treated rats. Since recent evidence suggests that Aβ-(1-42) plays a more important role than Aβ-(1-40) in the pathology of AD (Jarrett & Lansbury, 1993; Iwatsubo et al., 1994), rats were continuously infused with Aβ-(1-42) into the cerebral ventricle. Control rats were infused with non-toxic reverse fragment Aβ-(40-1). We confirmed that the vehicle itself failed to induce any behavioural and neurochemical changes at this flow rate (Nitta et al., 1994).
Methods
Animals
The rats used in the present study were males of the Wistar strain (7 weeks old; Charles River Japan Inc., Yokohama, Japan) weighing 250±20 g at the beginning of experiments. They were housed in groups of two or three in a temperature- and light-controlled room (23°C; 12-h light cycle starting at 09 h 00 min) and had free access to food and water, except during the behavioural experiments. All experiments were performed in accordance with the Guidelines for Animal Experiments of the Nagoya University School of Medicine, the Guiding Principles for the Care and Use of Laboratory Animals approved by the Japanese Pharmacological Society, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Surgery
Rats were anaesthetized with pentobarbital (50 mg kg−1) i.p., and the infusion cannula for Aβ was implanted into the right ventricle (A: −0.3, L: 1.2, V: 4.5), according to the atlas of Paxinos & Watson (1986). Continuous infusion of Aβ-(1-42) (300 pmol day−1) was maintained for at least 2 weeks by attaching an infusion cannula to a mini-osmotic pump (Alzet 2002; Alza, Palo Alto, CA, U.S.A.) (Nitta et al., 1994). The control rats were infused with Aβ-(40-1). We have confirmed that the vehicle by itself has no effect on learning behaviour at this flow rate (Nitta et al., 1994; 1997).
Drug administration and experimental design
The experimental schedule is shown in Figure 1. The behavioural tests and the administration of nefiracetam were started on day 7 after the start of Aβ-(1-42) infusion. The behavioural tests were carried out sequentially. Nefiracetam at doses of 1, 3 and 10 mg kg−1 or saline was administered orally to Aβ-(1-42)-infused rats for 14 consecutive days, 1 h before the behavioural test throughout the experimental period. Aβ-(40-1)-infused control rats were administered with saline. Each group consisted of eight to nine rats. On day 20 after the start of Aβ infusion, rats were killed 1 h after the last administration of nefiracetam for the measurement of ChAT activity and the contents of monoamines and their metabolites in the brain.
Figure 1.
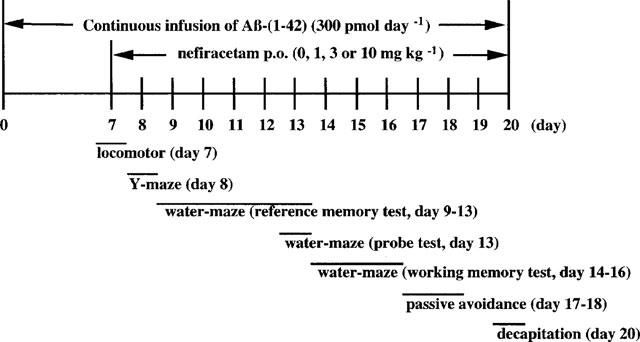
Experimental schedule.
Measurement of locomotor activity
Locomotor activity was measured on day 7 after the start of Aβ infusion. The experimental apparatus consisted of a locomotor cage (25×42×20 cm), with photobeams placed 2 cm above the floor at 1-inch intervals along two sides of the cage (Colombus Instruments, U.S.A.). Locomotor activity was measured during a 10-min period (Fuji et al., 1993).
Y-maze task
The Y-maze task in rats was carried out on day 8 after the start of Aβ infusion, as described previously (Maurice et al., 1994). The experimental apparatus consisted of a black-painted Y-maze made of plywood. Each arm of the Y-maze was 35 cm long, 25 cm high and 10 cm wide and positioned at an equal angle. Each rat was placed at the end of one arm and allowed to move freely through the maze during an 8-min session. The sequence of arm entries was recorded manually. A spontaneous alternation behaviour, which is regarded as a measure of spatial memory (Maurice et al., 1994; Yamada et al., 1996), was defined as entry into all three arms on consecutive choices in overlapping triplet sets. The per cent spontaneous alternation behaviour was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries−2)×100.
Water maze task
The water maze task (Morris, 1984), with some modification (Tanaka et al., 1998), was carried out from day 9 to 16 after the start of Aβ infusion. The experimental apparatus consisted of a circular water tank (140 cm in diameter and 45 cm high). A transparent platform (10 cm in diameter and 25 cm high) was set inside the tank, which was filled, to a height of 27 cm, with water of temperature approximately 23°C; the surface of the platform was 2 cm below the surface of the water. The pool was located in a large test room, in which there were many cues external to the maze (e.g. pictures, lamps, etc.). The position of the cues remained unchanged throughout the water maze task.
Reference memory test:
For each training trial, the rat was put into the pool at one of five starting positions, the sequence of the positions being selected randomly. The platform was located in a constant position throughout the test period in the middle of one quadrant, equidistant from the centre and edge of the pool. In each training session, the latency to escape onto the hidden platform was recorded. If the rat found the platform, it was allowed to remain there for 15 s and was then returned to its home cage. If the rat was unable to find the platform within 90 s, the training was terminated and a maximum score of 90 s was assigned. Training was conducted for 5 consecutive days, twice a day, from day 9 to day 13 after the start of Aβ infusion.
Probe test:
Immediately after the tenth training trial on day 13 after the start of Aβ infusion, the platform was removed from the pool and animals were tested on a 30 s spatial probe trial. The time spent in the platform-quadrant where the platform had been located during training was measured.
Working memory (repeated acquisition) test:
Working memory test was conducted for three consecutive days from day 14 to day 16 after the start of Aβ infusion, and consisted of five trials (one session) per day. The working memory test was procedurally similar to standard water maze test training, except that the platform location was changed for each session. Since the platform position was changed daily, this task evaluated working memory (Frick et al., 1995). For each trial, the rat was put into the pool at one of five starting positions, the sequence of the positions being selected randomly. The first trial of each session was an informative sample trial in which the rat was allowed to swim to the platform in its new location and to remain there for 15 s. The rat was then placed in a home cage for an intertrial interval of 1 min. The platform remained in the same location throughout the remaining four trials of the day. Spatial working memory was regarded as the mean escape latency of the second to fifth trials. The ability of working memory in each rat was assessed by the mean performance for three consecutive days from day 14 to day 16.
