

Abstract
Peroxynitrite, a potent oxidant formed by the reaction of nitric oxide and superoxide causes thymocyte necrosis, in part, via activation of the nuclear enzyme poly(ADP-ribose) synthetase (PARS). The cytotoxic PARS pathway initiated by DNA strand breaks and excessive PARS activation has been shown to deplete cellular energy pools, leading to cell necrosis. Here we have investigated the effect of tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN) a heavy metal chelator on peroxynitrite-induced cytotoxicity.
TPEN (10 μM) abolished cell death induced by authentic peroxynitrite (25 μM) and the peroxynitrite generating agent 3-morpholinosidnonimine (SIN-1, 250 μM). Preincubation of TPEN with equimolar Zn2+ but not Ca2+ or Mg2+ blocked the cytoprotective effect of the chelator.
TPEN (10 μM) markedly reduced the peroxynitrite-induced decrease of mitochondrial transmembrane potential, secondary superoxide production and mitochondrial membrane damage, indicating that it acts proximal to mitochondrial alterations.
Although TPEN (1–300 μM) did not scavenge peroxynitrite, it inhibited PARS activation in a dose-dependent manner.
The cytoprotective effect of TPEN is only partly mediated via PARS inhibition, as the chelator also protected PARS-deficient thymocytes from peroxynitrite-induced death.
While being cytoprotective against peroxynitrite-induced necrotic death, TPEN (10 μM), similar to other agents that inhibit PARS, enhanced apoptosis (at 5–6 h after exposure), as characterized by phosphatydilserine exposure, caspase activation and DNA fragmentation.
In conclusion, the current data demonstrate that TPEN, most likely by zinc chelation, exerts protective effects against peroxynitrite-induced necrosis. Its effects are, in part, mediated by inhibition of PARS.
Keywords: Peroxynitrite, cytotoxicity, zinc, TPEN, poly (ADP-ribose) synthetase
Introduction
Peroxynitrite, a potent oxidant formed in the near diffusion-limited reaction of superoxide and nitric oxide (Beckman et al., 1994; Beckman & Koppenol, 1996) is a major mediator of tissue injury in various forms of shock, inflammation and ischaemia-reperfusion (Zingarelli et al., 1997b; Szabó, 1996). The cytotoxic effect of peroxynitrite is mainly due to the activation of poly(ADP-ribose) synthetase (PARS) (Zingarelli et al., 1996; Szabó et al., 1996a,1996b; 1998; Virág et al., 1998b). PARS is a DNA nick sensor enzyme which, upon activation by DNA single strand breaks, cleaves NAD to nicotinamide and ADP-ribose and transfers poly(ADP-ribose) adducts to DNA and proteins (Szabó et al., 1996a). PARS is thought to play a role in maintaining genome integrity and may facilitate DNA repair (de Murcia et al., 1986; 1997; Trucco et al., 1998). Excessive PARS activation, however, depletes cellular energy pools and causes cell death (Cochrane, 1991; Szabó et al., 1996a; 1998; Virág et al., 1998b). The pathophysiological role of the PARS pathway has been demonstrated in various disease states (Szabó et al., 1996b; 1997a,1997c; 1998; for review see: Szabó, 1998; Szabó & Dawson, 1998).
We have previously shown that PARS activation is responsible for the dissipation of mitochondrial membrane potential, secondary superoxide production, mitochondrial membrane damage and the breakdown of plasma membrane integrity in peroxynitrite-treated thymocytes (Virág et al., 1998a). Furthermore, we have demonstrated that in the absence of PARS, peroxynitrite induced cell-death is diverted from necrotic to apoptotic death as indicated by increased DNA fragmentation, phosphatidylserine exposure and caspase activation (Virág et al., 1998b).
Here we have investigated the effect of tetrakis-(2-pyridylmethyl)ethylenediamine (TPEN), a zinc chelator, on the peroxynitrite-induced cytotoxicity. As PARS activation has been proposed to play a role in peroxynitrite-induced cell death, we also set out to investigate the effect of TPEN on PARS activation.
Methods
Animals
PARS-deficient and wild type mice (breeding pairs: kind gifts of Dr Z.Q. Wang, Inst. Molecular Pathology, Vienna, Austria) were bred at the animal care facility of the Children's Hospital Medical Center and were used at 4–6 weeks of age. Animals received food and water ad libitum, and lighting was maintained on a 12 h cycle.
