

Abstract
The ability of μ-opioid receptor agonists to activate G-proteins in the spinal cord of μ-opioid receptor knockout mice was examined by monitoring the binding to membranes of the non-hydrolyzable analogue of GTP, guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS).
In the receptor binding study, Scatchard analysis of [3H][D-Ala2,NHPhe4,Gly-ol]enkephalin ([3H]DAMGO; μ-opioid receptor ligand) binding revealed that the heterozygous μ-knockout mice displayed approximately 40% reduction in the number of μ-receptors as compared to the wild-type mice. The homozygous μ-knockout mice showed no detectable μ-binding sites.
The newly isolated μ-opioid peptides endomorphin-1 and -2, the synthetic selective μ-opioid receptor agonist DAMGO and the prototype of μ-opioid receptor agonist morphine each produced concentration-dependent increases in [35S]GTPγS binding in wild-type mice. This stimulation was reduced by 55–70% of the wild-type level in heterozygous, and virtually eliminated in homozygous knockout mice.
No differences in the [35S]GTPγS binding stimulated by specific δ1- ([D-Pen2,5]enkephalin), δ2- ([D-Ala2]deltorphin II) or κ1- (U50,488H) opioid receptor agonists were noted in mice of any of the three genotypes.
The data clearly indicate that μ-opioid receptor gene products play a key role in G-protein activation by endomorphins, DAMGO and morphine in the mouse spinal cord. They support the idea that μ-opioid receptor densities could be rate-limiting steps in the G-protein activation by μ-opioid receptor agonists in the spinal cord. These thus indicate a limited physiological μ-receptor reserve. Furthermore, little change in δ1-, δ2- or κ1-opioid receptor-G-protein complex appears to accompany μ-opioid receptor gene deletions in this region.
Keywords: knockout mice, endomorphins, G-proteins, homologous recombination, opioid peptides, μ-opioid receptors, signal transduction, spinal cord
Introduction
The μ-opioid receptor is expressed by neurons in several central nervous system regions, including the spinal cord dorsal horn. Its occupancy with agonist drugs such as morphine and agonist peptides produces many of the effects of opiate drugs, including much of the profound analgesic response attributed to the receptors localized in the spinal cord. Many synthetic opioids and derivatives of opium have reasonably-good affinities and selectivities for this receptor. Morphine and [D-Ala2,NHPhe4,Gly-ol]enkephalin (DAMGO) are useful ligands for probing μ-opioid receptor function. However, selective endogenous ligands for this receptor had not been clearly defined until recent successes of Zadina and colleagues (1997) who isolated two high-affinity μ-opioid receptor selective peptides ligands, endomorphin-1 (Tyr-Pro-Trp-Phe-NH2) and -2 (Tyr-Pro-Phe-Phe-NH2), from mammalian brain.
Recent cloning and expression studies have revealed that the μ-opioid receptor, and the related δ- and κ-opioid receptors are seven-transmembrane domain receptors whose actions are mediated through activation of heterotrimeric guanine nucleotide binding proteins (G-proteins) of several classes, including Gi- and Go-proteins (Chen et al., 1993; Law, 1995). The activation of G-proteins by the μ-opioid receptor can be measured by assessing agonist stimulation of membrane binding of the non-hydrolyzable analogue of GTP, guanosine-5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) (Traynor & Nahorski, 1995; Sim et al., 1995, 1996; Selley et al., 1997; Shimohira et al., 1997; Narita & Tseng, 1998). [35S]GTPγS addition results in accumulation of a stable Gα-[35S]GTPγS complex in brain membranes. It has been recently reported that endomorphins increased [35S]GTPγS binding in rat brain (Sim et al., 1998), mouse spinal cord (Narita et al., 1998), SH-SY5Y human neuroblastoma cells (Harrison et al., 1998) and C6 rat glioma cells stably expressing a cloned rat μ receptor (Alt et al., 1998). μ-Opioid agonist-stimulated [35S]GTPγS binding in isolated membranes thus provide a functional measurement of agonist occupation of μ-opioid receptors and its efficacy in leading to activation of G-proteins.