Multiple-trial passive avoidance task
The multiple-trial passive avoidance task was carried out on day 17 and 18 after the start of Aβ infusion, as described previously (Yamada et al., 1996). The experimental apparatus consisted of two compartments (25×15×15 cm high), one illuminated and one dark, both equipped with a grid floor. The two compartments were separated by a guillotine door. In the acquisition trial, each rat was placed in the illuminated compartment; when the animal entered the dark compartment, the door was closed and an inescapable footshock (0.3 mA, 5 s) was delivered through the grid floor. The rat was removed after receiving the footshock and was placed back into the light compartment by the experimenter. The door was again opened 30 s later to start the next trial. Training continued in this manner until the rat stayed in the light compartment for 120 s on a single trial. In the retention trial, given 24 h after the acquisition test, the rat was again placed in the illuminated compartment and the time taken to enter the dark compartment was measured as step-through latency. When the rat did not enter for at least 300 s, a score of 300 s was assigned.
Measurement of ChAT activity and monoamine and metabolite content
Rats were killed 1 h after the last administration of nefiracetam on day 20 after the start of Aβ-(1-42) infusion. The brains were dissected into four regions, the frontal cortex, parietal cortex, striatum and the hippocampus, after which they were rapidly frozen and stored in a deep freezer at −80°C until assayed. Measurement of ChAT activity was carried out as reported previously (Kaneda & Nagatsu, 1985). The content of dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-hydroxytryptamine (5-HT), 5-hydroxyindoleacetic acid (5-HIAA), and noradrenaline was determined by a high-performance liquid chromatography system with an electrochemical detector (Eicom, Kyoto, Japan), as described by Yamada et al. (1995b).
Drugs
Aβ-(1-42) and Aβ-(40-1) were obtained from Bachem (Torrance, CA, U.S.A.), and dissolved in 35% acetonitrile containing 0.1% trifluoroacetic acid at a concentration of 25 μM. Nefiracetam [N-(2,6-dimethylphenyl)-2-(2-oxo-1-pyrrolidinyl) acetamide, DM-9384] was kindly provided by Daiichi Pharmaceutical Co. Ltd. (Tokyo, Japan), and dissolved in saline.
Statistical analysis
Results were expressed as means±s.e.mean. Statistical significance between Aβ-(40-1)-treated control and Aβ-(1-42)-treated groups was determined by Student's t-test for locomotor activity, number of arm entries in the Y-maze task, number of training trials in the passive avoidance task, and the neurochemical data. Mann-Whitney U-test was also used for the two group comparison in spontaneous alternation beha-viour in Y-maze, performance in water maze task and step-through latency in the passive avoidance task. Statistical signi-ficance in the behavioural and neurochemical effects of nefir-acetam was determined by one-way analysis of variance (AN-OVA) or the Kruskal-Wallis test, followed by Bonferroni's test. Two-way ANOVA was also conducted for analysing data of the training trials of the water maze.
Results
Effects of nefiracetam on locomotor activity in the Aβ-(1-42)-treated rats
There was no difference in locomotor activity counts on day 7 after the start of Aβ infusion between Aβ-(40-1)-infused control (1734±21 counts/10 min, n=8) and the Aβ-(1-42)-infused group (1708±38 counts/10 min, n=9) (P=0.9112). The activity counts in the Aβ-(1-42)-infused rats that received an oral administration of nefiracetam at doses of 1, 3 and 10 mg kg−1 prior to the measurement of locomotor activity, were 1842±21, 2089±19, and 1817±37 counts/10 min, respectively. One-way ANOVA revealed that there was no statistical difference in locomotor activity counts between vehicle-treated and nefiracetam-treated Aβ-(1-42)-infused rats [F(3,30)=0.835, P=0.4853].
Effects of nefiracetam on spontaneous alternation behaviour in the Aβ-1-42)-treated rats
As shown in Figure 2A, the spontaneous alternation behaviour in the Aβ-(1-42)-infused group was significantly lower than that in the Aβ-(40-1)-infused control group (P<0.01). There was a statistical difference in spontaneous alternation behaviour between vehicle-treated and nefiracetam-treated Aβ-(1-42)-infused rats [KW=8.395, P=0.0385]. Post-hoc analysis revealed that nefiracetam at a dose of 10 mg kg−1 significantly attenuated the impairment of this behaviour induced by Aβ-(1-42) (P<0.05). The number of arm entries did not differ between the five different treatment groups of rats (Figure 2B), indicating that changes in alternation behaviour were not due to generalized exploratory, locomotor or motivational effects.
Figure 2.
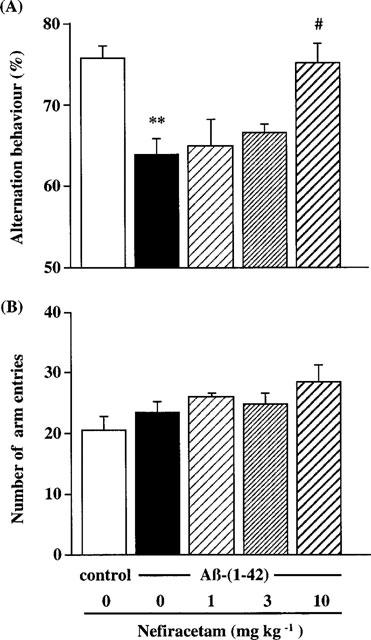
Effects of nefiracetam on spontaneous alternation behaviour in the Aβ-(1-42)-treated rats. Spontaneous alternation behaviour (A) and the number of arm entries (B) during an 8-min session in the Y-maze task were measured on day 8 after the start of Aβ infusion. Daily oral administration of nefiracetam was started on day 7, and spontaneous alternation behaviour was measured 1 h after the administration. Columns indicate means±s.e.mean (n=8–9). **P<0.01 vs Aβ-(40-1)-treated control rats. #P<0.05 vs Aβ-(1-42)-treated rats.