Thymocyte preparation and peroxynitrite treatment
Thymi from 4–6-week-old male mice were aseptically removed and placed into ice cold RPMI media supplemented with 10% v v−1 foetal calf serum, glutamine (10 mM), HEPES (10 mM), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin. Single cell suspensions were prepared by sieving the organs through a stainless wire mesh. Cells isolated this way were routinely 95% viable, as assessed by Trypan blue exclusion assay. Thymocytes (106 cells in 0.5 ml medium) were seeded in 24-well plates. Peroxynitrite was diluted in phosphate buffered saline (PBS) (pH 8.9) and added to the cells in a bolus of 50 μl. Thymocytes were then incubated for various times (20 min for PARS assay, 3 h for the measurement of mitochondrial parameters, 4 h for propidium iodide and Annexin V staining or 6 h for DNA fragmentation and caspase activation). Decomposed peroxy nitrite (incubated for 30 min at pH 7.0) served as control, and failed to influence any of the parameters studied. In another set of studies, the morpholinosidnonimine compound SIN-1 (250 μM) was used to simultaneously generate NO and superoxide, which then combines to peroxynitrite and induced thymocyte death (see also Virág et al., 1998a).
Measurement of mitochondrial membrane potential, superoxide production and cardiolipin content
The mitochondrial membrane potential was quantitated by the flow cytometric analysis of 3,3′dihexyloxacarbocyanine iodide [DiOC6(3)]-stained cells (Zamzami et al., 1995). Intramitochondrial generation of reactive oxygen intermediates was determined by analysing with flow cytometry the superoxide-induced conversion of the oxidant-sensitive dye, dihydroethidium to ethidium (Zamzami et al., 1995). Mitochondrial membrane damage was determined by measuring the cardiolipin degradation, as described (Zamzami et al., 1995). The fluorochrome 10-N nonyl-acridine orange (NAO) stoichiometrically interacts with cardiolipin (1 : 2), the cellular distribution of which is restricted to mitochondria.
Flow cytometry
Thymocytes were stained with 5 μg ml−1 PI, 3,3′dihexyloxacarbocyanine iodide [DiOC6(3)] (40 nM), hydroethidine (HE) (2 μM), 10-N nonyl-acridine orange (NAO) (100 nM) for 15 min at 37°C, washed once with PBS and analysed with a FacsCalibur flow cytometer as described (Virág et al., 1998a). For the measurement of mitochondrial parameters, forward and side scatters were gated on the major population of normal-sized cells. For the cytotoxicity assay, the percentage of PI-positive cells was calculated from the total (ungated) population.
Samples processed for Annexin V-FITC/propidium iodide staining (Vermes et al., 1995) were washed with PBS and 105 cells (in 100 μl) were stained with 5 μl Annexin V-FITC and 5 μg ml−1 propidium iodide (PI) in annexin binding buffer: (in mM) HEPES (pH 7.4) 10, NaCl 140, CaCl2 2.5) at room temperature. After 15 min, 400 μl annexin binding buffer was added to the samples which were then immediately analysed with a FacsCalibur flow cytometer (Becton-Dickinson, San Jose, CA, USA).
Dihydrorhodamine assay
The peroxynitrite-dependent oxidation of dihydrorhodamine 123 to rhodamine 123 was measured based on the principles of the method previously described (Szabó et al., 1995). Briefly, peroxynitrite (5 μM) was added into phosphate-buffered saline containing 10 μM dihydrorhodamine 123, in the absence or presence of TPEN (3–300 μM). After a 10 min incubation at 22°C, the fluorescence of rhodamine 123 was measured using a Perkin-Elmer fluorimeter (Model LS50B; Perkin-Elmer, Norwalk, CT, USA) at an excitation wavelength of 500 nm, emission wavelength of 536 nm (slit widths 2.5 and 3.0 nm, respectively). In control, reverse-order experiments we have confirmed that TPEN neither showed fluorescence at the above wavelengths, nor was the inhibition of fluorescence by the compounds due to reduction of the rhodamine 123 fluorescence (data not shown).
Cytochrome c oxidation
The peroxynitrite-dependent oxidation of cytochrome c2+, was measured as described (Szabó et al., 1997b). Cytochrome c was reduced by sodium dithionite immediately before use and purified by chromatography on Sephadex G-25 using potassium phosphate (100 mM) plus DTPA, pH 7.2 (0.1 mM) as the elution buffer. The concentration of cytochrome c2+ was determined spectrophotometrically at 550 nm in the same buffer (ε=21 mM−1 cm−1). Cytochrome c2+ oxidation (50 μM) yields upon addition of peroxynitrite (25 μM initial concentration after mixing) were assessed by incubation of reaction mixtures in potassium phosphate (100 mM) plus DTPA, pH 7.2 (0.1 mM) at 22°C for 3 min in the absence or presence of TPEN (1–300 μM). Oxidation of cytochrome c2+ was followed at 550 nm using a Beckman DU 640 spectrophotometer (Fullerton, CA, USA). In control, reverse-order experiments we have confirmed that TPEN did not interfere with the spectrophotometric measurements at the above wavelengths. Moreover, in control experiments we have confirmed that the compound tested does not reduce cytochrome c3+.
