

Abstract
The effect of interleukin-8 (IL-8) and growth-related oncogene α (GROα) on [35S]-guanosine 5′-O-(3-thiotriphosphate) ([35S]GTPγS) binding, forskolin-stimulated cyclic AMP accumulation and cytosolic calcium concentration were determined in recombinant CHO cells expressing HA-tagged CXC-chemokine receptors 1 and 2 (CXCR1 and CXCR2).
Radioligand binding assays confirmed that the binding profiles of the recombinant receptors were similar to those of the native proteins. IL-8 displaced [125I]-IL-8 binding to CXCR1 and CXCR2 with pKi values of 8.89±0.05 and 9.27±0.03, respectively. GROα, a selective CXCR2 ligand, had a pKi value of 9.66±0.39 at CXCR2 but a pKi>8 at CXCR1. Calcium mobilization experiments were also consistent with previous reports on native receptors.
Activation of both receptors resulted in stimulation of [35S]GTPγS binding and inhibition of adenylyl cyclase.
A comparison of the functional data at CXCR1 showed that a similar potency order (IL-8>>GROα) was obtained in all three assays. However, at CXCR2 whilst the potency orders for calcium mobilization and inhibition of adenylyl cyclase were similar (IL-8⩾GROα), the order was reversed for stimulation of [35S]GTPγS binding (GROα>IL-8).
All of the functional responses at both receptors were inhibited by pertussis toxin (PTX), suggesting coupling to a Gi/Go protein. However, the calcium mobilization induced by IL-8 at CXCR1 was not fully inhibited by PTX, suggesting an interaction with a G-protein of the Gq family. Our results with pertussis toxin also suggested that, in the [35S]GTPγS binding assay, CXCR1 displays some constitutive activity.
Thus, we have characterized the binding and several functional responses at HA-tagged CXCRs 1 and 2 and have shown that their pharmacology agrees well with that of the native receptors. We also have preliminary evidence that CXCR1 displays constitutive activity in our cell line and that CXCR2 may traffic between different PTX sensitive G-proteins.
Keywords: Interleukin-8, GROα, chemokine receptors, [35S]GTPγS binding
Introduction
The chemokines are a group of proteins which act as leukocyte chemotactic factors. They can be divided into two main families on the basis of the relative positions of the first two of four conserved cysteine residues (Power & Wells, 1996). In the CXC chemokines, these cysteines are separated by an intervening amino acid residue, whilst in the CC-chemokines they are adjacent. Interleukin-8 (IL-8) and the related peptide, growth-related oncogene α (GROα) (also termed melanocyte growth stimulatory activity, MGSA), are members of the CXC-chemokine family and are potent chemotactic factors for neutrophils (Baggiolini & Clark-Lewis, 1992). The effects of IL-8 on human neutrophils are mediated by two receptors, CXC-chemokine receptors 1 and 2 (CXCR1 and 2); previously known as IL-8 receptors A and B. CXCR1 is highly selective for IL-8, virtually all of the other CXC-chemokines having much lower binding affinities and functional potencies at this receptor (e.g. Lee et al., 1992; Petersen et al., 1994; Ahuja & Murphy, 1996). CXCR2 is less ligand selective than CXCR1, being a receptor for IL-8 and a number of other CXC-chemokines (including GROα), all of which contain the sequence glu-leu-arg (ELR) immediately preceding the first of the conserved cysteine residues (Loetscher et al., 1994; Petersen et al., 1994; Ahuja & Murphy, 1996). CXC-chemokines without this ELR sequence appear to lack activity at CXCR1 and CXCR2 (Petersen et al., 1996; Clark-Lewis et al., 1993; Proost et al., 1993).
The functional significance of the presence of two receptors for IL-8 on human neutrophils is as yet unclear, for example mouse neutrophils possess only one CXCR (Lee et al., 1995), and there appears to be a large degree of overlap between the signalling pathways activated by the two receptors. In recombinant systems, activation of either receptor induces chemotaxis and calcium mobilization (Lee et al., 1992; Loetscher et al., 1994; Wu et al., 1996). Both receptors have also been shown to be able to couple to both pertussis toxin (PTX) sensitive and insensitive G-proteins (of the Gq family) when co-transfected into COS7 cells (Wu et al., 1993). However, responses to IL-8 and GROα in human neutrophils, including chemotaxis and calcium mobilization, are markedly inhibited by PTX (Bacon & Camp, 1990; Thelen et al., 1988; Wuyts et al., 1997), suggesting either that the PTX sensitive pathways are predominant or that there is synergy between the PTX sensitive and insensitive signalling mechanisms. Interestingly, in neutrophils, whilst both IL-8 and GROα induce calcium mobilization and chemotaxis (Walz et al., 1991; Schroder et al., 1990), only IL-8 is able to activate phospholipase D (L'Heureux et al., 1995), implying some signalling specificity for CXCR1 (or its co-activation with CXCR2). Also, calcium mobilization in response to IL-8 and GROα appears to occur through different mechanisms: GROα has been reported to cause the influx of extracellular calcium as well as release of calcium from intracellular stores (Damaj et al., 1996b), whilst in the same study IL-8 was only able to elicit intracellular store release. One study using blocking antibodies selective for either CXCR1 or CXCR2 has suggested that the major receptor for IL-8 on the human neutrophil is CXCR1 (Hammond et al., 1995). In this study, an anti-CXCR2 antibody virtually abolished chemotaxis to GROα, whilst only weakly affecting that to IL-8, whereas an anti-CXCR1 antibody markedly inhibited chemotaxis in response to IL-8 but had no significant effect on that to GROα.