Knockout mice with μ-opioid receptor gene deletions have been successfully developed by homologous recombination (Matthes et al., 1996; Sora et al., 1997b). These mice display profound gene-dose-dependent reductions in morphine analgesia in tests including tests of ‘spinal' analgesia. The availability of transgenic μ-opioid receptor knockout mice allows us to determine the extent to which the μ-opioid receptor gene products are necessary for the expression of physiological actions by these newly discovered opioid peptides and alkaloids, including G-protein activation. The present study was thus designed to investigate the effects of μ-opioid receptor agonists including newly isolated opioid peptides endomorphin-1 and -2 on [35S]GTPγS binding to spinal cord membranes obtained from mice expressing normal (wild-type), half-normal (heterozygous), and absent (homozygous) μ-opioid receptor complements. We discuss the data in light of the concepts of receptor reserve for μ-opioid receptors and the possible compensatory changes in the functions of other opioid receptor types in the presence of μ-opioid receptor gene deletions in this region.
Methods
Animals
μ-Opioid receptor knockout transgenic mice were maintained on C57BL/6 and 129Sv mixed genetic backgrounds as described (Sora et al., 1997a,1997b). Animals were housed five per cage in a room maintained at 22±0.5°C with an alternating 12 h light-dark cycle. Food and water were available ad libitum.
Membrane preparation
The spinal cord was removed from wild-type, and heterozygous and homozygous μ-opioid receptor knockout mice after decapitation. The spinal cord was homogenized in 20 volumes of ice-cold Tris buffer containing Tris-HCl (pH 7.4) (50 mM) for the μ-opioid receptor binding assay or ice-cold Tris-Mg2+ buffer containing Tris-HCl (pH 7.4) 50 mM, MgCl2 5 mM, and EGTA 1 mM for the [35S]GTPγS binding assay. The homogenate was centrifuged at 48,000×g at 4°C for 10 min. The pellets were resuspended in Tris buffer or [35S]GTPγS binding assay buffer containing Tris-HCl (pH 7.4) 50 mM, MgCl2 5 mM, EGTA 1 mM, and NaCl 100 mM and recentrifuged at 48,000×g at 4°C for 10 min. The final pellets were resuspended in each assay buffer and stored at −70°C until experiments.
μ-Opioid receptor binding assay
The μ-opioid receptor binding assays were carried out in duplicate with [tyrosyl-3,5-3H(N)]-DAMGO ([3H]DAMGO; 55.3 Ci mmol−1; NEN, Boston, MA, U.S.A.) at 0.2–20 nM in a final volume of 1.0 ml which contained Tris-HCl (pH 7.4) buffer (50 mM) and 0.1 ml of the homogenated membrane fraction. The amount of membrane protein used in each assay was in the range of 120–170 μg, as determined by the method of Lowry et al. (1951). The test tubes were incubated for 120 min at 25°C. The specific binding was defined as the difference in binding observed in the absence and presence of 10−5 M unlabelled naloxone. The incubations were terminated by collecting the membranes on Whatman GF/B filters using a Brandel cell harvester (Model M-24, Brandel, MD, U.S.A.). The filters were then washed three times with 5 ml Tris-HCl buffer (pH 7.4) at 4°C and transferred to scintillation vials. Then, 0.5 ml of Soluene-350 (Packard Instrument Company, Inc., Meriden, CT, U.S.A.) and 4 ml of Hionic Fluor Cocktail (Packard Instrument Company) were added to the vials. After a 12 h equilibration period, the radioactivity in the samples was determined in liquid scintillation analyzer (Model 1600 CA, Packard Instrument Company). Values for Scatchard analysis represent the means±s.e.mean of three independent determinations.
[35S]GTPγS binding assay
The reaction was initiated by the addition of membrane suspension (30–80 μg ml−1 of membrane proteins) into the assay buffer with the opioid receptor agonists, GDP 30 μM and [35S]GTPγS (50 pM) (1000 Ci mmol−1; Amersham, Arlington Heights, IL, U.S.A.). The suspensions were incubated at 25°C for 120 min and the reaction was terminated by filtering through Whatman GF/B glass filters using a Brandel cell harvester. The filters were then washed three times with 5 ml of Tris-HCl buffer (pH 7.4) and transferred to scintillation counting vials containing scintillation cocktail (0.5 ml of Soluene-350 and 4 ml of Hionic Fluor Cocktail). The radioactivity in the samples was determined with a liquid scintillation analyzer. Non-specific binding was measured in the presence of 10 μM unlabelled GTPγS.