Measurement of cellular PARS activity
Thymocytes (107 cells in 1 ml culture medium) were treated with peroxynitrite. After 20 min, cells were spun, medium was aspirated and cells were resuspended in 0.5 ml assay buffer (in mM) HEPES (pH 7.5) 56, KCl 28, NaCl 28, MgCl2 2, 0.01% w v−1 digitonin and 0.125 μM 3H-NAD (0.5 μCi ml−1)]. PARS activity was then measured as previously described (Virág et al., 1998b). Briefly, following incubation (10 min at 37°C), 200 μl ice cold 50% w v−1 TCA was added and samples incubated for 4 h at 4°C. Samples were then spun (10,000×g, 10 min) and pellets washed twice with ice cold 5% w v−1 TCA and solubilized overnight in 250 μl 2% w v−1 SDS/0.1 N NaOH at 37°C. Contents of the tubes were added to 6.5 ml ScintiSafe Plus scintillation liquid (Fisher Scientific) and radioactivity was determined using a liquid scintillation counter (Wallac, Gaithersburg, MD, USA).
Detection of internucleosomal DNA fragmentation of thymocytes
Thymocytes were pretreated with TPEN for 20 min and then treated with peroxynitrite (10–80 μM). After 6 h, cells were washed once with cold PBS and pellets resuspended in sample buffer (10 mM Tris, pH 8.0, 5% v v−1 glycerol, 0.05% w v−1 bromophenol blue, 5 mg ml−1 RNase). DNA fragmentation was detected as described (Eastman, 1995). Agarose (2% w v−1) was poured on a horizontal gel support. After solidification of the gel the top part (above the comb) was replaced with 1% w v−1 agarose containing 2% w v−1 SDS and 64 μg ml−1 proteinase K. Cells (2×106) were loaded in 20 μl sample buffer. Electrophoresis was carried out at 25 V for 12 h and the gel was stained with 2 μg ml−1 ethidium bromide for 1 h.
Measurement of caspase 3-like activity
Caspase activity was measured by the cleavage of the fluorogenic tetrapeptide-amino-4-methylcoumarine conjugate (DEVD-AMC) as described (Vanags et al., 1997). Unless otherwise indicated cells (4–10×106) were harvested 6 h after peroxynitrite treatment, washed once in PBS and then lysed in a lysis buffer: (in mM) HEPES 10, 0.1% w v−1 CHAPS, dithiothreitol 5, EDTA 2, 10 μg ml−1 aprotinin, 20 μg ml−1 leupeptin, 10 μg ml−1 pepstatin A and PMSF, pH 7.25 (1 mM), for 10 min on ice. Cell lysates and substrates (50 μM) were combined in triplicate in the caspase reaction buffer: HEPES (100 mM), 10% w v−1 sucrose, dithiothreitol (5 mM), 0.1% w v−1 CHAPS, pH 7.25 in the presence or absence of 10 μM of the tetrapeptide caspase 3 inhibitor N-acetyl-aspartyl-glutamyl-valyl-aspartyl-aldehyde (DEVD-CHO) and samples were incubated at 37°C for 60 min. AMC liberation was determined with a Perkin-Elmer fluorimeter using 380 nm excitation and 460 mission wavelength. Data are given as absolute fluorescence units.
Statistical analysis
All values in the figures and text are expressed as mean±standard deviation (S.D.) of n observations; n⩾3. Data sets were examined by analysis of variance and individual group means were then compared with Bonferroni-'s post hoc test. A P value less than 0.05 was considered statistically significant. When the results are presented as representative gels, or flow cytometry analyses, results similar to the ones shown were obtained in at least three different experiments.