Effects of nefiracetam on performance of the water maze task in the Aβ-(1-42)-treated rats
Changes in escape latency time taken to find the hidden platform produced by training trials in each group of rats are shown in Figure 3A. Two-way ANOVA with all treatment groups revealed significant main effects of group [F(4,370)=3.437, P=0.0089] and training [F(9,370)=86.354, P<0.0001], but not group by trial interactions [F(36,370)= 0.9808, P>0.05). Post hoc analysis indicated that performance in the Aβ-(1-42)-infused group was significantly impaired, compared to that in the Aβ-(1-40)-infused control group (P<0.0188). Repeated daily administration of nefiracetam at a dose of 3 mg kg−1 significantly ameliorated the impairment of performance caused by Aβ-(1-42) (P<0.001) (Figure 3A). Nefiracetam at 10 mg kg−1 also improved performance in the Aβ-(1-42)-treated rats, but the effect was not significant (P=0.0514).
Figure 3.
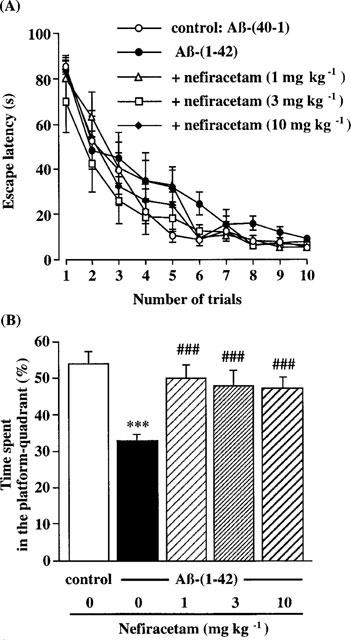
Effects of nefiracetam on performance in the training trials (A) and in the probe trial (B) of the water maze task in the Aβ-(1-42)-treated rats. The training trials were carried out on days 9–13 after the start of Aβ infusion. The probe trial was carried out on day 13 after the start of Aβ infusion, immediately after the tenth training trial. Daily oral administration of nefiracetam was started on day 7, and the training was carried out 1 h after the administration. Symbols indicate the means±s.e.mean (n=8–9). ***P<0.001 vs Aβ-(40-1)-treated control rats. ###P<0.001 vs Aβ-(1-42)-treated rats.
A 30-s spatial probe trial was carried out on day 13 after the start of Aβ-(1-42) infusion, following the tenth training trial (Figure 3B). The Aβ-(1-42)-infused group, compared with the Aβ-(40-1)-treated control group, showed a significant decrease in the time spent in the quadrant in which the platform had been located (platform-quadrant) during training (control group: 53.8±3.9%, Aβ-(1-42)-infused group: 33.0±2.4%) (Figure 3B). There was a significant group effect on the time spent in the platform-quadrant between the vehicle- and nefiracetam-treated Aβ-(1-42)-infused groups [KW=11.098, P<0.0112]. Post-hoc analysis revealed that all doses of nefiracetam examined in the present study significantly increased the time spent in the platform-quadrant (P<0.01), relative to the Aβ-(40-1)-treated control group.
The escape latencies in the first trial (sample trial) and in the second to fifth trials (test trials) of the working memory test are shown in Figure 4A and B, respectively. Although there was no difference in the escape latency in the sample trial (Figure 4A), a marked difference in the test trials was observed between Aβ-(40-1)-infused control and Aβ-(1-42)-treated groups (P<0.001) (Figure 4B). Nefiracetam significantly and dose-dependently ameliorated the impairment of performance in the test trials in the Aβ-(1-42)-infused rats (P<0.001) (Figure 4B), without affecting the escape latency in the sample trial (Figure 4A).
Figure 4.

Effects of nefiracetam on performance in the sample trial (A) and in the test trials (B) of the working memory test of the water maze task in the Aβ-(1-42)-treated rats. The working memory test (five trials per day) was carried out on days 14–16 after the start of Aβ infusion. Daily oral administration of nefiracetam was started on day 7, and the test was carried out 1 h after the administration. Values are the means±s.e.mean (n=8–9). ***P<0.001 vs Aβ-(40-1)-treated control rats. ###P<0.001 vs Aβ-(1-42)-treated rats.
Effects of nefiracetam on performance of the multi-trial passive avoidance task in the Aβ-(1-42)-treated rats
In the acquisition trial, there was no difference in the step-through latency (Figure 5A) or the number of training trials (Figure 5B) among the five treatment groups. Furthermore, there was no apparent difference in vocalization of rats in each treatment group when they received a footshock.
Figure 5.
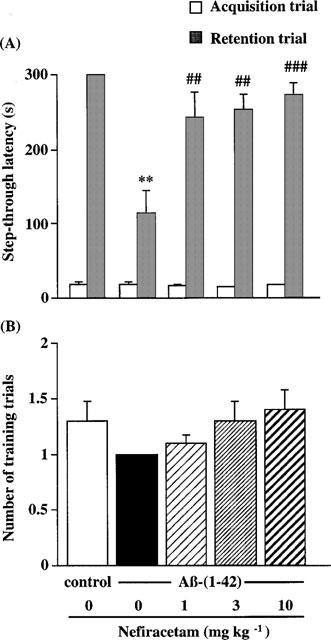
Effects of nefiracetam on step-through latency (A) and the number of training trials (B) in the multiple-trial passive avoidance task in the Aβ-(1-42)-treated rats. The task was carried out on days 17–18 after the start of Aβ infusion. Daily oral administration of nefiracetam was started on day 7, and the passive avoidance task was carried out 1 h after the administration. Values are the means±s.e.mean (n=8–9). **P<0.01 vs Aβ-(40-1)-treated control rats. ##P<0.01, ###P<0.001 vs Aβ-(1-42)-treated rats.
In the retention trial, however, a significant reduction of the step-through latency was observed in the Aβ-(1-42)-treated groups compared with the Aβ-(40-1)-treated control group (P<0.01). The deficits of retention of the passive avoidance learning in the Aβ-(1-42)-treated rats were improved by the treatment with nefiracetam at doses of 1, 3, and 10 mg kg−1 [KW=10.110, P=0.0177].
