

Abstract
The thienopyridine clopidogrel is a specific inhibitor of ADP-induced platelet aggregation ex vivo. No direct effects of clopidogrel (⩽100 μM) on platelet aggregation in vitro have been described so far.
Possible in vitro antiaggregatory effects (turbidimetry) of clopidogrel were studied in human platelet-rich plasma and in washed platelets.
Incubation of platelet-rich plasma with clopidogrel (⩽100 μM) for up to 8 h did not result in any inhibition of ADP (6 μM)-induced platelet aggregation.
Incubation of washed platelets with clopidogrel resulted in a time- (maximum effects after 30 min) and concentration-dependent (IC50 1.9±0.3 μM) inhibition of ADP (6 μM)-induced platelet aggregation. Clopidogrel (30 μM) did not inhibit collagen (2.5 μg ml−1)-, U46619 (1 μM)- or thrombin (0.1 u ml−1)-induced platelet aggregation. The inhibition of ADP-induced aggregation by clopidogrel (30 μM) was insurmountable indicating a non-equilibrium antagonism of ADP actions. The R enantiomer SR 25989 C (30 μM) was significantly less active than clopidogrel (30 μM) in inhibiting platelet aggregation (32±5 % vs 70±1 % inhibition, P<0.05, n=5).
In washed platelets, clopidogrel (⩽30 μM) did not significantly reverse the inhibition of prostaglandin E1 (1 μM)-induced platelet cyclic AMP formation by ADP (6 μM).
The antiaggregatory effects of clopidogrel were unchanged when the compound was removed from the platelet suspension. However, platelet inhibition by clopidogrel was completely abolished when albumin (350 mg ml−1) was present in the test buffer.
It is concluded that clopidogrel specifically inhibits ADP-induced aggregation of washed platelets in vitro without hepatic bioactivation. Inhibition of ADP-induced platelet aggregation by clopidogrel in vitro occurs in the absence of measurable effects on the reversal of PGE1-stimulated cyclic AMP by ADP.
Keywords: Clopidogrel, SR 25989 C, platelets, aggregation, shape change, ADP
Introduction
Clopidogrel is an antiplatelet compound that has recently been shown to be effective in the secondary prevention of cardiovascular complications of atherosclerosis (CAPRIE Steering Committee, 1996). The thienopyridines clopidogrel and ticlopidine are specific inhibitors of ADP-induced platelet aggregation but not of ADP-induced shape change or Ca2+ transients (for reviews see Coukell & Markham, 1997; Schrör, 1998). Although the exact mechanisms are not known, treatment with clopidogrel reduces the binding of ADP or stable ADP analogues to high-affinity binding sites on platelets (Mills et al., 1992; Savi et al., 1994b; 1997; Gachet et al., 1995). The ADP receptors that are modified by clopidogrel are also believed to mediate the inhibition of prostaglandin I2- or E1-induced cyclic AMP formation by ADP (Defreyn et al., 1991; Mills et al., 1992).
In platelet-rich plasma, clopidogrel (⩽100 μM) does not inhibit platelet aggregation in vitro (Herbert, 1994). In rats, the in vivo activity of clopidogrel has been proposed to be dependent on hepatic biotransformation to an active metabolite (Savi et al., 1992). In these studies, clopidogrel (40 mg kg−1) was less effective in hepatectomized rats as compared to normal control rats. In addition, clopidogrel did inhibit platelet aggregation in isolated, blood-perfused rat livers. The bioactivation of clopidogrel has been suggested to be mediated by the hepatic cytochrome P450 system (Savi et al., 1994a).
However, an active metabolite of clopidogrel has not been published so far and the need for hepatic activation of clopidogrel has not been demonstrated in humans. In this context, it is noteworthy that in animal studies much higher doses of clopidogrel were used as compared to the standard dosage for humans (75 mg d−1). Thus, different mechanisms may account for platelet inhibition in humans as compared to animal studies.
This study demonstrates that clopidogrel specifically inhibits ADP-induced aggregation of washed human platelets in vitro without the need of hepatic bioactivation. Inhibition of ADP-induced platelet aggregation by clopidogrel in vitro occurs in the absence of measurable effects on the reversal of PGE1-stimulated cyclic AMP by ADP.
Methods
Preparation of washed human platelets
Washed human platelets were prepared as previously described (Weber et al., 1993). Fresh citrated blood was obtained from a local blood-bank. Platelet-rich plasma was prepared by centrifugation at 250×g for 10 min at room temperature. The pH was adjusted to 6.5 with acidic citrate dextrose (Biotest, Frankfurt, Germany). The platelets were washed twice in a buffer (pH 6.5) containing [mM]: NaCl 113, Na2HPO4 4, NaH2PO4 24, KH2PO4 4, supplemented with 0.05 u ml−1 apyrase (Grade V, Sigma, Deisenhofen, Germany), 5 mM glucose and 50 nM prostaglandin E1. Washed platelets were resuspended in HEPES-buffered Tyrode solution (pH 7.4) of the following composition [mM]: NaCl 134, NaHCO3 12, KCl 2.9, NaH2PO4 0.36, MgCl2 1, CaCl2 2, HEPES 5, supplemented with 5 mM glucose. When indicated, clopidogrel (0.3–30 μM) or vehicle was incubated with the platelets for 2.5–120 min at 37°C prior to stimulation. In some experiments, platelet-rich plasma was pre-incubated with acetylsalicylic acid (10 μM) for 15 min.