Drugs
The drugs used in the present studies were: Tyr-Pro-Trp-Phe-NH2 (endomorphin-1, Tocris Cookson Inc., Ballwin, MO, U.S.A.); Tyr-Pro-Phe-Phe-NH2 (endomorphin-2, Tocris Cookson Inc.); [D-Ala2,NHPhe4,Gly-ol]enkephalin (DAMGO, Bachem California, Torrance, CA, U.S.A.); morphine (Mallinckrodt Chemical Works, St. Louis, MO, U.S.A.), [D-Pen2,5]enkephalin (Bachem California), Tyr-D-Ala-Phe-Glu-Val-Val-Gly-NH2 ([D-Ala2] deltorphin II, Molecular Research Laboratories, Durham, NC); trans(±)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl] benzeneacetamide (U50, 488H, Research Biochemicals International, Natick, MA, U.S.A.); naloxone (Research Biochemicals International); guanylyl-5′-O-(γ-thio)-triphosphate (GTPγS, Research Biochemicals International); and guanosine-5′-O-(2-thio)-diphosphate (GDP, Sigma Chemical Company, St. Louis, MO, U.S.A.).
Statistical analysis
The data were expressed as means±s.e.mean. The binding data for the determination of the density (Bmax) and affinity (Kd) of binding sites were evaluated by a computer-assisted analysis, EDBA and LIGAND (Biosoft, Cambridge, U.K.). Student's t-test was used for the statistical analysis of Bmax and Kd values. Comparisons of all other data were performed via ANOVA followed by Newman-Keuls' test (Tallarida & Murray, 1987).
Results
The spinal cord membranes obtained from wild-type, heterozygous and homozygous μ-opioid receptor knockout mice were incubated with a range of [3H]DAMGO concentrations (0.2–20 nM) for 2 h at 25°C. Figure 1 illustrates Scatchard plots for three genotypes. A mean Bmax value of 61.6±4.9 fmol mg−1 protein with a Kd of 1.1±0.1 nM was found in the wild-type mice. The membranes obtained from heterozygous μ-opioid receptor knockout mice displayed a 41.3% reduction in the number of [3H]DAMGO binding sites (36.1±3.6 fmol mg−1 protein; P<0.01, as compared to the wild-type mice) with no change in affinity (1.1±0.1 nM). The membranes obtained from homozygous μ-opioid receptor knockout mice showed no detectable [3H]DAMGO binding sites.
Figure 1.
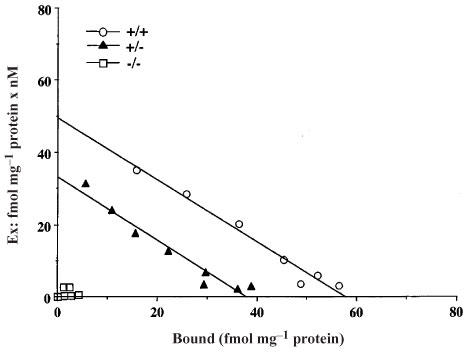
Scatchard analysis of [3H]DAMGO binding to spinal cord membranes obtained from wild-type (+/+), heterozygous (+/−) and homozygous (−/−) μ-opioid receptor knockout mice. Membranes were incubated with [3H]DAMGO at 0.2–20 nM in a final volume of 1.0 ml which contained Tris-HCl (pH 7.4) 50 mM buffer and 0.1 ml of the homogenated membrane fraction (120–170 μg) for 120 min at 25°C. The specific binding was defined as the difference in binding observed in the absence and presence of 10−5 M unlabelled naloxone. A representative experiment that was replicated three times is shown.
Both endomorphins-1 and -2 (0.001–10 μM) produced concentration-dependent increases in [35S]GTPγS binding to spinal cord membranes obtained from wild-type mice (Figure 2a and b). Maximal stimulation was 63.1±5.3 and 61.6±2.5%, respectively, using 10 μM peptide concentrations. As shown in Figure 3, the synthetic selective μ-opioid receptor agonist DAMGO and the prototype of μ-opioid receptor agonist morphine also stimulated [35S]GTPγS binding in a concentration-dependent manner and produced a maximal stimulation of 94.4±9.6 and 65.1±1.3%, respectively, at 10 μM.