Effects of nefiracetam on choline acetyltransferase activity in the Aβ-(1-42)-treated rats
Table 1 shows that continuous infusion of Aβ-(1-42) had no significant effect on ChAT activity in the hippocampus, frontal cortex or the striatum. Repeated oral administration of nefiracetam at a dose of 3 mg kg−1 for 14 days caused a significant increase (25%) in ChAT activity in the hippocampus, but not in other areas of the brain such as the frontal cortex and striatum, of the Aβ-(1-42)-treated rats. Nefiracetam, at doses of 1 and 10 mg kg−1, failed to affect ChAT activity in any of the brain areas examined.
Table 1.
Effects of nefiracetam on the ChAT activity in the brains of rats continuously infused with Aβ-(1-42) into the cerebral ventricle

Effects of nefiracetam on monoamine and metabolite content in the Aβ-(1-42)-treated rats
Table 2 shows the effects of nefiracetam on dopamine metabolism in the brains of rats continuously infused with Aβ-(1-42) into the cerebral ventricle. It is evident that continuous infusion of Aβ-(1-42) had no significant effect on dopamine, DOPAC and HVA levels in the frontal and parietal cortex, striatum and hippocampus (Table 2). There were also no differences in 5-HT, 5-HIAA and noradrenaline contents in any of these areas of the brain between Aβ-(40-1)-infused control and Aβ-(1-42)-treated animals (data not shown).
Table 2.
Effects of nefiracetam on dopamine metabolism in the brains of rats continuously infused with Aβ-(1-42) into the cerebral ventricle
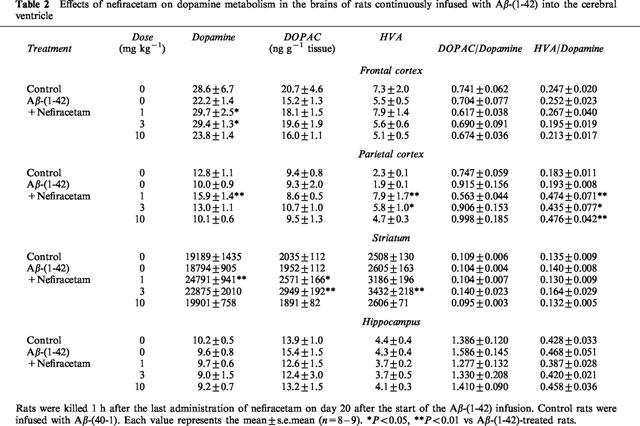
Nefiracetam significantly increased dopamine content in the frontal and parietal cortex, and striatum, but not hippocampus, in the Aβ-(1-42)-treated rats. The effect was most significant at the lowest dose (1 mg kg−1). Nefiracetam also increased DOPAC levels in the parietal cortex, and DOPAC and HVA levels in the striatum at doses of 1 and 3 mg kg−1. Fur-thermore, this compound increased the DOPAC : dopamine ratio in the parietal cortex, by more than 2 fold, at the dose range of 1–10 mg kg−1. Nefiracetam did not affect the content of 5-HT, 5-HIAA or noradrenaline in any areas of the brain examined in the Aβ-(1-42)-treated animals (data not shown).
Discussion
Previous studies, including those from our laboratory, have demonstrated that intracerebral injection of Aβ causes brain dysfunction as evidenced by neurodegeneration and an impairment of learning and memory (Kowall et al., 1991; Flood et al., 1994; Nitta et al., 1994; 1997; Giovannelli et al., 1995; Maurice et al., 1996; Yamada et al., 1998) although neurotoxic effects of Aβ in vivo have been somewhat controversial (Clemens & Stephenson, 1992; Games et al., 1992; Winkler et al., 1994). In the present study, therefore, we extensively investigated behavioural changes in Aβ-(1-42)-treated rats.
We observed that continuous infusion of Aβ-(1-42) into the cerebral ventricle caused an impairment of spontaneous alternation behaviour in a Y-maze and performance in a water maze task, both of which are considered to involve spatial memory. Retention of long-term passive avoidance learning was also impaired by the continuous infusion of Aβ-(1-42). The feature of learning and memory impairments in rats induced by continuous infusion of Aβ-(1-42) is, in general, similar to that found in rats infused with Aβ-(1-40) (Nitta et al., 1994; 1997; Yamada et al., 1998; Tanaka et al., 1998). However, ChAT activities in the brains of Aβ-(1-42)-infused rats did not differ from those in the Aβ-(40-1)-infused control rats whereas continuous infusion of Aβ-(1-40) decreased the hippocampal ChAT activity (Nitta et al., 1994; 1997; Yamada et al., 1998). Therefore, there may be some difference in the neurotoxicity in vivo caused by Aβ-(1-40) and Aβ-(1-42).
It is unlikely that the impairment of performance of the Aβ-(1-42)-treated rats in learning and memory tasks is due to changes in motivation or sensorimotor function since different motivation is involved in these behavioural tasks, and different skill is required for better performance in each task. Actually, the locomotor activity and the number of total arm entries in the Y-maze task in the Aβ-(1-42)-treated rats did not differ from those in the Aβ-(40-1)-treated control rats, indicating no changes in motor function and exploratory activity. There were also no differences between the Aβ-(40-1)-treated control and Aβ-(1-42)-treated rats in the sensitivity to electric footshock in the acquisition trial of the passive avoidance task and the escape latency onto the submerged platform in the first training trial and the sample trial of working memory test in the water maze task. These results indicate no changes in shock sensitivity and swimming ability. Therefore, it is likely that impairment of performance in the Aβ-(1-42)-treated rats is due to learning and memory deficits.
Nefiracetam, at a dose range of 1–10 mg kg−1, significantly ameliorated impairment of spontaneous alternation behaviour in the Y-maze task, performance in the water maze task, and shortened step-through latency in the retention trial of passive avoidance task in the Aβ-(1-42)-treated rats. This compound did not affect locomotor activity, sensitivity to electric footshock in the acquisition trial of the passive avoidance task or the escape latency onto the submerged platform in the first training trial and the sample trial of working memory test in the water maze task in the Aβ-(1-42)-treated rats. Therefore, it is unlikely that the observed improvement by nefiracetam of performance in various cognitive tasks is due to changes in sensorimotor function and/or motivation in the Aβ-(1-42)-treated rats. Rather, it is plausible that nefiracetam ameliorates learning and memory deficits caused by the continuous infusion of Aβ-(1-42) into the cerebral ventricle in rats.