Platelet aggregation and shape change
Platelet function was measured as previously described (Weber et al., 1993). Briefly, 400 μl of washed platelet suspension or 400 μl of platelet-rich plasma were incubated with 90 μl of test buffer (HEPES-buffered Tyrode, see above) supplemented with 1.5 mg ml−1 fibrinogen (final concentration 0.3 mg ml−1) in a 2-channel aggregometer (Labor, Hamburg, Germany) for 2 min at 37°C. Platelets were stimulated by adding 10 μl of the respective stimulus (0.2–600 μM ADP, 2.5 μg ml−1 collagen, 1 μM U46619, or 0.1 u ml−1 thrombin, respectively). Changes in light transmission were recorded during constant stirring of the samples (1,200 r.p.m., 37°C). Aggregation responses were assessed by measuring the maximum change of light transmission (% of control) and the change of light transmission (% of control) 4 min after addition of the stimulus. For shape change measurements, washed platelets were resuspended in Ca2+-free test buffer. 400 μl of washed platelet suspension were incubated with 90 μl of test buffer supplemented with 10 mM EGTA (final concentration 2 mM) in a 2-channel aggregometer (Labor, Hamburg, Germany) for 2 min at 37°C. Platelets were stimulated by adding 10 μl of the stimulus (0.2–20 μM ADP). Shape change responses were assessed by measuring the decrease in light transmission.
Determination of cyclic AMP in washed platelets
Washed platelets (480 μl) were incubated in an aggregometer for 1 min at constant stirring (1,200 r.p.m., 37°C). Then, 1 μM prostaglandin E1 (10 μl) or buffer (10 μl) were added for 1 min. Subsequently, the platelets were stimulated with 6 μM ADP (10 μl). After 1 min, the reaction was terminated by adding 100 μl 25% HClO4. The samples were neutralized with 125 μl 2 M K2CO3 and centrifuged at 10,000×g for 5 min. Cyclic AMP was determined in the supernatants by radioimmunoassay as previously described (Schröder & Schrör, 1993).
Materials
Acidic citrate-dextrose (Biostabil®, Biotest, Frankfurt, Germany); ADP (Boehringer, Mannheim, Germany); clopidogrel, SR 25989 C (Sanofi Recherche, Toulouse, France); collagen (Collagenreagent Horm®, Nycomed, München, Germany); [3H]-cyclic AMP (New England Nuclear, Dreieich, Germany), U46619 (Upjohn Diagnostics, Heppenheim, Germany). α-Thrombin was a gift from Dr J. Stürzebecher, Erfurt, Germany. All other reagents were obtained from Sigma (Deisenhofen, Germany).
Statistics
Data are means±s.e.mean from n experiments. Statistical analysis was performed using two-tailed Student's t-test for paired or unpaired data, as applicable. P<0.05 was considered significant.
Results
Clopidogrel does not inhibit platelet aggregation in platelet-rich plasma
When clopidogrel (⩽100 μM) was incubated with platelet-rich plasma, no inhibition of ADP (6 μM)-induced platelet aggregation was observed. Even an incubation of clopidogrel with platelet-rich plasma for up to 8 h did not result in any antiaggregatory activity of the compound (not shown).
Clopidogrel selectively inhibits ADP-induced aggregation in washed platelets
In contrast, when clopidogrel (0.3–30 μM) was incubated with washed platelets for 1 h, a concentration-dependent inhibition of ADP (6 μM)-induced platelet aggregation was seen (Figure 1). Incubation of washed platelets with clopidogrel resulted in a reduction of the maximum aggregation amplitude. In addition, there was a marked effect on the reversibility of ADP-induced aggregation in clopidogrel-treated platelets. Thus, both, maximum aggregation amplitudes as well as aggregation amplitudes 4 min after addition of the stimulus, were used for quantification of clopidogrel effects (Figure 2). Data analysis revealed a significant (P<0.05) inhibition of ADP-induced platelet aggregation by ⩾0.3 μM clopidogrel. From the data presented in Figure 2B, an IC50 value of 1.9±0.3 μM was calculated. Importantly, the inhibitory effects of clopidogrel on aggregation of washed platelets were selective for ADP. Platelet aggregation, induced by collagen (2.5 μg ml−1), the thromboxane A2 mimetic U46619 (1 μM) or thrombin (0.1 u ml−1), was not significantly inhibited by clopidogrel (30 μM) (Figure 2). Interestingly, in the aggregation experiments, ADP (6 μM)-induced platelet shape change did not seem to be affected by clopidogrel. However, because platelet aggregation interferes with the measurement of platelet shape change, possible effects of clopidogrel on platelet shape change were studied in a separate series of experiments, performed in the presence of EGTA. In these studies, a complete inhibition of platelet shape change by clopidogrel was observed at low concentations of ADP (0.2 μM) (Figure 3). At higher ADP concentrations (2 μM, 20 μM), the inhibitory effects of clopidogrel on ADP-induced platelet shape change were less obvious (Figure 3).