Whilst the signalling pathways activated in response to stimulation of CXCR1 and CXCR2 have been studied to some extent, there have been few reports in which systematic pharmacological comparisons have been made between agonists at either receptor. In the present study we have examined the binding of IL-8 and GROα to haemagglutinin (HA)-tagged CXCR1 and CXCR2 expressed in Chinese hamster ovary (CHO) cells and have compared the abilities of these agonists to elicit a number of functional responses. Calcium mobilization was measured as this assay has been widely used to study the activation of CXCRs (see above) and was, therefore, used to assess the authenticity of the pharmacology of the tagged receptors. Also, calcium mobilization is thought to be involved in a number of neutrophil responses (Maxfield, 1993, Sengelov et al., 1993). We have also determined the ability of the agonists to inhibit forskolin-stimulated cyclic adenosine monophosphate (cyclic AMP) accumulation and to stimulate [35S]-guanosine 5′-O-(3-thiotri-phosphate) (GTPγS) binding. CXCRs have been shown to couple to Gi proteins (Wu et al., 1993; Damaj et al., 1996a) and would thus be expected to inhibit forskolin-stimulated cyclic AMP accumulation in CHO cells. The inhibition of adenylyl cyclase is thought to be mediated by the α-subunit of Gi proteins whilst PTX sensitive calcium mobilization is generally thought to involve the activation of phospholipase C-β isoforms by the βγ-subunit (Morris & Scarlata, 1996). The measurement of both of these responses will, therefore, allow the comparison of two signal transduction events occurring at different points along the transduction pathways arising from the activation of the same G-protein. Stimulation of [35S]GTPγS binding measures one of the first signalling events initiated by G-protein coupled receptors and it was, therefore, of interest to compare it with the other two functional assays. This assay also has the advantage of being a functional assay which can be performed on membrane preparations in a radioligand binding format.
Methods
Cell culture
CHO-K1 cells were transfected with cDNA encoding CXCR1 or CXCR2 (derived from human neutrophils) using previously published methods (Solari et al., 1997). The cells were grown at 37°C in a 5% CO2 atmosphere in a 1 : 1 mixture of Dulbecco's modified Eagle medium and HAMS F-12 nutrient mixture (DMEM F-12) containing 10% foetal bovine serum (FBS), 2 mM L-glutamine and 300 μg ml−1 geneticin (to maintain selection pressure). Cells were passaged every 3–4 days. Untransfected CHO cells were grown in the same medium but in the absence of geneticin.
Measurement of increases in cytosolic calcium concentration
Confluent 175-cm2 flasks of cells were harvested by treatment with 5 mM ethylenediaminetetraacetic acid (EDTA) in phosphate buffered saline ((mM) NaCl 137, KCl 2.7, KH2PO4 1.5, Na2HPO4 8) (PBS-EDTA) followed by centrifugation at 500×g for 5 min. The cell pellet was resuspended in 10 ml HEPES-saline ((mM) NaCl 145, KCl 5, MgCl2 1, CaCl2 1, glucose 10, N-2-hydroxyethylpiperazine-N′-ethanesulphonic acid (HEPES) 10, 2 mg ml−1 bovine serum albumin (BSA), 2.5 mM probenecid (to prevent export of fura2 from the cells; Edelman et al., 1994), pH 7.4) and the resulting cell suspension was incubated at 37°C for 45 min in the presence of 4 μM fura2-AM. The fura2-loaded cells were then centrifuged at 500×g for 5 min and resuspended at 3×105 ml in HEPES-saline. The cells were maintained at 37°C and aliquots were transferred to a Perkin-Elmer LS50 fluorimeter for calcium measurement. The response was measured as the ratio of emitted light intensities at 510 nm after alternate excitation at 340 or 380 nm (Grynkiewicz et al., 1985). The fluorescence intensity ratios were calibrated as previously described (Hall & Hourani, 1993). In experiments using PTX, the medium in the culture flask was replaced with fresh medium or fresh medium containing 100 ng ml−1 PTX 16–20 h prior to harvesting the cells.
Responses were quantified as the maximal increase in cytosolic calcium concentration after addition of agonist. Concentration-response curves were analysed by fitting the data with a four parameter logistic equation to determine pEC50 values and maximal increases in cytosolic calcium concentration.
Measurement of cyclic AMP accumulation
Cells were seeded into 96-well plates and incubated (37°C) in culture medium for 16–20 h before the medium was replaced with DMEM F-12 (160 μl) containing 300 μM isobutylmethylxanthine (IBMX), a phosphodiesterase inhibitor. After incubation for 60 min at 37°C, forskolin (final concentration 10 μM, which had been shown in control experiments to be the EC50) and appropriate concentrations of chemokines were added in 40 μl of DMEM F-12 containing 0.5% dimethylsulphoxide (DMSO). The final DMSO concentration in all wells was 0.1%. Cyclic AMP was allowed to accumulate for 15 min, after which the assay was terminated by aspiration of the medium and addition of 200 μl ice-cold 95% ethanol. The cells were incubated at 4°C for 45 min and 25 μl aliquots of the ethanol extract were transferred to 96-well scintillation proximity assay (SPA) plates and dried under a stream of air. The dry extract was then dissolved in 25 μl of cyclic AMP assay buffer (as provided with the Amersham kit) and the cyclic AMP content was determined using an Amersham scintillation proximity assay (SPA) kit, following the manufacturer's instructions. The cyclic AMP content of each sample was corrected to give the amount of cyclic AMP (pmol) per well.
In experiments using PTX, the cells were seeded into 96-well plates and incubated for 4 h at 37°C in culture medium to allow them to adhere. The medium was then replaced with fresh medium alone or fresh medium containing 100 ng ml−1 of PTX and the cells incubated for a further 16 h and treated as described above.
Concentration-response curves were fitted with four-parameter logistic equations to obtain estimates of the pEC50, maximal inhibition and Hill coefficient. For pooling of concentration-response curves, the data were converted to percentage inhibitions of the forskolin control after subtraction of cyclic AMP accumulation in the presence of IBMX alone.
Measurement of GTPγS binding in cell membrane preparations
The optimization of this assay protocol has previously been reported (Hall et al., 1998).