Materials
Peroxynitrite was a kind gift of Dr H. Ischiropoulos (Inst. Environmental Medicine, University of Pennsylvania, PA, USA). 3-morpholinosidnonimine (SIN-1) was purchased from Calbiochem (San Diego, CA, USA). Tetrakis-(2-pyridyl-methyl)ethylenediamine (TPEN), 3,3′dihexyloxacarbocyanine iodide [DIOC6(3)], dihydroethidium (HE), nonyl-acridine orange (NAO), propidium iodide were obtained from Molecular Probes (Eugene, OR, USA). The tetrapeptide substrate (DEVD-AMC) and inhibitor (DEVD-CHO) of caspase 3 were purchased from Biomol (Plymouth Meeting, PA, USA). Proteinase K was obtained from Life Technologies (Grand Island, NY, USA). Annexin V-FITC was from Pharmingen (San Diego, CA, USA). Tris, magnesium chloride, analytical test filter funnels and Scintisafe scintillation cocktail were from Fisher Scientific (Pittsburgh, PA, USA). 3H-NAD was purchased from DuPont NEN (Boston, MA, USA). All the other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
Results
TPEN protects from peroxynitrite-induced cytotoxicity
Treatment of wild-type thymocytes with authentic peroxynitrite (20 μM) or the peroxynitrite releasing agent SIN-1 (250 μM) resulted in cell death, as determined by the uptake of the cell-impermeable fluorescent dye propidium iodide (Figure 1). Pretreatment of the cells with TPEN (10 μM) abolished peroxynitrite-induced cytotoxicity. Preincubation of TPEN with equimolar zinc chloride (ZnCl2) but not calcium chloride (CaCl2) or magnesium chloride (MgCl2) neutralized the protective effect of TPEN, indicating that the effect of TPEN is not related to calcium or magnesium chelation but may result from the chelation of zinc.
Figure 1.

Thymocytes were pretreated with either TPEN (10 μM) or TPEN (10 μM) in the presence or absence of equimolar CaCl2, MgCl2 or ZnCl2 for 30 min and then treated with the indicated concentration of peroxynitrite (ONOO) (A) or SIN-1 (B). After 4 h, cells were stained with propidium iodide and analysed by flow cytometry. Percentage number of PI positive cells±s.d. of triplicate samples are shown. **indicates a significant (P<0.01) cytotoxic effect of peroxynitrite and ##indicates a significant (P<0.01) protection against cytotoxicity.
TPEN does not scavenge peroxynitrite
TPEN at concentrations of 1–300 μM failed to affect the oxidation of cytochrome c by peroxynitrite (Figure 2A) indicating that the cytoprotective effect of the chelator is not due to a potential peroxynitrite scavenging activity. Similarly, the lack of peroxynitrite-scavenging effect of TPEN has also been confirmed with the dihydrorhodamine assay (Figure 2B): TPEN did not affect the oxidation of dihydrorhodamine 1,2,3 to rhodamine 1,2,3.
Figure 2.

The oxidation of cytochrome c (A) and dihydrorhodamine (B) by peroxynitrite was determined in the presence of various concentrations of TPEN. The peroxynitrite-induced increase in dihydrorhodamine (DHR) fluorescence or decrease in cytochrome c absorbance is shown. Data are given as mean±s.d. of triplicate experiments.
TPEN acts proximal to mitochondrial alterations
A central event of the cell death process is the collapse of mitochondrial membrane potential, followed by the production of superoxide anion and the loss of cardiolipin (for review see Kroemer et al., 1997). Our previous work has demonstrated that the same sequence of events occur during peroxynitrite-induced cell death (Virág et al., 1998a). We have also provided evidence that these mitochondrial alterations can be abolished by inactivation of PARS. Here we have determined whether the protective effect of TPEN is proximal or distal to mitochondrial alterations. The peroxynitrite or SIN-1-induced decrease of mitochondrial potential indicated by the decreased DiOC6(3) staining was prevented by TPEN (10 μM) pretreatment (Figure 3A). Similarly, peroxynitrite or SIN-1 induced secondary oxyradical production as measured by HE staining (Figure 3B) as well as the loss of mitochondrial cardiolipin indicated by reduced NAO staining were also markedly inhibited by TPEN (10 μM) (Figure 3C). Prevention of peroxynitrite-induced mitochondrial function alteration suggests that TPEN acts proximal to the mitochondrial phase during peroxynitrite-induced cell death.
Figure 3.