Figure 2.
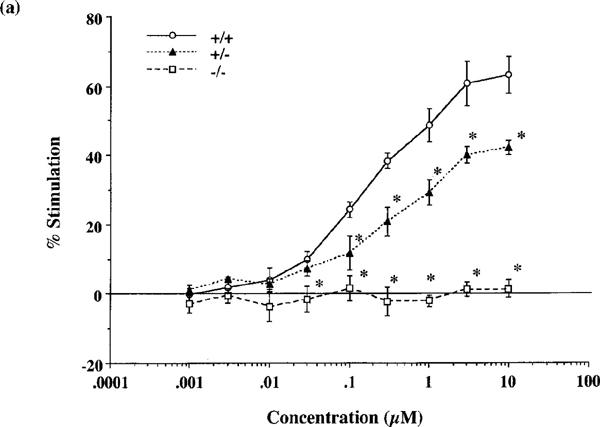
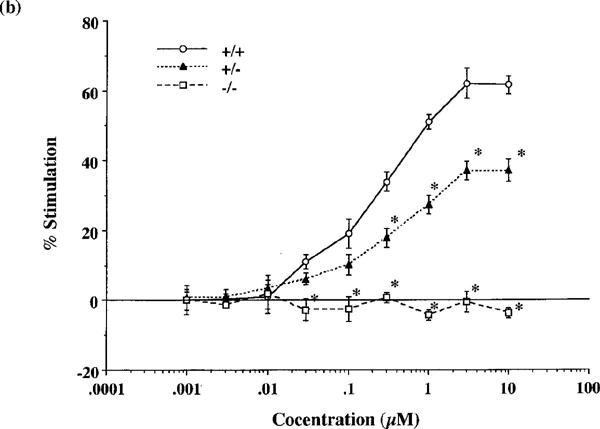
Comparison of the stimulation of [35S]GTPγS binding to spinal cord membranes of wild-type (+/+), and heterozygous (+/−) and homozygous (−/−) μ-opioid receptor knockout mice by endomorphin-1 (a) and -2 (b). Membranes were incubated with [35S]GTPγS (50 pM) and GDP (30 μM) with and without various concentrations of endomorphin-1 or -2 for 120 min at 25°C. The data are expressed as the percentage of basal [35S]GTPγS (50 pM) binding measured in the presence of GDP and absence of agonist, and represent the means±s.e.mean from at least three separate experiments. *P<0.05 vs wild-type mice.
Figure 3.
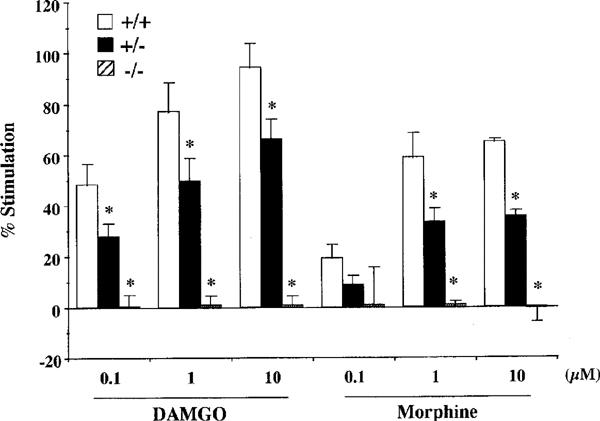
Effect of DAMGO and morphine on [35S]GTPγS binding to spinal cord membranes of wild-type (+/+), and heterozygous (+/−) and homozygous (−/−) μ-opioid receptor knockout mice. Membranes were incubated with [35S]GTPγS (50 pM) and GDP (30 μM) with and without different concentrations of DAMGO or morphine. The data are expressed as the percentage of basal [35S]GTPγS (50 pM) binding measured in the presence of GDP and absence of agonist, and represent the means±s.e.mean from at least three separate experiments. *P<0.05 vs wild-type mice.
In heterozygous μ-opioid receptor knockout mice, [35S]GTPγS binding stimulated by either endomorphin-1, -2, DAMGO or morphine was markedly decreased to 67, 60, 70 or 55% of the stimulation observed in wild-type mice, respectively. In homozygous mice, no stimulation of [35S]GTPγS binding stimulated by either endomorphin-1, -2, DAMGO or morphine could be detected (Figures 2a, b and 3).