For preparation of cell membranes, cells were harvested from confluent 850-cm2 roller bottles by treatment with PBS-EDTA and centrifugation at 500×g for 5 min. The cell pellet was resuspended in homogenization buffer ((mM) HEPES 20, EDTA 1, [ethylene-bis(oxyethylenenitrilo)]tetraacetic acid (EGTA) 1, MgCl2 6, pH 7.4) and the cells left to swell on ice for 20 min before homogenization in a Waring blender (3×15 s, with 2 min incubation on ice between each bout). The homogenate was centrifuged at 1000×g for 10 min and the resulting supernatant was centrifuged at 48,000×g for 20 min at 4°C. The supernatant was discarded and the pellet was resuspended in 5 ml homogenization buffer per roller bottle. The membrane suspension was then frozen in aliquots at −80°C until required. The protein concentration was determined by the Bradford method using BSA as a standard.
For measurement of agonist-stimulated GTPγS binding, membranes from CHO-CXCR1 (25 μg protein ml−1) or CHO-CXCR2 (50 μg protein ml−1) were incubated in GTPγS binding buffer ((mM) HEPES 20, MgCl2 10, NaCl 100, pH 7.4) at 30°C for 20 min in the presence of GDP (10 μM). Agonists were added and followed 10 min later by [35S]GTPγS (100 pM). The total volume was 250 μl. The reaction was allowed to proceed for 20 min, with shaking, before the mixture was harvested through Whatman GF/C filters on a Brandel cell harvester. The filters were washed four times with 1 ml of ice cold milliQ water, transferred to scintillation vials, soaked in 4 ml of scintillation fluid overnight and the bound radioactivity determined by liquid scintillation counting. Non-specific binding was determined in the presence of 100 μM GTP. In experiments using PTX, cells were treated with 100 ng ml−1 of toxin for 16 h prior to membrane preparation.
After subtraction of non-specific binding, the raw d.p.m. data were fitted with four parameter logistic equations. When the data were pooled, the [35S]GTPγS bound in the presence of agonist was divided by that in the absence of agonist and is presented as a percentage of the basal.
Radioligand binding assays
Membranes were prepared as described for GTPγS binding assays above. The radioligand binding experiments were performed using a SPA protocol. Membranes (10 or 100 μg protein for CXCR1 or CXCR2, respectively) were mixed with 0.5 mg wheat-germ agglutinin (WGA) SPA bead and increasing concentrations of [125I]IL-8 (for saturation binding assays) or 50 pM [125I]IL-8 and increasing concentrations of unlabelled IL-8 or GROα (for competition experiments). The plates were incubated at room temperature for 8 h and counted on a Microbeta plate counter. The total assay volume was 100 μl. Non-specific binding was determined in the presence of 100 or 10 nM unlabelled IL-8 for CXCR1 and CXCR2, respectively. Saturation binding data were analysed by directly fitting the sum of a rectangular hyperbola and a straight line to the total binding data. Competition binding data were analysed by fitting the data with a four parameter logistic equation.
Statistics
All statistical comparisons were made using Student's t-test unpaired, P values less than 0.05 were considered significant.
Materials
IL-8 was obtained from the Geneva Biomedical Research Institute. GROα was purchased from Peprotech EC Ltd. Fatty acid-free BSA, DMEM F-12, EDTA, EGTA, FBS, fura2-AM, guanine nucleotides, HEPES, IBMX, PTX and probenecid were obtained from Sigma. [35S]GTPγS (1250 Ci mmol−1) was obtained from NEN, [125I]IL-8 (2000 Ci mmol−1), Biotrak SPA cyclic AMP assay kits and WGA SPA beads were from Amersham International. SPA plates were from Wallac. Geneticin was from Gibco BRL and L-glutamine was from Hyclone. Protein assay dye reagent was obtained from BioRad. All other reagents were AnalaR grade from BDH.
Results
Calcium mobilization
There was no significant difference between the potencies of IL-8 at CXCR1 and CXCR2 in the calcium mobilization assay (Figure 1, Table 1), however, IL-8 did induce a significantly greater maximal increase in calcium concentration in the CHO-CXCR1 cells (Figure 1, Table 1). GROα was a full agonist at CXCR2 (Figure 1b, Table 2), inducing a maximal increase in calcium concentration which was not significantly different from that of IL-8 in these cells. The potency of GROα at CXCR2 was not significantly different from that of IL-8. However, GROα had a low potency at CXCR1, only consistently mobilizing calcium at concentrations greater than 100 nM (Figure 1a). Treatment with PTX (100 ng ml−1, 16 h) effectively abolished calcium mobilization in response to IL-8 in the CHO-CXCR2 cells (Figure 1d). However, PTX-treated CHO-CXCR1 cells were still responsive to IL-8 (Figure 1c) with a pEC50 of 7.20±0.11 (n=3), which was more than 60 fold less potent than in untreated cells, and a significantly reduced maximal increase in cytosolic calcium concentration (180±44 nM, n=3). The effect of IL-8 (up to 10 μM) in untransfected CHO cells was indistinguishable from that of the vehicle (water) control (data not shown).
Figure 1.

Effect of IL-8 and GROα on cytosolic calcium concentration in CHO-CXCR1 (a) or CHO-CXCR2 (b) cells or of IL-8 in control or PTX-treated CHO-CXCR1 (c) or CHO-CXCR2 (d). Values are the means±s.e.mean of three separate determinations. Vertical bars show the s.e.mean. The curves plotted through the data points are four parameter logistic equations fitted to the mean data.
Table 1.
The pEC50 or pKi values, Emax values and Hill coefficients (nH) for IL-8 in the functional and binding assays at CXCR1 and CXCR2

Table 2.