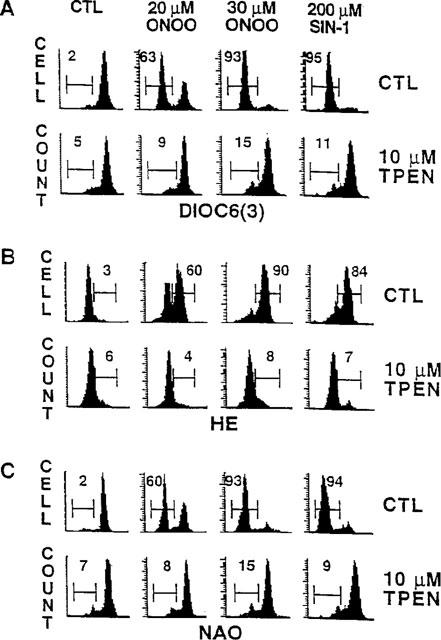
Thymocytes were either left untreated or were pretreated with TPEN (10 μM). Cells were then exposed to peroxynitrite or SIN-1 and incubated for 3 h. Thymocytes were then stained with DiOC6(3), hydroethidine (HE) or nonyl-acridine orange (NAO) for the measurement of mitochondrial membrane potential (A), superoxide production (B) and mitochondrial membrane damage (C), respectively. Histograms presented are representatives of three different experiments. Values shown indicate percentage of cells displaying decreased mitochondrial membrane potential, increased superoxide production and decreased mitochondrial membrane damage.
TPEN inhibits PARS activation
Since TPEN, similar to compounds that inhibit PARS, blocked peroxynitrite cytotoxicity at a step proximal to mitochondrial perturbations, we have examined the effect of TPEN on PARS activation. TPEN inhibited peroxynitrite-induced PARS activation (Figure 4) in a dose-dependent manner. At the concentration of TPEN used throughout the current study (10 μM), the chelator completely blocked peroxynitrite-induced PARS activity. Higher concentrations of TPEN reduced PARS activity below the baseline levels (Figure 4).
Figure 4.

Thymocytes were pretreated for 30 min with TPEN. Cells were then stimulated with peroxynitrite (40 μM) for 20 min and PARS activity was determined with the 3H-NAD assay. Data are given as mean±s.d. of triplicate experiments. **indicates a significant (P<0.01) inhibition of PARS activity.
Effect of TPEN on peroxynitrite-induced caspase 3 activation and DNA fragmentation
Our recent work has shown that the cytoprotection provided by PARS inhibition results in a shift from necrosis toward apoptotic cell death (Virág et al., 1998b). In the absence of PARS, peroxynitrite (10–80 μM) induced a dose-dependent increase in caspase 3 activity and DNA fragmentation. In the wild type cells, however, only low concentrations (10–20 μM) of peroxynitrite caused DNA fragmentation. At higher doses (40–80 μM) of peroxynitrite, PARS activation led to necrosis without DNA fragmentation (Virág et al., 1998b). Here we have investigated the effect of TPEN on two apoptotic parameters of peroxynitrite-treated cells: caspase activation and DNA fragmentation. In line with the PARS inhibitory effect of TPEN, the chelator prevented the inhibition of DNA fragmentation observed at higher doses (40–80 μM) of peroxynitrite (Figure 5A). TPEN alone (in the absence of peroxynitrite treatment) also induced DNA fragmentation in thymocytes, with DNA laddering first detectable 4 h after TPEN treatment (Figure 5C).
Figure 5.

Wild type (W.T.) and PARS knock out (K.O.) thymocytes were either left untreated or pretreated with TPEN (10 μM). Thymocytes were then exposed to the indicated concentrations (μM) of peroxynitrite and incubated for 6 h. DNA fragmentation was visualized by agarose gel electrophoresis (A) (TPEN pretreatment is indicated by ‘+' sign) and caspase-3 like activity was measured by DEVD-AMC cleavage (B). The time course of TPEN-induced DNA cleavage (C) and DEVD-ase activity (D) has also been determined 0, 2, 4 and 6 h after TPEN exposure. Data of caspase activity are given as mean±s.d. of triplicate measurements. *, **indicate significant (*P<0.05, **P<0.01) difference between vehicle and TPEN treated samples. #, # # indicate a significant increase of caspase activity by peroxynitrite when compared to the activity in untreated samples at time 0 (#P<0.05 and ##P<0.01).
Peroxynitrite caused a dose-dependent increase in caspase activity peaking at 40 μM peroxynitrite (Figure 5B). In addition, TPEN alone also induced a marked increase in caspase-3 like activity, starting at 2 h after exposure (Figure 5D).