Figure 1.
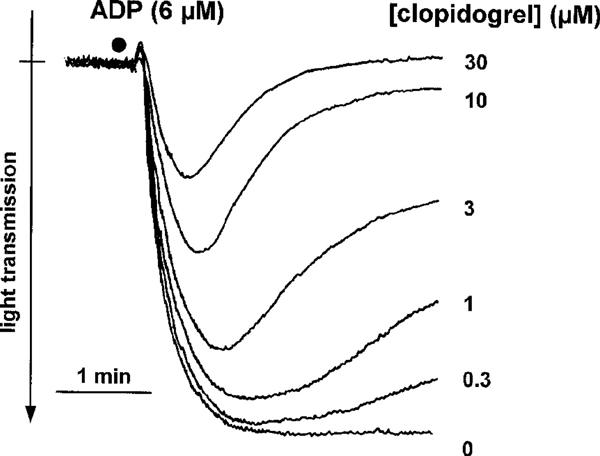
Representative original tracings demonstrating the effects of clopidogrel (0.3–30 μM) on ADP (6 μM)-induced aggregation and shape change of washed human platelets.
Figure 2.
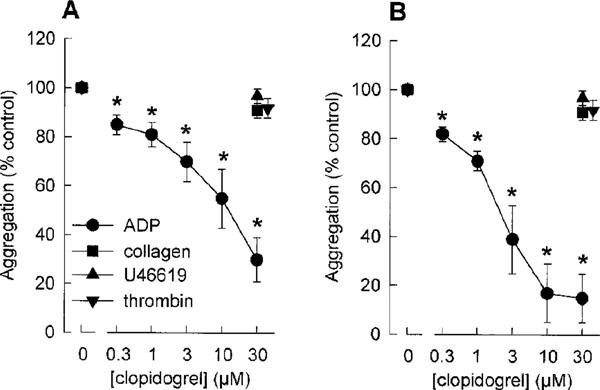
Effects of clopidogrel (0.3–30 μM) on ADP (6 μM)-, collagen (2.5 μg ml−1), U46619 (1 μM) or thrombin (0.1 u ml−1)-induced aggregation of washed human platelets. The aggregation was assessed by measuring the maximum change of light transmission (A) and the change of light transmission 4 min after addition of the stimulus (B). Data are given as % of aggregation response induced by the respective agonists without clopidogrel (control=100%). (means±s.e.mean of n=5–6 independent experiments, *P<0.05 vs control, Student's t-test for paired data).
Figure 3.
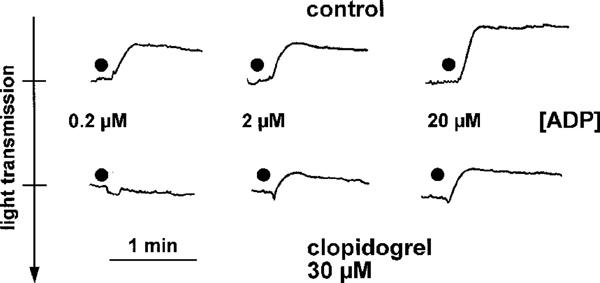
Original tracings demonstrating the effects clopidogrel (30 μM) on ADP (0.2–20 μM)-induced platelet shape change. The experiments were carried out in a Ca2+- and fibrinogen-free medium supplemented with 2 mM EGTA. Shown is one representative experiment of n=3 with similar results.
The inhibitory effects on platelet aggregation are restricted to apyrase-sensitive mechanisms
In order to further characterize if the in vitro actions of clopidogrel are specific for ADP-mediated processes, the antiaggregatory effects of clopidogrel were compared with apyrase, an ADP/ATP-degrading enzyme. In control experiments, apyrase (1 u ml−1) has been shown to completely inhibit ADP-induced platelet aggregation. The effects of apyrase on collagen (2.5 μg ml−1)- and U46619 (1 μM)-induced platelet aggregation were studied. Similar to clopidogrel, (see Figure 2), apyrase (1 u ml−1) did not inhibit collagen (2.5 μg ml−1)- or U46619 (1 μM)-induced aggregation of washed platelets (not shown). Therefore, we have repeated the experiments with acetylsalicylic acid (10 μM) pre-treated platelets. In this system, apyrase significantly (P<0.05) inhibited collagen-induced platelet aggregation, indicating that ADP is an essential amplification mechanism for collagen-induced aggregation of acetyl-salicylic acid-treated platelets (Figure 4). Interestingly, clopidogrel (30 μM) also significantly reduced the aggregation of acetylsalicylic acid-treated platelets to an extent similar to the effects of apyrase (Figure 4). In contrast, when the effects of apyrase and clopidogrel, respectively, on U46619-induced aggregation of acetylsalicylic acid-treated platelets were studied, apyrase did not inhibit U46619-induced platelet aggregation, indicating that the thromboxane A2 mimetic is capable of stimulating platelet aggregation independently of ADP-mediated amplification mechanisms even after cyclooxygenase inhibition. Consistently, there was no inhibition of U46619-induced aggregation of acetylsalicylic acid-treated platelets by clopidogrel (30 μM). Thus, antiaggregatory actions of clopidogrel can only be observed in apyrase-sensitive systems. These data provide further evidence for the ADP selectivity of clopidogrel actions in vitro.