The pEC50 or pKi values, Emax values and Hill coefficients (nH) for GROα in the functional and binding assays at CXCR1 and CXCR2
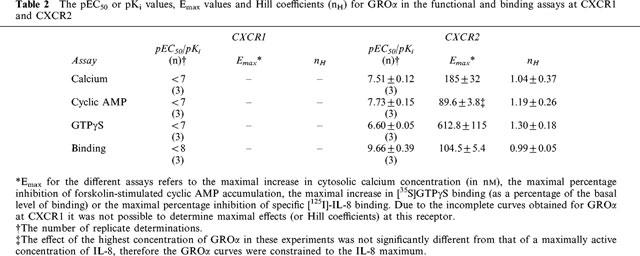
Cyclic AMP accumulation
IL-8 potently inhibited forskolin-stimulated cyclic AMP accumulation in both CHO cell lines, causing greater than 90% inhibition in both cases (Figure 2a, b, Table 1). IL-8 was slightly (3.3 fold) but significantly more potent in CHO-CXCR1 than in the CXCR2 cells. In the CXCR1 cells, GROα showed no significant inhibitory activity at up to 100 nM (Figure 2a), but it caused a potent inhibition of forskolin-stimulated cyclic AMP accumulation in the CXCR2 cells (Figure 2b, Table 2). The potency and maximal inhibitory effect of GROα in CXCR2 cells were not significantly different from those of IL-8. Treatment of either cell line with PTX completely abolished IL-8 mediated inhibition of forskolin-stimulated cyclic AMP accumulation. In the PTX treated cells, there appears to be a slight stimulation of cyclic AMP production at high concentrations of IL-8 (Figure 2c and d), however, this effect was not statistically significant. Also, in the absence of forskolin, IL-8 (up to 1 μM) was without effect on cyclic AMP levels in PTX-treated cells (data not shown). There was no effect of IL-8 on forskolin-stimulated cyclic AMP accumulation in untransfected CHO cells (data not shown).
Figure 2.
Effect of IL-8 and GROα on forskolin-stimulated cyclic AMP accumulation in CHO-CXCR1 (a) or CHO-CXCR2 (b) cells or of IL-8 in control or PTX-treated CHO-CXCR1 (c) or CHO-CXCR2 (d). Values are the means±s.e.mean of 3–11 separate determinations in duplicate. Vertical bars show the s.e.mean. The curves plotted through the data points are four parameter logistic equations fitted to the mean data.
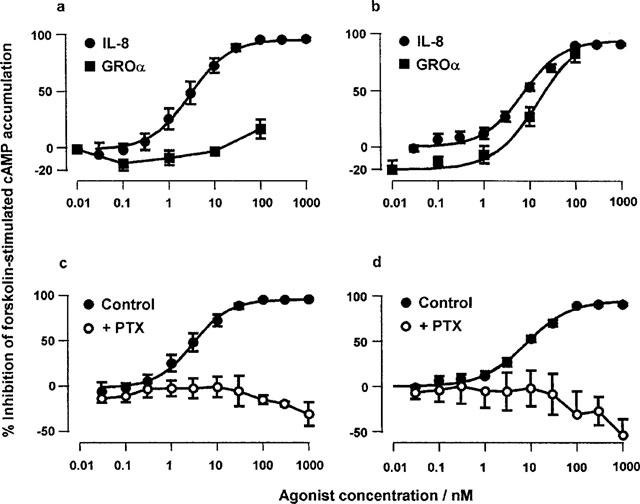
GTPγS binding
IL-8 induced marked increases in GTPγS binding in membranes prepared from both CHO-CXCR1 and -CXCR2 cells (Figure 3a, b, Table 1). IL-8 was 320 fold more potent at CXCR1 than CXCR2 in this assay. However, interestingly, the maximal stimulation of binding in CHO-CXCR2 membranes was significantly greater than that in CHO-CXCR1. GROα was significantly (approximately 3 fold) more potent than IL-8 at CXCR2, but was more than two orders of magnitude less potent than IL-8 in CXCR1 membranes (Figure 3a, b, Table 2). In the CXCR2 membranes, the maximal response to GROα was not well defined, however, the maximal response from the fitted curves was not significantly different from that of IL-8 suggesting that GROα was a full agonist. No response to IL-8 was seen in PTX-treated membranes from either cell line (Figure 3c and d). Interestingly, there was a significantly lower basal level of [35S]GTPγS binding in the PTX-treated CXCR1 membranes than in the untreated control membranes. There was no response to IL-8 in membranes from untransfected CHO cells (not shown).
Figure 3.
Effect of IL-8 and GROα on the binding of [35S]GTPγS to membranes from CHO-CXCR1 (a) or CHO-CXCR2 (b) cells or of IL-8 in control or PTX-treated CHO-CXCR1 (c) or CHO-CXCR2 (d) membranes. Values are the means±s.e.mean of 3–9 separate determinations in duplicate. Vertical bars show the s.e.mean. In (c) and (d) the values are presented as a percentage of the basal in the PTX-treated membranes. The curves plotted through the data points are four parameter logistic equations fitted to the mean data.
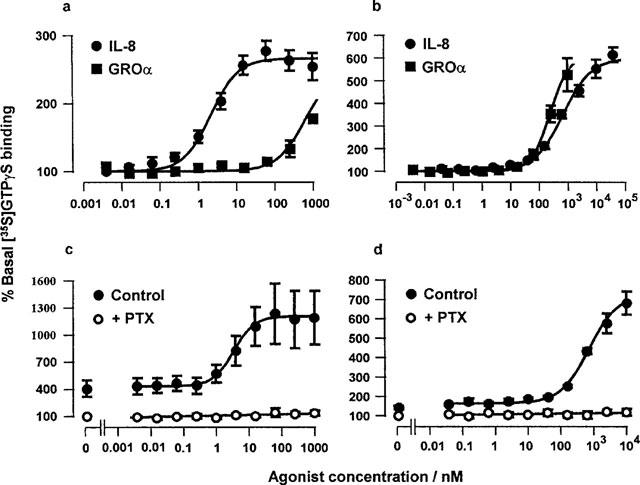
Radioligand binding
[125I]IL-8 bound saturably to membranes from both CHO-CXCR1 and -CXCR2 (data not shown). At CXCR1 the pKD was 9.64±0.03, the Hill coefficient was 1.17±0.01 and the maximal level of binding (Bmax) was estimated as 20±2 pmol.mg protein−1; at CXCR2 the pKD was 9.41±0.03, the Hill coefficient was 0.95±0.02 and the Bmax was estimated as 1.1±0.1 pmol.mg protein−1. These Bmax estimates indicate that the CHO-CXCR1 cell line was expressing an 18 fold greater density of receptors than CHO-CXCR2.