The mechanism of learning and memory dysfunction in the Aβ-(1-42)-treated rats is unknown at present. We have previously demonstrated, by using in vivo brain dialysis, that high potassium- and nicotine-induced increase in ACh and dopamine release in the hippocampus/cerebral cortex and the striatum, respectively, is markedly impaired by the continuous infusion of Aβ-(1-40) although the basal levels of these neurotransmitters in the Aβ-(1-40)-infused rats did not differ from those in vehicle-infused control animals (Itoh et al., 1996). Harkany et al. (1998) showed that bilateral injection of β-amyloid(Phe(SO3H)24)25-35 peptide, a metabolically stable analogue of Aβ-(25-35), into the rat nucleus basalis magnocellularis caused a reduction of cortical acetylcholinesterase-positive projections. Maurice et al. (1996) demonstrated that tacrine, a cholinesterase inhibitor, and (−)-nicotine improved the Aβ-(25-35)-induced impairment of spontaneous alternation behaviour in a Y-maze, passive avoidance learning, and water maze learning in mice. An in vitro experiment has shown that Aβ-(25-35) activates tau protein kinase 1/glycogen synthase kinase 3β, leading to an inactivation of pyruvate dehydrogenase, and causes an impairment of ACh synthesis without affecting ChAT activity (Hoshi et al., 1996). In light of these previous findings, we consider that dysfunctions of cholinergic and dopaminergic neuronal systems are responsible, at least in part for the Aβ-induced learning and memory deficits.
Nefiracetam activates VSCC in vitro (Yoshii & Watabe, 1994) and the effect is involved in the amelioration by nefiracetam of scopolamine-induced memory impairment (Yamada et al., 1994). Accordingly, it is plausible that nefiracetam ameliorates the Aβ-(1-42)-induced learning and memory deficits by activating VSCC, and thereby stimulates release of ACh and dopamine in the brain. Indeed, it has been shown that nefiracetam stimulates the turnover of ACh (Abe et al., 1994; Kawajiri et al., 1994), dopamine (Abe et al., 1992; 1994) and GABA (Watabe et al., 1993), as well as ACh release in the frontal cortex (Sakurai et al., 1998). In the present study, we have demonstrated that nefiracetam can increase dopamine turnover in the cerebral cortex and striatum in the Aβ-treated rats.
One might argue against our hypothesis that the amelioration by nefiracetam of Aβ-(1-42)-induced learning and memory deficits is due to the activation of VSCC, since accumulating evidence suggests that high concentration of Aβ destabilizes intracellular calcium mobilization (Fraser et al., 1997). Some studies have suggested that calcium influx through VSCC contributes to Aβ-induced neurotoxicity in vitro and that VSCC antagonists, by reducing such influx, are able to attenuate Aβ-induced neurotoxicity (Weiss et al., 1994; Abe & Kimura, 1996).
We do not argue against the calcium hypothesis of the Aβ-induced neurotoxicity. However, ChAT activities in the brains of Aβ-(1-42)-treated rats did not differ from those in Aβ-(40-1)-treated control rats, suggesting that memory deficits in the Aβ-(1-42)-treated rats are not directly associated with cholinergic neurotoxicity. Furthermore, nefiracetam did not improve nor potentiate the Aβ-(1-42)-induced learning and memory impairments if daily administration of this compound began 3 days before the start of Aβ-(1-42) infusion (unpublished observation). In such experiments, nefiracetam was administered p.o. 1 h after the behavioural test. Therefore, it is unlikely that the ameliorating effects of nefiracetam observed in the present study are due to its neuroprotective effect against Aβ-(1-42). Rather it is possible that amelioration by nefiracetam of Aβ-(1-42)-induced learning and memory impairments is associated with its acute memory-enhancing effect (Nabeshima, 1994).
It has been suggested that low concentrations of soluble Aβ can potently induce cholinergic and dopaminergic hypoactivity (Itoh et al., 1996) that is not dependent on concurrent neurotoxicity (for review see Auld et al., 1998). Accordingly, we speculate that learning and memory deficits observed in the Aβ-(1-42)-treated rats are accompanied by hypoactivity of cholinergic and dopaminergic neurons, which is not directly related to the neurodegeneration and that nefiracetam can ameliorate learning and memory impairments by stimulating ACh (Sakurai et al., 1998) and dopamine release through the activation of VSCC.
In addition, nefiracetam modulates GABAA (Huang et al., 1996) or nicotinic ACh receptor currents (Nishizaki et al., 1998), stimulates ChAT and glutamate decarboxylase activities (Shiotani et al., 1992; Tanaka et al., 1992; Watabe et al., 1993), and muscarinic ACh receptor binding (Nabeshima et al., 1991b). Therefore, these effects may also contribute to the amelioration by nefiracetam of Aβ-(1-42)-induced learning and memory deficits. In fact, we observed an increase in the hippocampal ChAT activity in the Aβ-(1-42)-treated rats which had received repeated oral administration of nefiracetam at a dose of 3 mg kg−1 for 14 days although the effect was not dose-related.
In conclusion, we have demonstrated in the present study that a novel cognition enhancing drug, nefiracetam, ameliorates learning and memory deficits in rats which had previously received a continuous infusion of Aβ-(1-42) into the cerebral ventricle. Our findings suggest that nefiracetam may be useful for the treatment of patients with AD and that clinical trials of nefiracetam for the treatment of AD are warranted.
Acknowledgments
We are grateful to Daiichi Pharmaceutical Co. Ltd. (Tokyo, Japan) for the generous donation of nefiracetam. This study was supported, in part, by Grants-in-Aid for Science Research from the Ministry of Education, Science, and Culture of Japan (No. 07557009, 08457027, 10897005 and 97450) and by an SRF Grant for Biomedical Research.