Figure 4.
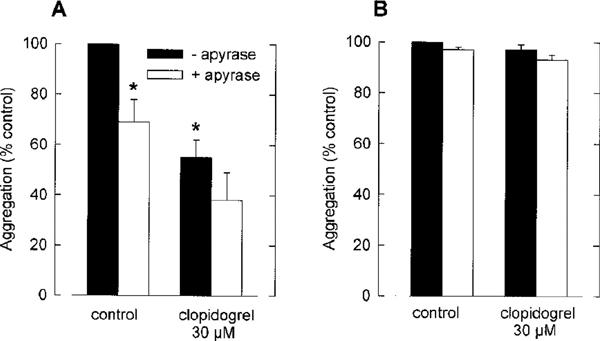
Effects of clopidogrel (30 μM) on collagen (2.5 μg ml−1) (A) and U46619 (1 μM) (B)-induced aggregation of acetylsalicylic acid (10 μM)-treated washed human platelets. Experiments were carried out in the absence and presence of apyrase (1 u ml−1). The aggregation was assessed by measuring the maximum change of light transmission. Data are given as % of aggregation response induced by the respective agonists without clopidogrel (control=100%). (means±s.e.mean of n=4 independent experiments, *P<0.05 vs control, Student's t-test for paired data).
The inhibition of ADP-induced aggregation by clopidogrel is insurmountable
In order to further characterize the effects of clopidogrel (30 μM, 60 min) on ADP-induced platelet aggregation in vitro, ADP concentration-effect curves had been constructed (Figure 5). In these experiments, the maximum response to ADP was reduced by clopidogrel indicating a non-equilibrium antagonism of ADP actions.
Figure 5.
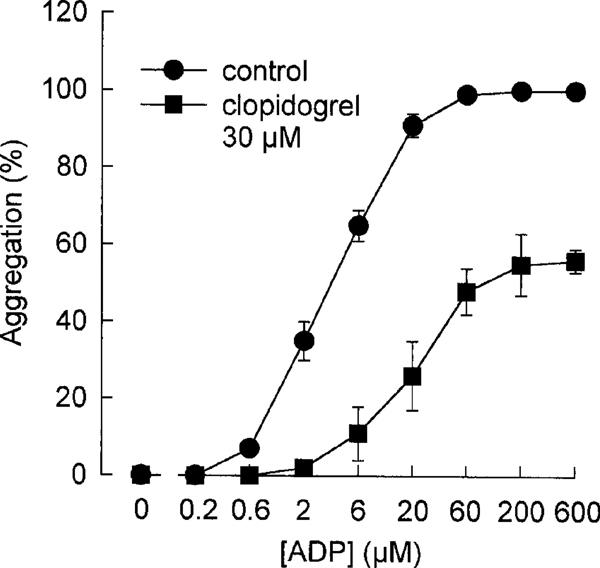
ADP (0.2–600 μM)-induced platelet aggregation in control and in clopidogrel (30 μM)-treated washed platelets. Data are given as % of maximum aggregation response (control=100%). (means ±s.e.mean of n=3 independent experiments).
The antiaggregatory effects of clopidogrel are dependent on the incubation time but not on the presence of the compound in the test medium
The inhibition of ADP-induced platelet aggregation was time-dependent (Figure 6). Measurable effects could be detected after 2.5 min. However, there was an increase in the inhibitory effects of clopidogrel upon further incubation. Maximum and stable inhibition was observed after 30 min (Figure 6). In order to study if the presence of clopidogrel is required for the antiaggregatory effects of the compound, clopidogrel (30 μM, 60 min) pre-incubated washed platelets were washed again and resuspended in clopidogrel-free buffer. Untreated control platelets were processed in the same way and served as controls. In these experiments, the inhibitory effects of clopidogrel were not changed (not shown).
Figure 6.
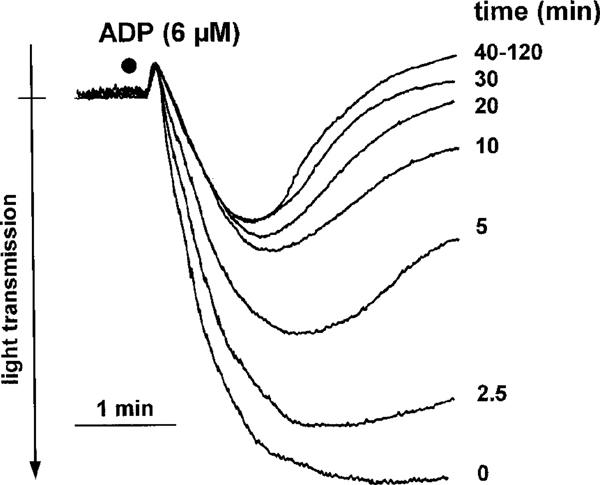
Original tracings demonstrating the time course of the inhibitory actions of clopidogrel (30 μM) on ADP (6 μM)-induced platelet aggregation. Shown is one representative experiment of n=4 with similar results.