No significant changes in δ1-, δ2- or κ1-opioid receptor functions were noted in μ-opioid receptor knockout mice. The δ1-opioid receptor agonist [D-Pen2,5]enkephalin (DPDPE), the δ2-opioid receptor agonist [D-Ala2]deltorphin II and the κ1-opioid receptor agonist U-50,488H both produced robust stimulation of [35S]GTPγS binding. As shown in Figure 4, DPDPE, [D-Ala2]deltorphin II and U50,488H (10 μM) produced 17.6±2.5, 31.6±8.6 and 7.3±5.9% stimulation, respectively, in wild-type mice. The levels of [35S]GTPγS binding stimulated by DPDPE, [D-Ala2]deltorphin II or U50,488H in both heterozygous and homozygous μ-opioid receptor knockout mice were similar to those found in wild-type mice.
Figure 4.
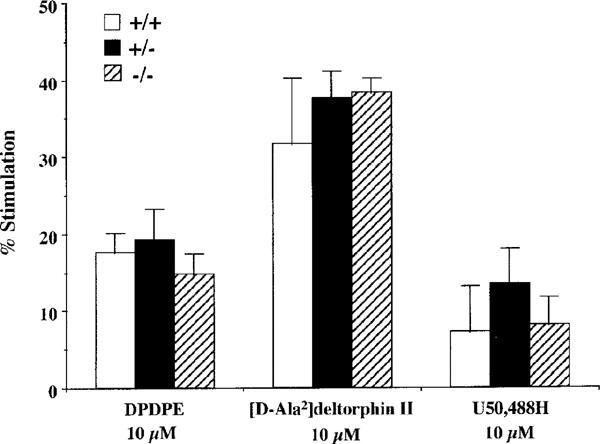
No differences in the [35S]GTPγS binding stimulated by specific δ1- (DPDPE), δ2- ([D-Ala2]deltorphin II) or κ1- (U50,488H) opioid receptor agonists in the μ-opioid receptor knockout mice. The spinal cord membranes obtained from wild-type (+/+), and heterozygous (+/−) and homozygous (−/−) μ-opioid receptor knockout mice were incubated with [35S]GTPγS (50 pM) and GDP (30 μM) with and without opioid agonists. The data are expressed as the percentage of basal [35S]GTPγS (50 pM) binding measured in the presence of GDP and absence of agonist, and represent the means±s.e.mean from at least three separate experiments.
Discussion
The knockout mice used in the present study display gene dose-dependent reductions in the levels of μ-opioid receptor expression in the spinal cord. Heterozygous knockout animals manifest 40–50% reductions in μ-opioid receptor densities, whereas homozygous knockout mice display no detectable μ-opioid receptor binding or immunoreactivity (this study; Sora et al., 1997b). Analyses of these animals provides direct evidence for μ-opioid receptor involvement in the expression of physiological actions by μ-opioid receptor agonists. The data from these studies documents that disruption of the mouse μ-opioid receptor gene ablates the G-protein activation in the mouse spinal cord by endomorphin-1 and -2 found in wild-type mice. Elimination of μ-opioid receptor expression in mutant mice virtually abolished both DAMGO- and morphine-stimulated [35S]GTPγS binding. This agrees with our previous findings that μ-opioid receptor agonist-stimulated [35S]GTPγS binding was completely blocked by μ-opioid receptor antagonists (Narita & Tseng, 1998). These findings clearly indicate that μ-opioid receptor gene products are the molecular target of endomorphins, DAMGO and morphine relevant to activation of G-proteins in the mouse spinal cord.
Regional differences in the relationships between μ-opioid receptor densities and pharmacological efficacies produced by μ-opioid receptor agonists could result from different patterns of the μ-opioid receptor reserve (Chavkin & Goldstein, 1984). Studies using heterozygous mice with half of the normal, wild-type μ-opioid receptor densities can provide information about physiologic μ-opioid receptor reserves. Previous analgesic tests in knockout mice suggest that even 50% reductions in spinal μ-opioid receptor densities alter baseline nociception and agonist-induced changes in nociceptive responses in the tail-flick assay, which largely reflects spinal antinociceptive influences (Sora et al., 1997b). In the present study, μ-opioid receptor agonist-stimulated [35S]GTPγS binding was reduced by 55–70% in heterozygous mice, and virtually eliminated in homozygous knockout mice. Both this behavioural result and the current biochemical data thus support the idea that μ-opioid receptors are rate-limiting steps in the biochemical and physiological spinal cord pathways stimulated by endogenous and exogenous μ-opioid receptor agonists. Both lines of evidence indicate a limited physiological ‘μ-opioid receptor reserve' in these spinal cord mechanisms.