The data from competition binding assays with IL-8 and GROα are summarized in Tables 1 and 2 and illustrated in Figure 4. The affinity of IL-8 was greater at CXCR2 than CXCR1. The affinity of GROα was similar to that of IL-8 at CXCR2 but markedly less at CXCR1.
Figure 4.
Competition binding curves for IL-8 and GROα versus [125I]IL-8 in membranes from CHO-CXCR1 (a) or CHO-CXCR2 (b). Values are the means±s.e.mean of three separate determinations. Vertical bars show the s.e.mean. The curves plotted through the data points are four parameter logistic equations fitted to the mean data.
Discussion
In this study we have compared the ability of IL-8 receptors to interact with two commonly measured signalling systems, calcium mobilization and cyclic AMP production, and their ability to stimulate [35S]GTPγS binding, one of the earliest measurable consequences of receptor activation.
[125I]IL-8 bound saturably to both cell lines with affinities in the nanomolar range. The estimated Bmax values should be treated with caution as they were obtained using an agonist radioligand and in a SPA format. However, the data do suggest a higher density of receptor in the CXCR1 compared to the CXCR2 cells. Competition binding experiments confirmed that the affinities of the recombinant receptors for IL-8 and GROα were similar to those reported by others both in recombinant cell lines and in neutrophils (Ahuja & Murphy, 1996; Lee et al., 1992; Loetscher et al., 1994; Wu et al., 1996), all of which were in the low nanomolar range. Similarly, the potency orders of these peptides (IL-8>>GROα at CXCR1; IL-8≈amp;GRO-α at CXCR2) in our calcium mobilization experiments are consistent with their potencies for the mobilization of calcium and the induction of chemotaxis in recombinant cells (Ahuja et al., 1996; Loetscher et al., 1994). Both of these observations suggest that the HA-tagged receptors in these cell lines exhibit native pharmacology.
IL-8-induced calcium mobilization at both receptors was sensitive to PTX. This is consistent with the effect of PTX in neutrophils and indicates that the calcium mobilization in our cell lines was predominantly mediated by Gi/Go proteins (and presumably therefore by the βγ subunit). There was, however, a residual calcium mobilization in response to IL-8 after PTX treatment in CHO-CXCR1. This is unlikely to be due to the use of an insufficiently high concentration of toxin as the concentration used (100 ng ml−1) was sufficient to fully block the inhibition of cyclic AMP accumulation and stimulation of [35S]GTPγS binding in these cells. The PTX insensitive component of the response is likely therefore to represent an interaction with a PTX-insensitive class of G-protein, presumably of the Gq family. The ability of both CXCR1 and CXCR2 to interact with several members of the Gq family, when co-expressed in COS7 cells, has been reported (Wu et al., 1996). It is not clear why CXCR2 was unable to couple to the PTX insensitive pathway in our cell line. It may represent a difference in the coupling of the two receptors in CHO cells, however, CHO-CXCR2 expresses a lower density of receptor than CHO-CXCR1. If the receptors interact with relatively low efficiency with this pathway (as seems to be the case from the much lower EC50 of IL-8 at CXCR1 after PTX treatment), then the level of receptor present in CHO-CXCR2 may be insufficient to activate it. The maximal response to IL-8 in CHO-CXCR1 was larger than that in CHO-CXCR2 (see Table 1) but it is interesting to note that the size of the PTX-sensitive component of the response in the CXCR1 cells (∼184 nM) is similar to that induced by IL-8 in CHO-CXCR2 (175±43 nM), which was fully PTX-sensitive. This may imply that the PTX-sensitive pathways activated by these two receptors are similar.
The relative agonist potencies in the cyclic AMP accumulation assays were similar to those in the calcium mobilization assays. However, in contrast with the calcium response, inhibition of adenylyl cyclase by IL-8 was abolished by PTX in both cell lines (there was no PTX insensitive component in CHO-CXCR1) and the maximal levels of inhibition induced by IL-8 were similar. In both cell lines, the potencies of IL-8 (and GROα in CHO-CXCR2) in the cyclic AMP accumulation assays were very similar to those in the calcium mobilization assays. The relative potencies for inhibition of adenylyl cyclase and calcium mobilization in response to Gi-coupled receptor activation seem to vary depending on the cell line (Dickenson & Hill, 1995; Sharp et al., 1996; Schoeffter et al., 1997). However, in CHO cells, stimulation of the endogenous 5-HT1B-like receptor activates these two pathways with equal potency (Dickenson & Hill, 1995) which is consistent with our data.
The effects of the peptides in CHO-CXCR1 membranes in the [35S]GTPγS binding assay were also similar to those in the other two functional assays. In CXCR2 membranes, however, the potency order of the peptides was reversed, GROα was ∼3 fold more potent at stimulating [35S]GTPγS binding than IL-8. This difference in potency is relatively small but it is highly statistically significant (P<0.01). One possible explanation is that the G-proteins measured in the [35S]GTPγS binding assay are distinct from those which mediate the downstream responses. The difference in agonist potency series between [35S]GTPγS binding and the downstream responses would then imply some form of agonist trafficking (Kenakin, 1995). Another possibility is that the G-proteins interact less efficiently with the downstream effectors when the receptor is activated by GROα than when it is activated by IL-8. It has recently been shown, for the β2 adrenoceptor, that an agonist was able to stimulate the activity of adenylyl cyclase even when the G-protein pool was saturated with guanine nucleotides (Ugur & Onaran, 1997) implying that an agonist can influence the activity of signalling pathways by a mechanism additional to catalysis of nucleotide exchange. Different agonists would not necessarily have similar orders of efficacy for these two effects resulting potentially in differences in potency orders between [35S]GTPγS binding and downstream effects. However, further experiments would be necessary to distinguish between and provide firm support for either of these hypotheses.