The antiaggregatory effects of clopidogrel are stereoselective
The in vitro antiaggregatory effects of clopidogrel were also compared with the R enantiomer SR 25989 C. In these experiments, SR 25989 C (30 μM) was significantly less active as compared to clopidogrel (30 μM) with respect to the inhibition of ADP (6 μM)-induced platelet aggregation (32±5% vs 70±1% inhibition, P<0.05, Student's t-test for paired data, n=5), indicating a relative stereoselectivity of clopidogrel effects.
Clopidogrel does not reverse the inhibitory effects of ADP on prostaglandin E1-induced cyclic AMP formation
Stimulation of washed platelets with prostaglandin E1 (1 μM) resulted in a significant (P<0.05) increase in cyclic AMP levels (Figure 5). When ADP (6 μM) was added to prostaglandin E1-stimulated platelets, the stimulatory effects on cyclic AMP formation were completely abolished. Pretreatment of the platelets with clopidogrel (3–30 μM) for 1 h did not significantly reverse the ADP-mediated inhibition of platelet adenylate cyclase. The data are summarized in Figure 7.
Figure 7.
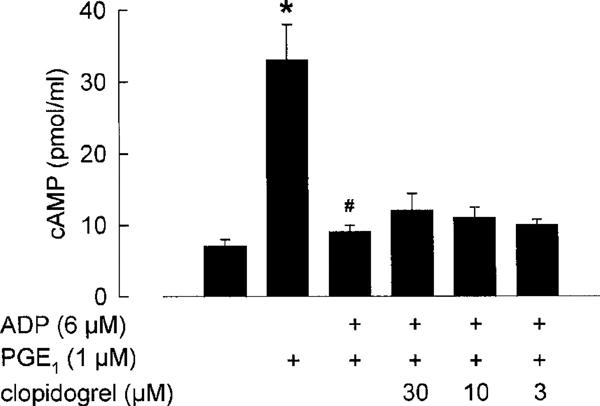
Effects of clopidogrel (3–30 μM) on the inhibitory effects of ADP (6 μM) on prostaglandin E1 (PGE1, 1 μM)-stimulated platelet adenylate cyclase. (means±s.e.mean of n=4 independent experiments, *P<0.05 vs control, #P<0.05 vs PGE1, Student's t-test for unpaired data).
Protein prevents in vitro antiaggregatory actions of clopidogrel in platelet-rich plasma
Finally, we have tested the hypothesis that proteins, present in platelet-rich plasma, might prevent the in vitro antiaggregatory actions of clopidogrel. Therefore, experiments were carried out, using washed human platelets in test buffer supplemented with albumin (350 mg ml−1). When protein was present in the buffer, the antiaggregatory effects of clopidogrel (30 μM) were completely abolished (not shown). Interestingly, when washed platelets were preincubated with clopidogrel for 1 h, subsequent addition of albumin was not able to reverse the inhibition of platelet aggregation by clopidogrel (not shown).
Discussion
The thienopyridine clopidogrel is believed to be inactive in vitro and it has been suggested that its activity is dependent on hepatic biotransformation (for review see Coukell & Markham, 1997). The present study is the first to demonstrate direct inhibition of platelet aggregation by clopidogrel in vitro. The in vitro inhibition of platelet aggregation by clopidogrel is selective for ADP and does not require hepatic bioactivation.
In line with previous reports (for review see Herbert, 1994), we were not able to detect any direct antiaggregatory effects of clopidogrel (⩽100 μM) in platelet-rich plasma. However, when washed human platelets were studied, a concentration-dependent inhibition of ADP-induced platelet aggregation by clopidogrel was observed. Several lines of evidence indicate that the observed in vitro antiaggregatory effects of clopidogrel are due to a specific mechanism that is similar to the ex vivo inhibition of platelet aggregation by the compound.