Effect of TPEN on phosphatidylserine exposure
Peroxynitrite-induced necrotic and apoptotic cell death was accompanied by the appearance of phosphatidylserine in the outer membrane leaflet, as indicated by Annexin V-FITC binding. Annexin V binding in the absence of PI uptake indicates early apoptotic cells whereas Annexin V/PI double positive cells represent a primary necrotic or late apoptotic population (Virág et al., 1998b). Since phosphatidylserine exposure appears very early in the cytotoxic process, Annexin V/PI double-staining allows a more sensitive detection of cell death. Annexin V-FITC/PI double staining revealed that TPEN blocked the breakdown of membrane integrity (PI uptake) both at 3 and 5 h after peroxynitrite treatment. (Figure 6A and B, respectively). TPEN-induced phosphatidylserine exposure could be detected as early as 3 h after peroxynitrite treatment and further increased at 5 h. TPEN markedly antagonized the cytotoxic effect of peroxynitrite at 3 h (Figure 6A). Even at 5 h, when TPEN itself began to induce cytotoxicity, the zinc chelator protected against peroxynitrite-induced cytotoxicity. For example, considering Annexin V/PI double positive cells as dead, Annexin V single positive cells as committed to die, and double negative cells as viable, at 5 h, TPEN induced 40+19=59% and SIN-1 caused 22+63=85% toxicity. In TPEN pretreated cells exposed to SIN-1, however, only 26+11=37% cytotoxicity could be detected (Figure 6).
Figure 6.

Thymocytes were either left untreated or were pretreated with TPEN (10 μM). Cells were then treated with peroxynitrite or SIN-1 and incubated for 3 h (A) or 5 h (B) followed by Annexin V-FITC/propidium iodide double staining. Numbers indicate percentage of cells in lower right (Annexin V single positive) and upper right (Annexin V/propidium iodide double positive) quadrant. Dot plots shown are representative of three independent experiments.
PARS-independent effect of TPEN in peroxynitrite-induced cytotoxicity
Since PARS-deficient thymocytes were resistant to peroxynitrite-induced cytotoxicity (Virág et al., 1998a,1998b), in order to achieve a similar degree of cell death in these cells, four times higher doses of peroxynitrite (80 μM) were required. TPEN provided significant protection against peroxynitrite-induced cytotoxicity in the PARS-deficient thymocytes (Figure 7), indicating that the chelator also exerts cytoprotective effects independent of PARS inhibition. Similarly to wild type cells, 6 h after TPEN treatment PARS deficient thymocytes showed caspase activation and DNA fragmentation. Peroxynitrite caused DNA fragmentation at the dose of 20–80 μM (Figure 7B), whereas at 160 μM no DNA fragmentation could be detected (not shown). DEVD-ase activity was also increased following treatment with 20–80 μM peroxynitrite (Figure 7C). However, peroxynitrite-induced caspase activation and DNA fragmentation in PARS-deficient cells was unaffected by TPEN pretreatment.
Figure 7.

PARS-deficient thymocytes were pretreated with TPEN (10 μM) for 30 min and then treated with the peroxynitrite (ONOO) (80 μM). After 4 h, cells were stained with propidium iodide and analysed by flow cytometry (A). Percentage number of PI positive cells±s.d. of triplicate samples are shown. **indicates significant (P<0.01) increase in cytotoxicity. #, ## indicate significant (#P<0.05 and ##P<0.01, respectively) protection against cytotoxicity. The effect of TPEN (10 μM) on peroxynitrite (20–160 μM)-induced DNA fragmentation (B) (TPEN pretreatment is indicated by ‘+' sign) and caspase-3 like activity (C) was also investigated 6 h after peroxynitrite treatment. **indicates significant (P<0.01) difference between vehicle and TPEN treated samples. #, ## indicate a significant increase of caspase activity by peroxynitrite (#P<0.05 and ##P<0.01).
Discussion
Cytoprotective effect of TPEN via PARS inhibition
The present study demonstrates that peroxynitrite-induced thymocyte necrosis can be reduced by chelation of intracellular Zn2+. We have previously shown that, in our experimental system, peroxynitrite-induced PARS activation is responsible for the necrotic death of thymocytes (Virág et al., 1998a,1998b). Thus, it appeared plausible to hypothesize that the zinc chelator directly or indirectly inhibits PARS activation. In our current work we provide evidence that TPEN is a potent inhibitor of PARS activation. Considering that PARS is an enzyme with two zinc finger domains localized in the DNA binding subunit of the enzyme (Mazen et al., 1989; Menissier-de Murcia et al., 1989), it is plausible to hypothesize that TPEN acts via binding to zinc ions in the zinc finger domain of PARS, thereby inhibiting the binding of the enzyme to the single-strand break sites in the DNA.
The mode of the modulation of peroxynitrite-induced cytotoxicity by TPEN (suppression of necrosis, and enhancement of apoptotic DNA fragmentation), is similar to the effect of various PARS inhibitors (see Virág et al., 1998a,1998b). Thus, it is logical to propose that the mode of action of TPEN, in intact cells, is related to inhibition of cellular PARS activity. As discussed earlier (Virág et al., 1998a,1998b), in the absence of functional PARS, the cells are protected against necrotic death by energy failure, but, at the same time, may have preserved sufficient cellular energy reserves to complete the process of apoptosis, which is a known energy-dependent process.