It is possible that gene deletion strategies would produce compensatory changes in other receptor types associated with the deleted receptor. Quantitative autoradiography has revealed trends toward changes (10–15%) for δ- and κ-opioid receptor binding in some brain regions of μ-opioid receptor knockout mice (Kitchen et al., 1997). In the present study, however, we did not find any significant changes in δ1-, δ2- or κ1-opioid receptor agonist-stimulated [35S]GTPγS binding to spinal cord membranes in μ-opioid receptor knockout mice compared to wild-type mice. Matthes et al. (1998) have recently reported that mice lacking μ-opioid receptors do not display significant changes in either δ1-, δ2- or κ1-opioid receptor agonist-activated G-proteins in the brain. These findings suggest that functional coupling of δ1-, δ2- and κ1-opioid receptors to G-protein appears to be preserved in the process of μ-opioid receptor deletions. However, it should be also noted that some of antinociceptive actions of δ1- and δ2 -agonists when given intracerebroventricularly are less effective in μ-opioid receptor knockout mice than in wild-type mice (Sora et al., 1997a; Matthes et al., 1998). It is therefore worthwhile to examine more closely whether there could be physiological μ-receptor dependence of δ-receptor actions at the spinal cord level.
In conclusion, we have demonstrated profound gene-dose-dependent reductions in G-protein activation by μ-opioid receptor agonists including newly isolated peptides endomorphins in the mouse spinal cord. Furthermore, the preservation of δ1-, δ2- and κ1-opioid receptor signalling properties in the process of μ-opioid receptor deletions provides no evidence for cross-talk among opioid receptors at the cellular level.
Acknowledgments
This work was supported by U.S. Public Health Service Grant DA 03811 from the National Institute on Drug Abuse (NIDA), National Institutes of Health and the Intramural Research Program, NIDA.
Abbreviations
- DAMGO
[D-Ala2,NHPhe4,Gly-ol]enkephalin
- DPDPE
[D-Pen2,5]enkephalin
- GDP
guanosine-5′-O-(2-thio)-diphosphate
- GTPγS
guanylyl-5′-O-(γ-thio)-triphosphate
- U50,488H
trans(±)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]; benzeneacetamide
References
- ALT A., MANSOUR A., AKIL H., MEDZIHRADSKY F., TRAYNOR J.R., WOODS J.H. Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding by endogenous opioids acting at a cloned mu receptor. J. Pharmacol. Exp. Ther. 1998;286:282–288. [PubMed] [Google Scholar]
- CHAVKIN C., GOLDSTEIN A. Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc. Natl. Acad. Sci. U.S.A. 1984;81:7253–7257. doi: 10.1073/pnas.81.22.7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y., MESTEK A., LIU J., HURLEY J.A., YU L. Molecular cloning and functional expression of a μ-opioid receptor from the rat brain. Mol. Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- HARRISON L.M., KASTIN A.J., ZADINA J.E. Differential effects of endomorphin-1, endomorphin-2, and Tyr-W-MIF-1 on activation of G-proteins in SH-SY5Y human neuroblastoma membranes. Peptides. 1998;19:749–753. doi: 10.1016/s0196-9781(98)00022-9. [DOI] [PubMed] [Google Scholar]
- KITCHEN I., SLOWE S.J., MATTHES H.W.-D., KIEFFER B. Quantitative autoradiographic mapping of μ-, δ- and κ-opioid receptors in knockout mice lacking the μ-opioid receptor gene. Brain Res. 1997;778:73–88. doi: 10.1016/s0006-8993(97)00988-8. [DOI] [PubMed] [Google Scholar]