Abbreviations
- 5-HIAA
5-hydroxyindoleacetic acid
- Aβ
amyloid β-peptide
- ACh
acetylcholine
- AD
Alzheimer's disease
- ANOVA
one-way analysis of variance
- APP
β-amyloid precursor protein
- ChAT
choline acetyltransferase
- DOPAC
3,4-dihydroxyphenylacetic acid
- GABA
γ-aminobutyric acid
- HVA
homovanillic acid
- VSCC
voltage-sensitive calcium channels
References
- ABE K., KIMURA H. Amyloid β toxicity consists of a Ca2+-independent early phase and a Ca2+-dependent late phase. J. Neurochem. 1996;67:2074–2078. [PubMed] [Google Scholar]
- ABE E., MURAI S., SAITO H., MASUDA Y., ITOH T. Effects of nefiracetam, a novel pyrrolidone derivative, on brain monoamine metabolisms in mice. J. Neural. Transm. 1992;90:125–136. doi: 10.1007/BF01250794. [DOI] [PubMed] [Google Scholar]
- ABE E., MURAI S., SAITO H., MASUDA Y., ITOH T. Effects of nefiracetam on deficits in active avoidance response and hippocampal cholinergic and monoaminergic dysfunctions induced by AF64A in mice. J. Neural. Transm. 1994;95:179–193. doi: 10.1007/BF01271565. [DOI] [PubMed] [Google Scholar]
- AULD D.S., KAR S., QUIRION R. β-Amyloid peptides as direct cholinergic neuromodulators: a missing link. Trends Neurosci. 1998;21:43–49. doi: 10.1016/s0166-2236(97)01144-2. [DOI] [PubMed] [Google Scholar]
- CLEMENS J.A., STEPHENSON D.T. Implants containing beta-amyloid protein are not neurotoxic to young and old rat brain. Neurobiol. Aging. 1992;13:581–586. doi: 10.1016/0197-4580(92)90059-7. [DOI] [PubMed] [Google Scholar]
- FLOOD J.F., MORLEY J.E., ROBERTS E. An amyloid β-protein fragment, Aβ[12-28] equipotently impairs post-training memory processing when injected into different limbic structures. Brain Res. 1994;663:271–276. doi: 10.1016/0006-8993(94)91273-4. [DOI] [PubMed] [Google Scholar]
- FRASER S.P., SUY Y.-H., DJAMGOS M.B.A. Ionic effects of Alzheimer's disease β-amyloid precursor protein and its metabolic fragments. Trends Neurosci. 1997;20:67–72. doi: 10.1016/s0166-2236(96)10079-5. [DOI] [PubMed] [Google Scholar]
- FRICK K., BAXTER M.G., MARKOWSKA A.L., OLTON D.S., PRICE D.L. Age-related spatial reference and working memory deficits assessed in the water maze. Neurobiol. Aging. 1995;16:149–160. doi: 10.1016/0197-4580(94)00155-3. [DOI] [PubMed] [Google Scholar]
- FUJI K., HIRAMATSU M., KAMEYAMA T., NABESHIMA T. Effects of repeated administration of propentofylline on memory impairment produced by basal forebrain lesion in rats. Eur. J. Pharmacol. 1993;236:411–417. doi: 10.1016/0014-2999(93)90479-2. [DOI] [PubMed] [Google Scholar]
- GAMES D., ADAMS D., ALESSANDRINI R., BARBOUR R., BERTHELETTE P., BLACKWELL C., CARR T., CLEMENS J., DONALDSON T., GILLESPIE F., GUIDO T., HAGOPIAN S., JOHNSON-WOOD K., KHAN K., LEE M., LEIBOWITZ P., LIEBERBURG I., LITTLE S., MASLIAH E., MCCONLOGUE L., MONTOYA-ZAVALA M., MUCKE L., PAGANINI L., PENNIMAN E., POWER M., SCHENK D., SEUBERT P., SNYDER B., SORIANO F., TAN H., VITALE J., WADSWORTH S., WOLOZIN B., ZHAO J. Alzheimer-type neuropathology in transgenic mice overexpressing V717Fβ-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- GAMES D., KHAN K.M., SORIANO F.G., KEIM P.S., DAVIS D.L., BRYANT K., LIEBERBURG I. Lack of Alzheimer pathology after beta-amyloid protein injections in rat brain. Neurobiol. Aging. 1992;13:569–576. doi: 10.1016/0197-4580(92)90057-5. [DOI] [PubMed] [Google Scholar]
- GIOVANNELLI L., CASAMENTI F., SCALI C., BARTOLINI L., PEPEU G. Differential effects of amyloid peptides β-(1-40) and β-(25-35) injections into the rat nucleus basalis. Neuroscience. 1995;66:781–792. doi: 10.1016/0306-4522(94)00610-h. [DOI] [PubMed] [Google Scholar]
- HARKANY T., O'MAHONY S., KELLY J.P., SOÓS K., TÖRO I., PENKE B., LUITEN P.G.M., NYAKAS C., GULYA K., LEONARD B.E. β-Amyloid(Phe)SO3H)24)25-35 in rat nucleus basalis induces behavioural dysfunctions, impairs learning and memory and disrupts cortical cholinergic innervation. Behav. Brain Res. 1998;90:133–145. doi: 10.1016/s0166-4328(97)00091-0. [DOI] [PubMed] [Google Scholar]
- HIRAMATSU M., KOIDE T., ISHIHARA S., SHIOTANI T., KAMEYAMA T., NABESHIMA T. Involvement of the cholinergic system in the effects of nefiracetam (DM-9384) on carbon monoxide (CO)-induced acute and delayed amnesia. Eur. J. Pharmacol. 1992;216:279–285. doi: 10.1016/0014-2999(92)90371-a. [DOI] [PubMed] [Google Scholar]
- HOSHI M., TAKASHIMA A., NOGUCHI K., MURAYAMA M., SATO M., KONDO S., SAITO Y., ISHIGURO K., HOSHINO T., IMAHORI K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3β in brain. Proc. Natl. Acad. Sci. U.S.A. 1996;93:2719–2723. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG C.S., MA J.Y., MARSZALEC W., NARAHASHI T. Effects of the nootropic drug nefiracetam on the GABAA receptor-channel complex in dorsal root ganglion neurons. Neuropharmacology. 1996;35:1251–1261. doi: 10.1016/s0028-3908(96)00074-3. [DOI] [PubMed] [Google Scholar]