PARS-independent cytoprotection by TPEN
Our results, demonstrating that TPEN protects PARS deficient thymocytes against peroxynitrite-induced necrosis, indicate that, in addition to being an inhibitor of PARS activation, TPEN may also interfere with a yet undefined, PARS-independent cytotoxic pathway. A potential candidate for a target for TPEN in mediating PARS-independent cytoprotection may be another zinc finger protein family, namely the protein kinase C (PKC) family. Although the effect of peroxynitrite on kinase signalling pathways has not yet been investigated, superoxide and nitric oxide have been shown to be involved in N-methyl-D-aspartate receptor mediated PKC activation (Klann et al., 1998) and TPEN was reported to inhibit the translocation of PKC from the cytosolic to the membranous fraction in this model (Baba et al., 1991). Moreover, hydrogen peroxide, another oxidant known to induce PARS-activation has been shown to induce PKC activation in rat cardiac myocytes (Sabri et al., 1998), COS-7 cells (Konishi et al., 1997) and in Jurkat T cells (Whisler et al., 1995). Identification of the exact mechanism of peroxynitrite cytotoxicity including a possible involvement of PKC activation in this process, however, requires further investigation.
TPEN-induced apoptosis
While being cytoprotective against peroxynitrite-induced necrotic death via PARS inhibition, TPEN itself induces apoptosis in thymocytes, especially at longer times of exposure. It has been known that chelation of Zn2+ by TPEN causes apoptosis in thymocytes (McCabe Jr et al., 1993; Jiang et al., 1995), lymphocytes (Treves et al., 1994) and HaCaT keratinocytes (Parat et al., 1997) as evidenced by DNA fragmentation and appearance of apoptotic nuclear morphology (nuclear condensation). Although exogenous Zn2+ has been shown to prevent apoptosis by inhibiting caspases (Perry et al., 1997), the effect of Zn2+ chelation on caspase activation has not yet been examined. Here we have shown that during TPEN-induced apoptosis a markedly elevated caspase-3 like activity could be detected as measured by DEVD-AMC cleavage. Although the mechanism of caspase activation in the course of TPEN-induced programmed cell death requires further elucidation, we hypothesize that caspases may represent primary targets of TPEN for the initiation of apoptosis. Recently, a new family of endogenous apoptosis inhibitors called IAP-s (inhibitors of apoptosis proteins) have been described (Rothe et al., 1995). Some members of this family including XIAP, cIAP-1 and c-IAP-2 can bind and selectively inhibit caspases (Deveraux et al., 1997; Roy et al., 1997). A common structural element of these caspase-inhibiting IAP family proteins is a RING zinc finger domain. Although this domain is not directly involved in binding to and inhibiting caspases (Takahashi et al., 1998), it may serve as a regulator of IAP protein function and thus chelation of Zn2+ in the zinc finger domain may inhibit the function of caspase-inhibiting IAP family members.
Effect of TPEN on the mode of peroxynitrite-induced cell death
Low doses (10–25 μM) of peroxynitrite induce DNA fragmentation in thymocytes, whereas higher doses (40–80 μM) inhibit oligonucleosomal DNA cleavage (Virág et al., 1998b). This latter effect is mediated by PARS activation (Virág et al., 1998b). In line with the PARS-inhibitory effect of TPEN, the chelator reversed the inhibition of DNA fragmentation observed at high doses of peroxynitrite. Theoretically, it is possible that a very rapid TPEN-induced caspase activation (see above) ‘switches on' DNA fragmentation before PARS activation occurs which would result in inhibition of DNA laddering. However, the time-course of PARS activation (10–20 min) and TPEN-induced caspase activation (starting at 2 h) and DNA fragmentation (starting at 4 h) argues against this scenario and favours the hypothesis that TPEN reverses the peroxynitrite-induced inhibition of DNA cleavage due to its function as a PARS inhibitor.