- LAW P.-Y.G-proteins and opioid receptors' function The Pharmacology of Opioid Peptides 1995Amsterdam, Harwood Academic Publishers; 109–130.ed. Tseng, L.F. [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A.L., RANDALL R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- MATTHES H.W.D., MALDONADO R., SIMONIN F., VALVERDE O., SLOWE S., KITCHEN I., BEFORT K., DIERICH A., LE M.M., DOLLE P., TZAVARA E., HANOUNE J., ROQUES B.P., KIEFFER B.L. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- MATTHES H.W.D., SMADJA C., VALVERDE O., VONESCH J.-L., FOUTZ A.S., BOUDINOT E., DENAVIT-SAUBIÉ M., SEVERINI C., NEGRI L., ROQUES B.P., MALDONADO R., KIEFFER B.L. Activity of the δ-opioid receptor is partially reduced, whereas activity of the κ-receptor is maintained in mice lacking the μ-receptor. J. Neuroscience. 1998;18:7285–7295. doi: 10.1523/JNEUROSCI.18-18-07285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARITA M., MIZOGUCHI H., OJI G.S., TSENG E.L., SUGANUMA C., NAGASE H., TSENG L.F. Characterization of endomorphin-1 and -2 on [35S]GTPγS binding in the mouse spinal cord. Eur. J. Pharmacol. 1998;351:383–387. doi: 10.1016/s0014-2999(98)00395-1. [DOI] [PubMed] [Google Scholar]
- NARITA M., TSENG L.F. Evidence for the existence of the β-endorphin-sensitive ‘ε-opioid receptor' in the brain: the mechanisms of ε-mediated antinociception. Jpn. J. Pharmacol. 1998;76:233–253. doi: 10.1254/jjp.76.233. [DOI] [PubMed] [Google Scholar]
- SELLY D.E., SIM L.J., XIAO R., LIU, QIXU, CHILDERS S.R. μ-Opioid receptor-stimulated guanosin-5-O-(γ-thio)-triphosphate binding in rat thalamua and cultured cell lines: Signal transduction mechanisms underlying agonist efficacy. Mol. Pharmacol. 1997;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- SIM L.J., LIU Q., CHILDERS S.R., SELLEY D.E. Endomorphin-stimulated [35S]GTPγS binding in rat brain: Evidence for partial agonist activity at μ-opioid receptors. J. Neurochem. 1998;70:1567–1576. doi: 10.1046/j.1471-4159.1998.70041567.x. [DOI] [PubMed] [Google Scholar]
- SIM L.J., SELLY D.E., CHILDERS S.R. In vitro autoradiography of receptor-activated G-proteins in rat brain by agonist-stimulated guanylyl 5′-[γ-[35S]thio]-triphosphate binding. Proc. Natl. Acad. Sci. USA. 1995;92:7242–7246. doi: 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIM J.S., SELLY D.E., XIAO R., CHILDERS S.R. Differences in G-protein activation by μ- and δ-opioid, and cannabinoid, receptors in rat striatum. Eur. J. Pharmacol. 1996;307:97–105. doi: 10.1016/0014-2999(96)00211-7. [DOI] [PubMed] [Google Scholar]
- SHIMOHIRA I., TOKUYAMA S., HIMENO A., NIWA M., UEDA H. Characterization of nociceptin-stimulated in site [35S]GTPγS binding in comparison with opioid agonist-stimulated ones in brain regions of the mice. Neurosci. Lett. 1997;237:113–116. doi: 10.1016/s0304-3940(97)00807-0. [DOI] [PubMed] [Google Scholar]
- SORA I., FUNADA M., UHL G.R. The μ-opioid receptor is necessary for [D-Pen2,D-Pen5]enkephalin-induced analgesia. Eur. J. Pharmacol. 1997a;324:R1–R2. doi: 10.1016/s0014-2999(97)10016-4. [DOI] [PubMed] [Google Scholar]
- SORA I., TAKAHASHI N., FUNADA M., UJIKE H., REVAY R.S., DONOVAN D.M., MINER L.L., UHL G.R. Opiate receptor knockout mice define μ receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. U.S.A. 1997b;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of Pharmacological Calculation with Computer Programs. New York: Springer; 1987. [Google Scholar]
- TRAYNOR J.R., NAHORSKI S.R. Modulation by μ-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995;47:848–854. [PubMed] [Google Scholar]
- ZADINA J., HACKLER L., GE L.-J., KASTIN A.J. A potent and selective endogenous agonist for the μ-opiate receptor. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]