PTX abolished the effect of IL-8 on [35S]GTPγS binding in both cell lines. Additionally, in the CHO-CXCR1 membranes there was a marked reduction in the basal level of binding in the PTX-treated membranes (Figure 3c). A smaller effect, which was not statistically significant, was also seen in the CXCR2 membranes. This effect of PTX may suggest that there is some constitutive activity of CXCR1 in this cell line resulting in a significant level of G-protein activation in the absence of agonist. The significant effect in the CHO-CXCR1 membranes is consistent with the greater level of receptor expression in this cell line. It could be argued that the PTX sensitive basal activity in these membranes was due to the activity of other (endogenous) Gi coupled receptors in this cell line. However, if this were the case PTX might have been expected to have a similar magnitude of effect in CHO-CXCR2. The abolition by PTX of the [35S]GTPγS binding response in the CHO-CXCR1 cells also implies that the PTX-insensitive component of the calcium response at CXCR1 is mediated by a G-protein which is not detected in [35S]GTPγS binding assays or that it is not G-protein mediated.
In marked contrast to the other two functional responses, there was a very large difference in the potencies of IL-8 at CXCR1 and CXCR2 in the [35S]GTPγS binding assays. The potency of IL-8 at CXCR1 was very similar to that in the other assays whilst at CXCR2 it was some two orders of magnitude weaker. The close agreement of the pEC50 values for IL-8 in the three assay systems is somewhat surprising, particularly for the [35S]GTPγS binding assay at CXCR1. Each of the functional responses we have measured occur at different stages along the signal transduction pathway and would be expected to exhibit different levels of receptor reserve. [35S]GTPγS binding is a measure of nucleotide exchange on the G-protein and is one of the earliest measurable consequences of receptor activation. As this response occurs very early in the transduction pathway it would be expected to show the least amplification and agonists would therefore be expected to evoke this response with lower potencies than those further downstream. This was observed here for CXCR2 and has been observed for other receptors such as the D2 dopamine receptor (Gardner et al., 1997). The very similar pEC50 values for IL-8 at the three responses in the CXCR1 cells may suggest that CXCR1 interacts with a G-protein which is only poorly coupled to the downstream responses (particularly in comparison with CXCR2). This would result in a much smaller leftward shift in the dose-response curves between [35S]GTPγS binding and the downstream responses for CXCR1 than CXCR2. It would also suggest that the two receptors activate different G-proteins in the [35S]GTPγS binding assay.
However, this difference could also be due to the higher level of receptor expression in CHO-CXCR1 cells. From the effect of PTX in the [35S]GTPγS binding assays, CXCR1 may exhibit constitutive activity whilst CXCR2 does not. However, there did not seem to be any evidence of constitutive activity of CXCR1 in the whole cell assays, e.g. PTX treatment did not cause an increase in forskolin-stimulated cyclic AMP accumulation in CHO-CXCR1 cells. This may imply that, in the whole cell assays, the constitutive receptor activity had caused a homeostatic uncoupling of the G-protein from the effector pathways. The much lower levels of receptor in CXCR2 (which do not show significant constitutive activity in the [35S]GTPγS binding assay) would presumably not induce this uncoupling and would be expected to show a much greater level of amplification from G-protein to effectors, as indeed was observed. It is, of course, also possible that the G-proteins which are being measured in the [35S]GTPγS binding assay are not the G-proteins responsible for the downstream responses (see above). The constitutive activation of the G-protein would simply not be transmitted to the downstream effectors in this case. It may be possible to address some of these issues by performing adenylyl cyclase assays in membranes rather than in whole cells. This would allow the direct comparison of the adenylyl cyclase activity in membranes from the two cell lines after the various treatments, e.g. does PTX treatment affect the activity of adenylyl cyclase in the CXCR1 cells?
It is unwise to compare the functional potencies of the agonists in this study with their binding affinities as the latter were determined using an agonist radioligand. These affinities presumably therefore represent binding of the peptides to the high affinity state of the receptor whilst the low affinity state of the receptor is thought to be important for functional responses. For example, Gardner et al. (1997) were able to simultaneously measure agonist binding affinity and functional potency in a [35S]GTPγS binding assay and showed that for D2 dopamine receptors the affinities of agonists in the assay (which lay to the right of their functional potencies) corresponded to their low affinity states from competition binding assays.
In conclusion, we have characterized the binding and three functional responses of HA-tagged CXC-chemokine receptors 1 and 2 and shown that, in general, their pharmacology agrees well with that reported for the native receptors. The only anomaly was the stimulation of [35S]GTPγS binding at CXCR2 where the potency order of IL-8 and GROα was reversed. This may represent trafficking of the receptor between distinct signalling pathways in our cell lines for this receptor or may indicate subtle differences in the effects of the peptides at different stages of the activation process. We also have preliminary evidence that at relatively high expression levels CXCR1 displays constitutive coupling to PTX-sensitive G-proteins.
Acknowledgments
We would like to thank Tania Chernov-Rogan of Affymax Incorporated for providing the recombinant cell lines used in this study, Kerri-Ann Cartwright for technical assistance and Murray Mackinnon and Helen Connor for helpful discussions during the writing of this manuscript.