The inhibitory effects of clopidogrel were selective for ADP, because no inhibition of platelet aggregation was seen when collagen, thrombin, or the thromboxane A2 agonist U46619 were used as proaggregatory stimuli. In addition, the selectivity of clopidogrel for ADP-induced platelet activation was confirmed using apyrase, an ADP-degrading enzyme (Cattaneo et al., 1991). In these experiments, acetylsalicylic acid-treated platelets were sensitive to apyrase, when aggregation was stimulated with collagen. In contrast, U46619-induced aggregation of acetylsalicylic acid-treated platelets was not affected by apyrase. These data indicate that, when acetylsalicylic acid-treated platelets are used, collagen- but not U46619-induced platelet aggregation partially depends on endogenous ADP. Interestingly, in the presence of acetylsalicylic acid, clopidogrel inhibited collagen-induced platelet aggregation to an extent similar to apyrase. In contrast, there was no inhibition of U46619-induced aggregation of acetyl-salicylic acid-treated platelets by clopidogrel. Thus, in vitro antiaggregatory actions of clopidogrel are restricted to apyrase-sensitive systems and involve the suppression of the effects of released ADP. This is in line with previous reports on the ex vivo inhibition of platelet aggregation by the compound (Feliste et al., 1987). The demonstration of synergistic effects of clopidogrel and acetylsalicylic acid is interesting and corresponds to experimental and clinical data with ticlopidine (De Caterina et al., 1991; Neumann et al., 1997). In addition, in accordance to ex vivo data that demonstrate antiaggregatory effects of clopidogrel in washed platelets (Gachet et al., 1990), in vitro platelet inhibition persisted even when clopidogrel-treated platelets were washed again in order to remove any free compound from the platelet suspension. Thus, the antiaggregatory activity of clopidogrel was associated with the platelets and was not dependent on the presence of the compound in the test buffer. Since the time periods to study inhibition of washed human platelets are limited (washed platelet preparations are stable only for several hours), no statement about possible irreversibility of platelet inhibition by clopidogrel in vitro can be made. In line with previous ex vivo studies (Defreyn et al., 1991; Savi et al., 1992), clopidogrel (SR 25990 C) was significantly less effective in inhibiting ADP-induced platelet aggregation as compared to its levorotatory enantiomer (SR 25989 C). Finally, the inhibitory effects of clopidogrel on ADP-induced platelet aggregation were insurmountable. Thus, the in vitro actions of clopidogrel on human platelets involve a non-equilibrium antagonism, similar to the effects observed ex vivo (Mills et al., 1992). In line with the ex vivo data (Gachet et al., 1990; 1992; Mills et al., 1992), incubation of clopidogrel with washed platelets did not appear to inhibit ADP (6 μM)-induced platelet shape change. However, when shape change was measured separately, i.e. under conditions precluding aggregation, a marked inhibition of ADP (0.2 μM)-induced platelet shape change was seen. With higher ADP concentrations (2 μM, 20 μM), the inhibition of platelet shape change by clopidogrel was less obvious.
Taken together, several lines of evidence indicate that clopidogrel specifically and selectively inhibits ADP-induced platelet aggregation in vitro. These data are in contrast to the general assumption that hepatic biotransformation is required for antiaggregatory activity of clopidogrel (Savi et al., 1992; 1994a). However, this assumption is based on animal studies only, and the requirement of hepatic biotransformation in humans has not yet been demonstrated. Although several metabolites are known, a biologically active metabolite has not been published so far. Our data indicate that hepatic biotransformation is not required for platelet inhibition by clopidogrel. It is well possible that clopidogrel itself and not an unknown metabolite accounts for the antiaggregatory activity of the compound. This view is supported by early pharmacokinetic studies with ticlopidine, a sister-compound of clopidogrel. In these studies (Di Perri et al., 1991), a single dose (500 mg) was given to healthy volunteers and the time course of platelet inhibition was correlated with the plasma concentrations of ticlopidine as well as with the amount of cell-associated (unchanged) compound. The maximum concentration of erythrocyte- and neutrophil-associated ticlopidine clearly paralleled the peak plasma concentration of the compound. In contrast, maximum inhibition of platelet aggregation occurred several hours later and paralleled the concentration of platelet-associated (unchanged) compound. Thus, it has been speculated that a redistribution into a platelet-compartment, possibly via a specific uptake mechanism, can explain the delayed platelet inhibition by ticlopidine (Di Perri et al., 1991). Alternatively, it is also possible that platelets are capable to generate the putative active metabolite of clopidogrel. This possibility is supported by the time-dependence of platelet inhibitory actions of clopidogrel, as demonstrated in the present study. Clearly, further studies are needed to address this issue.
The second interesting point of our study is the finding that the in vitro inhibition of ADP-induced platelet aggregation by clopidogrel can be observed without a significant reversal of the inhibitory effects of ADP on platelet adenylate cyclase. It is well known that clopidogrel treatment selectively reduces the number of functional ADP receptors mediating the inhibition of stimulated adenylate cyclase (Defreyn et al., 1991; Mills et al., 1992; for review see Coukell & Markham, 1997). However, in a recent study, Savi et al. (1996) have proposed that cyclic AMP is not an essential messenger for ADP-induced platelet aggregation. Although clopidogrel reversed the inhibitory effects of ADP on platelet adenylate cyclase, this mechanism was not responsible for the ex vivo antiaggregatory actions of the compound (Savi et al., 1996). Our data support this conclusion and demonstrate that inhibition of ADP-induced platelet aggregation by clopidogrel can occur without a measurable alteration of cyclic AMP metabolism. However, it is well possible that the measurement of cyclic AMP by radioimmunoassay is not sensitive enough to detect small, though functionally relevant changes in platelet cyclic AMP concentrations. Furthermore, additional studies examining the effects of clopidogrel using a wider range of ADP concentrations are also required.
Finally, the discrepancy between the antiaggregatory effects of clopidogrel in washed platelets as compared to platelet-rich plasma needs to be addressed. Using platelet-rich plasma, we were not able to detect any in vitro inhibition of platelet aggregation by clopidogrel. This is in line with previous studies (for review see Coukell & Markham, 1997). In contrast, when washed human platelets were used, clopidogrel concentration-dependently inhibited platelet aggregation. There is an early study with ticlopidine, demonstrating no in vitro antiaggregatory effects in washed platelets (Di Minno et al., 1985). However, in this study, albumin was present in the test medium. We have, therefore, hypothesized that the presence of protein might explain the lack of platelet inhibition of clopidogrel in platelet-rich plasma. In fact, the addition of albumin to the test medium completely prevented the antiaggregatory actions of clopidogrel. Although these data can explain why in vitro antiaggregatory effects of clopidogrel have not yet been described, the question why clopidogrel is active in vivo, i.e. in the presence of high protein concentrations, remains open. However, it is known that several days of treatment with clopidogrel are needed to achieve maximum platelet inhibitory effects (Coukell & Markham, 1997). Thus, one might speculate that saturation of protein binding sites for clopidogrel might occur during this time. Alternatively, specific uptake or intercellular redistribution mechanisms (Di Perri et al., 1991) might explain the delayed onset of platelet inhibition in vivo.