- ITOH A., NITTA A., NADAI M., NISHIMURA K., HIROSE M., HASEGAWA T., NABESHIMA T. Dysfunction of cholinergic and dopaminergic neuronal systems in β-amyloid protein-infused rats. J. Neurochem. 1996;66:1113–1117. doi: 10.1046/j.1471-4159.1996.66031113.x. [DOI] [PubMed] [Google Scholar]
- IWATSUBO T., ODAKA A., SUZUKI N., MIZUSAWA H., NUKINA N., IHARA Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- JARRETT J.T., LANSBURY P.T., JR Seedling ‘one-dimensional crystallization' of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie. Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- JOHNSON-WOOD K., LEE M., MOTTER R., HU K., GORDON G., BARBOUR R., KHAN K., GORDON M., TAN H., GAMES D., LIEBERBURG I., SCHENK D., SEUBERT P., MCCONLOGUE L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANEDA N., NAGATSU T. Highly sensitive assay for choline acetyltransferase activity by high-performance liquid chromatography with electromechanical detection. J. Chromatogr. 1985;341:23–30. doi: 10.1016/s0378-4347(00)84006-2. [DOI] [PubMed] [Google Scholar]
- KANG J., LEMAIRE H.G., UNTERBECK A., SALBAUM J.M., MASTERS C.L., GRZESCHIK K.H., MULTHAUP G., BEYREUTHER K., MULLER-HILL B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- KAWAJIRI S., TANIGUCHI K., SAKURAI T., YAMAZAKI T. Nefiracetam enhances acetylcholine outflow from the frontal cortex: in vivo microdialysis study in the rat. J. Neural. Transm. 1994;98:15–22. doi: 10.1007/BF01277591. [DOI] [PubMed] [Google Scholar]
- KOH J.-Y., YANG L.L., COTMAN C.W. β-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 1990;533:315–320. doi: 10.1016/0006-8993(90)91355-k. [DOI] [PubMed] [Google Scholar]
- KOWALL W.N., BEAL F.M., BUSCIGLIO J., DUFFY K.L., YANKNER B.A. An in vivo model for the neurodegenerative effects of β-amyloid and protection by substance P. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATTSON M.P., CHENG B., DAVIS D., BRYANT K., LIEBERBURG I., RYDEL R.E. β-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAURICE T., HIRAMATSU M., ITOH J., KAMEYAMA T., HASEGAWA T., NABESHIMA T. Behavioural evidence for a modulating role of σ ligands in memory process. I. Attenuation of dizocilpine (MK-801)-induced amnesia. Brain Res. 1994;647:44–56. doi: 10.1016/0006-8993(94)91397-8. [DOI] [PubMed] [Google Scholar]
- MAURICE T., LOCKHART B.P., PRIVAT A. Amnesia induced by centrally administered β-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- MORRIS R.G.M. Developments of water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- NABESHIMA T. Ameliorating effects of nefiracetam (DM-9384) on brain dysfunction. Drugs Today. 1994;30:357–379. [Google Scholar]
- NABESHIMA T., ITOH A.Toxicity of β-amyloid protein: neurochemical, histological and behavioural changes Alzheimer's Disease: Biology, Diagnosis and Therapeutics 1997England: John Wiley & Sons Ltd; 623–630.eds Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, pp [Google Scholar]
- NABESHIMA T., NAKAYAMA S., ICHIHARA K., YAMADA K., SHIOTANI T., HASEGAWA T. Effects of nefiracetam on drug-induced impairment of latent learning in mice in a water finding task. Eur. J. Pharmacol. 1994;255:57–65. doi: 10.1016/0014-2999(94)90082-5. [DOI] [PubMed] [Google Scholar]
- NABESHIMA T., NITTA A. Memory impairment and neuronal dysfunction induced by β-amyloid protein in rats. Tohoku J. Exp. Med. 1994;174:241–249. doi: 10.1620/tjem.174.241. [DOI] [PubMed] [Google Scholar]
- NABESHIMA T., NODA Y., TOHYAMA K., ITOH J., KAMEYAMA T. Effects of DM-9384 in a model of amnesia based on animals with GABAergic neuronal dysfunctions. Eur. J. Pharmacol. 1990;178:143–149. doi: 10.1016/0014-2999(90)90469-m. [DOI] [PubMed] [Google Scholar]
- NABESHIMA T., OGAWA S., KAMEYAMA T., SHIOTANI T., TAKASU Y., SAKURAI T., HASEGAWA M., HASEGAWA T. Effects of DM-9384 and aniracetam on learning in normal and basal forebrain-lesioned rats. Res. Commun. Chem. Pathol. Pharmacol. 1991a;74:141–152. [Google Scholar]
- NABESHIMA T., TOHYAMA K., MURASE K., ISHIHARA S., KAMEYAMA T., YAMASAKI T., HATANAKA S., KOJIMA H., SAKURAI T., TAKASU Y., SHIOTANI T. Effects of DM-9384, a cyclic derivative of GABA, on amnesia and decrease in GABAA and muscarinic receptors induced by cycloheximide. J. Pharmacol. Exp. Ther. 1991b;257:271–275. [PubMed] [Google Scholar]
- NISHIZAKI T., MATSUOKA T., NOMURA T., SUMIKAWA K., SHIOTANI T., WATABE S., YOSHII M. Nefiracetam modulates acetylcholine receptor currents via two different signal transduction pathways. Mol. Pharmacol. 1998;53:1–5. doi: 10.1124/mol.53.1.1. [DOI] [PubMed] [Google Scholar]
- NITTA A., FUKUTA T., HASEGAWA T., NABESHIMA T. Continuous infusion of β-amyloid protein into cerebral ventricle induces learning impairment and neuronal and morphological degeneration. Jpn. J. Pharmacol. 1997;73:51–57. doi: 10.1254/jjp.73.51. [DOI] [PubMed] [Google Scholar]