Abbreviations
- Bmax
the maximal level of binding
- BSA
bovine serum albumin
- cyclic AMP
cyclic adenosine monophosphate
- CHO cells
Chinese hamster ovary cells
- CXCR1 and 2
CXC-chemokine receptors 1 and 2
- DMEM F-12
a 1 : 1 mixture of Dulbecco's modified Eagle medium and HAMS F-12 nutrient mixture
- DMSO
dimethylsulphoxide
- EDTA
ethylenediaminetetraacetic acid
- EGTA
[ethylene-bis(oxyethylenenitrilo)]tetraacetic acid
- FBS
foetal bovine serum
- GROα
growth-related oncogene α
- GTPγS
guanosine 5′-O-(3-thiotriphosphate)
- HA-tagged
haemagglutinin-tagged
- HEPES
N-2-hydroxyethylpiperazine-N′-ethanesulphonic acid
- IBMX
isobutylmethylxanthine
- IL-8
interleukin-8
- PBS
phosphate buffered saline
- PTX
pertussis toxin
- SPA
scintillation proximity assay
- WGA
wheat-germ agglutinin
References
- AHUJA S.K., LEE J.C., MURPHY P.M. CXC chemokines bind to unique sets of selectivity determinants that can function independently and are broadly distributed on multiple domains of human interleukin-8 receptor B. J. Biol. Chem. 1996;271:225–232. doi: 10.1074/jbc.271.1.225. [DOI] [PubMed] [Google Scholar]
- AHUJA S.K., MURPHY P.M. The CXC chemokines growth-related oncogene (GRO) α, GROβ, GROγ, neutrophil-activating peptide-2 and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not type A, human interleukin-8 receptor. J. Biol. Chem. 1996;271:20545–20550. doi: 10.1074/jbc.271.34.20545. [DOI] [PubMed] [Google Scholar]
- BACON K.B., CAMP R.D.R. Interleukin (IL)-8-induced in vitro human lymphocyte migration is inhibited by cholera and pertussis toxins and inhibitors of protein kinase C. Biochem. Biophys. Res. Commun. 1990;169:1099–1104. doi: 10.1016/0006-291x(90)92008-n. [DOI] [PubMed] [Google Scholar]
- BAGGIOLINI M., CLARK-LEWIS I. Interleukin-8, a chemo-tactic and inflammatory cytokine. FEBS Lett. 1992;307:97–101. doi: 10.1016/0014-5793(92)80909-z. [DOI] [PubMed] [Google Scholar]
- CLARK-LEWIS I., DEWALD B., GEISER T., MOSER B., BAGGIOLINI M. Platelet factor 4 binds to IL-8 receptors and activates neutrophils when its N-terminus is modified with glu-leu-arg. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3574–3577. doi: 10.1073/pnas.90.8.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAMAJ B.B., MCCOLL S.R., MAHANA W., CROUCH M.F., NACCACHE P.H. Physical association of G12α with interleukin-8 receptors. J. Biol. Chem. 1996a;271:12783–12789. doi: 10.1074/jbc.271.22.12783. [DOI] [PubMed] [Google Scholar]
- DAMAJ B.B., MCCOLL S.R., NEOTE K., HEBERT C.A., NACCACHE P.H. Diverging signal transduction pathways activated by interleukin 8 (IL-8) and related chemokines in human neutrophils: IL-8 and Gro-α differentially stimulate calcium influx through IL-8 receptors A and B. J. Biol. Chem. 1996b;271:20540–20544. doi: 10.1074/jbc.271.34.20540. [DOI] [PubMed] [Google Scholar]
- DICKENSON J.M., HILLS S.J. Coupling of an endogenous 5-HT1B-like receptor to increases in intracellular calcium through a pertussis toxin-sensitive mechanism in CHO-K1 cells. Br. J. Pharmacol. 1995;116:2889–2896. doi: 10.1111/j.1476-5381.1995.tb15941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDELMAN J.L., KAJIMURA M., WOLDEMUSSIE E., SACHS G. Differential effects of carbachol on calcium entry and release in CHO cells expressing m3 muscarinic receptor. Cell Calcium. 1994;16:181–193. doi: 10.1016/0143-4160(94)90021-3. [DOI] [PubMed] [Google Scholar]
- GARDNER B.R., HALL D.A., STRANGE P.G. Agonist action at D2(short) dopamine receptors determined in ligand binding and functional assays. J. Neurochem. 1997;69:2589–2598. doi: 10.1046/j.1471-4159.1997.69062589.x. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HALL D.A., BERESFORD I.J.M., GILES H.Characterisation of a [35S]GTPγS binding assay for chemokine CXC 1 and 2 (IL-8 A and B) receptors expressed on CHO cells Br. J. Pharmacol. 1998. in press [DOI] [PMC free article] [PubMed]
- HALL D.A., HOURANI S.M.O. Effect of analogues of adenine nucleotides on increases in intracellular calcium concentration mediated by P2T-purinoceptors on human blood platelets. Br. J. Pharmacol. 1993;108:728–733. doi: 10.1111/j.1476-5381.1993.tb12869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMMOND M.E.W., LAPOINTE G.R., FEUCHT P.H., HILT S., GALLEGOS C.A., GORDON C.A., GIEDLIN M.A., MULLENBACH G., TEKAMP-OLSON P. IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J. Immunol. 1995;155:1428–1433. [Google Scholar]
- KENAKIN T. Agonist-receptor efficacy II: Agonist trafficking of receptor signals. Trends Pharmacol. Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- LEE J., CACALANO G., CAMERATO T., TOY K., MOORE M.W., WOOD W.I. Chemokine binding and activities by the mouse IL-8 receptor. J. Immunol. 1995;155:2158–2164. [PubMed] [Google Scholar]
- LEE J., HORUK R., RICE G.C., BENNETT G.L., CAMERATE T., WOOD W.I. Characterisation of two high affinity human interleukin-8 receptors. J. Biol. Chem. 1992;267:16283–16287. [PubMed] [Google Scholar]