In summary, we demonstrate that clopidogrel specifically and selectively inhibits ADP-induced aggregation of washed platelets in vitro without hepatic bioactivation. It is difficult to speculate about the ADP receptor(s) involved in the antiaggregatory actions of clopidogrel in vitro. At least three different ADP receptors are expressed in platelets (Hechler et al., 1998; Geiger et al., 1998): (i) the Gq-coupled P2Y1 receptor which is thought to be involved in ADP-induced platelet shape change and aggregation; (ii) the Gi-coupled P2Y receptor, which has been termed ‘P2Ycyc' and which is thought to complete and to potentiate the initial P2Y1-mediated platelet activation; and (iii) P2X1, a receptor-operated Ca2+ channel the role of which is unknown. Hechler et al. (1998) speculate that the Gi-coupled P2Y receptor is involved in the antithrombotic actions of clopidogrel. A significant reversal of the inhibitory effects of ADP on platelet adenylate cyclase was not involved in the inhibition of ADP-induced platelet aggregation by clopidogrel in vitro. Thus, our data do not support the concept of a Gi-coupled P2Y receptor-mediated inhibition of platelet function by clopidogrel. On the other hand, the marked inhibition of ADP (0.2 μM)-induced platelet shape change by clopidogrel is also compatible with the concept that the P2Y1 receptor is the target of the compound. However, the present data do not allow definitive statements about the receptor(s) clopidogrel is acting on and further studies are needed to resolve this issue. The demonstration of an inhibition of platelet aggregation by clopidogrel in vitro should facilitate the investigation of many open questions regarding the mode of action of this exciting antiplatelet compound.
Acknowledgments
The authors gratefully acknowledge the competent secretarial help of Erika Lohmann.
Abbreviations
- ADP
adenosine 5′-diphosphate
- cyclic AMP
adenosine 3′:5′- cyclic monophosphate
- PGE1
prostaglandin E1
References
- CAPRIE STEERING COMMITTEE A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE) Lancet. 1996;348:1329–1339. doi: 10.1016/s0140-6736(96)09457-3. [DOI] [PubMed] [Google Scholar]
- CATTANEO M., AKKAWAT B., LECCHI A., CIMMINIELLO C., CAPITANIO A.M., MANUCCI P.M. Ticlopidine selectively inhibits human platelet responses to adenosine diphosphate. Thromb. Haemost. 1991;66:694–699. [PubMed] [Google Scholar]
- COUKELL A.J., MARKHAM A. Clopidogrel. Drugs. 1997;54:745–750. doi: 10.2165/00003495-199754050-00006. [DOI] [PubMed] [Google Scholar]
- DE CATERINA R., SICARI R., BERNINI W., LAZZERINI G., BUTI-STRATA G., GIANNESSI D. Benefit/risk profile of combined antiplatelet therapy with ticlopidine and aspirin. Thromb. Haemost. 1991;65:504–510. [PubMed] [Google Scholar]
- DEFREYN G., GACHET C., SAVI P., DRIOT F., CAZENAVE J.P., MAFFRAND J.P. Ticlopidine and clopidogrel (SR 45990 C) selectively neutralize ADP inhibition of PGE1-activated platelet cAMP formation in rats and rabbits. Thromb. Haemost. 1991;65:186–190. [PubMed] [Google Scholar]
- DI MINNO G., CERBONE A.M., MATTIOLI P.L., TURCO S., IOVINE C., MANCINI S. Functionally thrombasthenic state in normal patients following the administration of ticlopidine. J. Clin. Invest. 1985;75:328–338. doi: 10.1172/JCI111705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DI PERRI T., PASINI F.L., FRIGERIO C., BLARDI P., CENTINI F., MESSA G.L., GHEZZI A., VOLPI L. Pharmacodynamics of ticlopidine in man in relation to plasma and blood cell concentration. Eur. J. Clin. Pharmacol. 1991;41:429–434. doi: 10.1007/BF00626364. [DOI] [PubMed] [Google Scholar]
- FELISTE R., DELEBASSEE D., SIMON M.F., CHAP H., DEFREYN G., VALLEE E., DOUSTE-BLAZY L., MAFFRAND J.P. Broad spectrum anti-platelet activity of ticlopidine and PCR 4099 involves the suppression of the effects of released ADP. Thromb. Res. 1987;48:403–415. doi: 10.1016/0049-3848(87)90398-7. [DOI] [PubMed] [Google Scholar]