- NITTA A., ITOH A., HASEGAWA T., NABESHIMA T. β-Amyloid protein-induced Alzheimer's disease animal model. Neurosci. Lett. 1994;170:63–66. doi: 10.1016/0304-3940(94)90239-9. [DOI] [PubMed] [Google Scholar]
- PAXINOS G., WATSON C. The Rat Brain in the Stereotaxic Coordinates. New York: Academic Press; 1986. [Google Scholar]
- SAKURAI T., KATO T., MORI K., TAKANO E., WATABE S., NABESHIMA T. Nefiracetam elevates extracellular acetylcholine level in the frontal cortex of rats with cerebral dysfunctions: an in vivo microdialysis study. Neurosci. Lett. 1998;246:69–72. doi: 10.1016/s0304-3940(98)00244-4. [DOI] [PubMed] [Google Scholar]
- SHINODA I., FURUKAWA Y., FURUKAWA S. Stimulation of nerve growth factor synthesis/secretion by propentofylline in cultured mouse astroglial cells. Biochem. Pharmacol. 1990;49:1813–1816. doi: 10.1016/0006-2952(90)90130-d. [DOI] [PubMed] [Google Scholar]
- SHIOTANI T., TOHYAMA K., MURASE K., ISHIHARA S., KAMEYAMA T., YAMASAKI T., HATANAKA S., KOJIMA H., TAKASU Y., NABESHIMA T. Effects of nefiracetam, DM-9384, on amnesia and decrease in cholineacetyltransferase activity induced by cycloheximide. J. Neural. Transm. 1992;90:103–111. doi: 10.1007/BF01250792. [DOI] [PubMed] [Google Scholar]
- STURCHLER-PIERRAT C., ABRAMOWSKI D., DUKE M., WIEDERHOLD K.-H., MISTL C., ROTHACHER S., LEDERMANN B., BÜRKI K., FREY P., PAGANETTI P.A., WARIDEL C., CALHOUN M., JUCKER M., PROBST A., STAUFENBIEL M., SOMMER B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA S., WATABE S., KAKIHATA K., SAKURAI T., ENDO W., YAMAGUCHI H., ASHIDA S. Effects of the new cognition-enhancing agent nefiracetam in rats with cerebral embolism. Arzneim-Forsch/Drug Res. 1992;42:1274–1278. [PubMed] [Google Scholar]
- TANAKA T., YAMADA K., SENZAKI K., NARIMATSU H., NISHIMURA K., KAMEYAMA T., NABESHIMA T. NC-1900, an active fragment analog of arginine vasopressin, improves learning and memory deficits induced by β-amyloid protein in rats. Eur. J. Pharmacol. 1998;352:135–142. doi: 10.1016/s0014-2999(98)00344-6. [DOI] [PubMed] [Google Scholar]
- WATABE S., YAMAGUCHI H., ASHIDA S. DM-9384, a new cognition-enhancing agent, increases the turnover of components of the GABAergic system in the rat cerebral cortex. Eur. J. Pharmacol. 1993;238:303–309. doi: 10.1016/0014-2999(93)90861-b. [DOI] [PubMed] [Google Scholar]
- WEISS J.H., PIKE C.J., COTMAN C.W. Ca2+ channel blockers attenuate β-amyloid peptide toxicity to cortical neurons in culture. J. Neurochem. 1994;62:372–375. doi: 10.1046/j.1471-4159.1994.62010372.x. [DOI] [PubMed] [Google Scholar]
- WINKLER J., CONOER D.J., FRAUTSCHY S.A., BEHL C., WAITE J.J., COLE G.M., THAL L.J. Lack of long-term effects after β-amyloid protein injections in rat brain. Neurobiol. Aging. 1994;15:601–607. doi: 10.1016/0197-4580(94)00054-9. [DOI] [PubMed] [Google Scholar]
- YAMADA K., NABESHIMA T. Nefiracetam (DM-9384): a novel antiamnesic drug. CNS Drug Rev. 1996;2:322–342. [Google Scholar]
- YAMADA K., NAKAYAMA S., SHIOTANI T., HASEGAWA T., NABESHIMA T. Possible involvement of the activation of voltage-sensitive calcium channels in the ameliorating effects of nefiracetam on scopolamine-induced impairment of performance in a passive avoidance task. J. Pharmacol. Exp. Ther. 1994;270:881–892. [PubMed] [Google Scholar]
- YAMADA K., NITTA A., SAITO T., HU J., NABESHIMA T. Changes in ciliary neurotrophic factor content in the rat brain after continuous intracerebroventricular infusion of β-amyloid (1-40) protein. Neurosci. Lett. 1995a;201:155–158. doi: 10.1016/0304-3940(95)12161-7. [DOI] [PubMed] [Google Scholar]
- YAMADA K., NODA Y., NAKAYAMA S., KOMORI Y., SUGIHARA H., HASEGAWA T., NABESHIMA T. Role of nitric oxide in learning and memory and in monoamine metabolism in the rat brain. Br. J. Pharmacol. 1995b;115:852–858. doi: 10.1111/j.1476-5381.1995.tb15011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA K., NODA Y., HASEGAWA T., KOMORI Y., NIKAI T., SUGIHARA H., NABESHIMA T. The role of nitric oxide in dizocilpine-induced impairment of spontaneous alternation behaviour in mice. J. Pharmacol. Exp. Ther. 1996;276:460–466. [PubMed] [Google Scholar]
- YAMADA K., TANAKA T., SENZAKI K., KAMEYAMA T., NABESHIMA T. Propentofylline improves learning and memory deficits in rats induced by β-amyloid protein-(1-40) Eur. J. Pharmacol. 1998;349:15–22. doi: 10.1016/s0014-2999(98)00166-6. [DOI] [PubMed] [Google Scholar]
- YANKNER B.A., DUFFY L.K., KIRSCHNER D.A. Neurotrophic and neurotoxic effects of amyloid β protein: reversla by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- YOSHII M., WATABE S. Enhancement of neuronal calcium channel currents by the nootropic agent, nefiracetam (DM-9384), in NG108-15 cells. Brain Res. 1994;642:123–131. doi: 10.1016/0006-8993(94)90913-x. [DOI] [PubMed] [Google Scholar]