- L'HEUREUX G., BOURGOIN S., JEAN N., MCCOLL S.R., NACCACHE P.H. Diverging signal transduction pathways activated by interleukin-8 and related chemokines in human neutrophils: Interleukin-8, but not NAP-2 or GROα, stimulates phospholipase D activity. Blood. 1995;85:522–531. [PubMed] [Google Scholar]
- LOETSCHER P., SEITZ M., CLARK-LEWIS I., BAGGIOLINI M., MOSER B. Both interleukin-8 receptors independently mediate chemotaxis. Jurkat cells transfected with IL-8R1 or IL-8R2 migrate in response to IL-8, GROα and NAP-2. FEBS Lett. 1994;341:187–192. doi: 10.1016/0014-5793(94)80454-0. [DOI] [PubMed] [Google Scholar]
- MAXFIELD F.R. Regulation of leukocyte locomotion by Ca2+ Trends Cell Biol. 1993;3:386–391. doi: 10.1016/0962-8924(93)90088-i. [DOI] [PubMed] [Google Scholar]
- MORRIS A.J., SCARLATA S. Regulation of effectors by G-protein α- and βγ-subunits: Recent insights from studies of the phospholipase C-β isoenzymes. Biochem. Pharmacol. 1996;54:429–435. doi: 10.1016/s0006-2952(97)00032-4. [DOI] [PubMed] [Google Scholar]
- PETERSEN F., FLAD H.-D., BRANDT E. Neutrophil-activating peptides NAP-2 and IL-8 bind to the same sites on neutrophils but interact in different ways. Discrepancies in binding affinities, receptor densities and biologic effects. J. Immunol. 1994;152:2467–2478. [PubMed] [Google Scholar]
- PETERSEN F., LUDWIG A., FLAD H.D., BRANDT E. TNF-α renders human neutrophils responsive to platelet factor 4: Comparison of PF-4 and IL-8 reveal different activity profiles of the two chemokines. J. Immunol. 1996;156:1954–1962. [PubMed] [Google Scholar]
- POWER C.A., WELLS T.N.C. Cloning and characterisation of human chemokine receptors. Trends Pharmacol. Sci. 1996;17:209–213. doi: 10.1016/0165-6147(96)10019-5. [DOI] [PubMed] [Google Scholar]
- PROOST P., DE WOLF-PEETERS C., CONINGS R., OPDENAKKER G., BILLIAU A., VAN DAMME J. Identification of a novel granulocyte chemotactic peptide (GCP-2) from human tumour cells: In vitro and in vivo comparison with natural forms of GRO, IP-10 and IL-8. J. Immunol. 1993;150:1000–1010. [PubMed] [Google Scholar]
- SCHOEFFTER P., BOBIRNAC I., BODDECKE E., HOYER D. Inhibition of cAMP accumulation via recombinant human serotonin 5-HT1A receptors: Considerations on receptor effector coupling across systems. Neuropharmacol. 1997;36:429–437. doi: 10.1016/s0028-3908(97)00043-9. [DOI] [PubMed] [Google Scholar]
- SCHRODER J.-M., PERSOON N.L.M., CHRISTOPHERS E. Lipopolysaccharide-stimulated human monocytes secrete, apart from neutrophil-activating peptide 1/ interleukin 8, a second neutrophil activating protein: NH2-terminal amino acid sequence identity with melanocyte growth stimulatory activity. J. Exp. Med. 1990;171:1091–1100. doi: 10.1084/jem.171.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENGELOV H., KJELDSEN L., BORREGAARD N. Control of exocytosis in early neutrophil activation. J. Immunol. 1993;150:1535–1543. [PubMed] [Google Scholar]
- SHARP B.M., SHAHABI N.A., HEAGY W., MCALLEN K., BELL M., HUNTOON C., MCKEAN D.J. Dual signal transduction through delta opioid receptors in a transfected human T-cell line. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8294–8299. doi: 10.1073/pnas.93.16.8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOLARI R., OFFORD R.E., REMY S., AUBRY J.-P., WELLS T.N.C., WHITEHORN E., OUNG T., PROUDFOOT A.E.I. Receptor-mediated endocytosis of CC-chemokines. J. Biol. Chem. 1997;15:9617–9620. doi: 10.1074/jbc.272.15.9617. [DOI] [PubMed] [Google Scholar]
- THELEN M., PEVERI P., KERNEN P., VON TSCHARNER V., WALZ A., BAGGIOLINI M. Mechanism of neutrophil activation by NAF, a novel monocyte-derived peptide agonist. FASEB J. 1988;2:2702–2706. [PubMed] [Google Scholar]
- UGUR Ö., ONARAN H.O. Allosteric equilibrium model explains steady-state coupling of β-adrenergic receptors to adenylate cyclase in turkey erythrocyte membranes. Biochem. J. 1997;323:765–776. doi: 10.1042/bj3230765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALZ A., MELONI F., CLARK-LEWIS I., VON TSCHARNER V., BAGGIOLINI M. [Ca2+]i changes and respiratory burst in human neutrophils and monocytes induced by NAP1/interleukin-8, NAP-2, and gro/MGSA. J. Leukocyte Biol. 1991;50:279–286. doi: 10.1002/jlb.50.3.279. [DOI] [PubMed] [Google Scholar]
- WU D., LAROSA G.J., SIMON M.I. G-protein-coupled signal transduction pathways for IL-8. Science. 1993;261:101–103. doi: 10.1126/science.8316840. [DOI] [PubMed] [Google Scholar]
- WU L., RUFFING N., NEWMAN W., SOLER D., MACKAY C.R., QIN S. Discrete steps in binding and signalling of interleukin-8 with its receptor. J. Biol. Chem. 1996;271:31202–31209. doi: 10.1074/jbc.271.49.31202. [DOI] [PubMed] [Google Scholar]
- WUYTS A., VAN OSSELAER N., HAELENS A., SAMSON I., HERDEWIJN P., BEN-BURACH A., OPPENHEIM J.J., PROOST P., VAN DAMME J. Characterization of synthetic human granulocyte chemotactic protein 2: usage of chemokine receptors CXCR1 and CXCR2 and in vivo inflammatory properties. Biochem. 1997;36:2716–2723. doi: 10.1021/bi961999z. [DOI] [PubMed] [Google Scholar]