- GACHET C., CATTANEO M., OHLMANN P., HECHLER B., LECCHI A., CHEVALIER J., CASSEL D., MANNUCCI P.M., CAZENAVE J.P. Purinoceptors on blood platelets: further pharmacological and clinical evidence to suggest the presence of two ADP receptors. Br. J. Haematol. 1995;91:434–444. doi: 10.1111/j.1365-2141.1995.tb05319.x. [DOI] [PubMed] [Google Scholar]
- GACHET C., SAVI P., OHLMANN P., MAFFRAND J.-P., JAKOBS K.H., CAZENAVE J.-P. ADP receptor induced activation of guanine nucleotide binding proteins in rat platelet membranes–an effect selectively blocked by the thienopyridine clopidogrel. Thromb. Haemost. 1992;6:79–83. [PubMed] [Google Scholar]
- GACHET C., STIERLE A., CAZENAVE J.P., OHLMANN P., LANZA F., BOULOUX C., MAFFRAND J.P. The thienopyridine PCR 4099 selectively inhibits ADP-induced platelet aggregation and fibrinogen binding without modifying the membrane glycoprotein IIb-IIIa complex in rat and man. Biochem. Pharmacol. 1990;40:229–238. doi: 10.1016/0006-2952(90)90683-c. [DOI] [PubMed] [Google Scholar]
- GEIGER J., HÖNIG-LIEDL P., SCHANZENBÄCHER P., WALTER U. Ligand specifity and ticlopidine effects distinguish three human platelet ADP receptors. Eur. J. Pharmacol. 1998;351:235–246. doi: 10.1016/s0014-2999(98)00305-7. [DOI] [PubMed] [Google Scholar]
- HECHLER B., LEON C., VIAL C., VIGNE P., FRELIN C., CAZENAVE J.-P. , GACHET C. The P2Y1 receptor is necessary for adenosine 5′-diphosphate-induced platelet aggregation. Blood. 1998;92:152–159. [PubMed] [Google Scholar]
- HERBERT J-M. Clopidogrel and antiplatelet therapy. Expert Opin. Invest. Drug. 1994;3:449–455. [Google Scholar]
- MILLS D.C.B., PURI R., HU C.-J., MINNITI C., GRANA G., FREEDMAN M.D., COLMAN R.F., COLMAN R.W. Clopidogrel inhibits the binding of ADP analogues to the receptor mediating inhibition of platelet adenylate cyclase. Arterioscl. Thromb. 1992;12:430–436. doi: 10.1161/01.atv.12.4.430. [DOI] [PubMed] [Google Scholar]
- NEUMANN F.J., GAWAZ M., DICKFELD T., WEHNINGER A., WALTER H., BLASINI R., SCHÖMIG A. Antiplatelet effects of ticlopidine after coronary stenting. J. Am. Coll. Cardiol. 1997;29:1515–1519. doi: 10.1016/s0735-1097(97)00073-9. [DOI] [PubMed] [Google Scholar]
- SAVI P., BORNIA J., SALEL V., DELFAUD M., HERBERT J.-M. Characterization of P2x1 purinoceptors on rat platelets: effect of clopidogrel. Br. J. Haematol. 1997;98:880–886. doi: 10.1046/j.1365-2141.1997.3133126.x. [DOI] [PubMed] [Google Scholar]
- SAVI P., HERBERT J.-M., PFLIEGER A.M., DOL F., DELABASSEE D., COMBALBERT J., DEFREYN G., MAFFRAND J.P. Importance of hepatic metabolism in the antiaggregating activity of the thienopyridine clopidogrel. Biochem. Pharmacol. 1992;44:527–532. doi: 10.1016/0006-2952(92)90445-o. [DOI] [PubMed] [Google Scholar]
- SAVI P., COMBALBERT J., GAICH C., ROUCHON M.C., MAFFRAND J.P., BERGER Y., HERBERT J.-M. The antiaggregating activity of clopidogrel is due to metabolic activation by the hepatic cytochrome P450-1A. Thromb. Haemost. 1994a;72:313–317. [PubMed] [Google Scholar]
- SAVI P., LAPLACE M.C., MAFFRAND J.P., HERBERT J.-M. Binding of [3H]-2-methylthio ADP to rat platelets–effect of clopidogrel and ticlopidine. J. Pharmacol. Exp. Ther. 1994b;269:772–777. [PubMed] [Google Scholar]
- SAVI P., PFLIEGER A.M., HERBERT J.-M. cAMP is not an important messenger for ADP-induced platelet aggregation. Blood Coagul. Fibrinol. 1996;7:249–252. doi: 10.1097/00001721-199603000-00035. [DOI] [PubMed] [Google Scholar]
- SCHRÖDER H., SCHRÖR K. Prostacyclin-dependent cyclic AMP formation in endothelial cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 1993;347:101–104. doi: 10.1007/BF00168779. [DOI] [PubMed] [Google Scholar]
- SCHRÖR K.Clinical pharmacology of the adenosine diphosphate (ADP) receptor antagonist, clopidogrel Vasc. Med. 19983(in press) [DOI] [PubMed] [Google Scholar]
- WEBER A.-A., STROBACH H., SCHRÖR K. Direct inhibition of platelet function by organic nitrates via nitric oxide formation. Eur. J. Pharmacol. 1993;247:29–37. doi: 10.1016/0922-4106(93)90134-u. [DOI] [PubMed] [Google Scholar]