Conclusions and implications
Taken together, the conclusions of the present study are the following: (1) TPEN suppresses peroxynitrite-induced cell necrosis; (2) TPEN inhibits peroxynitrite-induced PARS activation and (3) TPEN also exerts PARS-independent cytoprotective effects. TPEN has previously been shown to protect spinal cord neurons from oxyradical-induced cytotoxicity (Michikawa et al., 1994) and prevent ATP depletion in hydrogen peroxide-treated Ac2F cells (Musicki & Behrman, 1993). Furthermore, treatment with TPEN proved beneficial in vivo in protecting from CCl4-induced liver injury (Moon et al., 1998), and from arrhythmia caused by myocardial ischaemia-reperfusion (Ferdinandy et al., 1998; Chevion, 1991). Regarding the cardioprotective effect of TPEN, it is noteworthy that in vivo peroxynitrite formation and PARS activation was proposed to mediate myocardial injury after ischaemia-reperfusion (Zingarelli et al., 1997a,1997b; 1998). Although the effect of TPEN on PARS activation in these experiments has not been investigated, it is conceivable that the protection against necrosis by TPEN in vivo is, at least in part, related to inhibition of PARS activation.
Acknowledgments
The authors wish to thank Mr Paul Hake for technical assistance, Dr Harry Ischiropoulos (Inst. Environmental Medicine, University of Pennsylvania, PA, U.S.A.) for generously donating authentic peroxynitrite, Dr Z.Q. Wang (Inst. Molecular Pathology, Vienna, Austria) for the PARS-deficient mice and Dr G.S. Scott for critically reading the manuscript. This work was supported by grants from the National Institutes of Health (R29GM54773 and R01HL59266) to C.S., L.V. was supported by a fellowship from the Ida and Zoltan Dobsa Foundation.
Abbreviations
- DiOC6(3)
3,3′dihexyloxacarbocyanine iodide
- HE
dyhydroethidium
- NAO
nonyl-acridine orange
- PARS
poly(ADP-ribose) synthase
- PI
propidium iodide
- PKC
protein kinase C
- SIN-1
3-morpholinosidnonimine
- TPEN
tetrakis-(2-pyridylmethyl)ethylenediamine
References
- BABA A., ETOH S., IWATA H. Inhibition of NMDA-induced protein kinase C translocation by a Zn2+ chelator: implication of intracellular Zn2+ Brain Res. 1991;557:103–108. doi: 10.1016/0006-8993(91)90121-b. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., CHEN J., ISCHIROPOULOS H., CROW J.P. Oxidative chemistry of peroxynitrite. Meth. Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., KOPPENOL W.H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- CHEVION M. Protection against free radical-induced and transition metal-mediated damage: the use of ‘pull' and ‘push' mechanisms. Free Radic. Res. Commun. 1991;12–13:691–696. doi: 10.3109/10715769109145848. [DOI] [PubMed] [Google Scholar]
- COCHRANE C. Mechanisms of oxidant injury of cells. Mol. Aspects Med. 1991;12:137–147. doi: 10.1016/0098-2997(91)90009-b. [DOI] [PubMed] [Google Scholar]
- DE MURCIA G., HULETSKY A., LAMARRE D., GAUDREAU A., POUYET J., DAUNE M., POIRIER G.G. Modulation of chromatin superstructure induced by poly(ADP-ribose)polimerase synthesis and degradation. J. Biol. Chem. 1986;261:7011–7017. [PubMed] [Google Scholar]
- DE MURCIA J.M., NIEDERGANG C., TRUCCO C., RICOUL M., DUTRILLAUX B., MARK M., OLIVER F.J., MASSON M., DIERICH A., LEMEUR M., WALTZINGER C., CHAMBON P., DE MURCIA G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVERAUX Q.L., TAKAHASHI R., SALVESEN G.S., REED J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- EASTMAN A. Assays of DNA fragmentation, endonucleases, and intracellular pH and Ca2+ associated with apoptosis. Methods Cell Biol. 1995;46:41–55. doi: 10.1016/s0091-679x(08)61923-8. [DOI] [PubMed] [Google Scholar]
- FERDINANDY P., APPELBAUM Y., CSONKA C., BLASIG I.E., TOSAKI A. Role of nitric oxide and TPEN, a potent metal chelator, in ischaemic and reperfused hearts. Clin. Exp. Pharmacol. Physiol. 1998;25:496–502. doi: 10.1111/j.1440-1681.1998.tb02242.x. [DOI] [PubMed] [Google Scholar]
- JIANG S., CHOW S.C., MCCABE M.J., JR, ORRENIUS S. Lack of Ca2+ involvement in thymocyte apoptosis induced by chelation of intracellular Zn2+ Lab. Invest. 1995;73:111–117. [PubMed] [Google Scholar]
- KLANN E., ROBERSON E.D., KNAPP L.T., SWEATT J.D. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J. Biol. Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- KONISHI H., TANAKA M., TAKEMURA Y., MATSUZAKI H., ONO Y., KIKKAWA U., NISHIZUKA Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KROEMER G., ZAMZAMI N., SUSIN S. A. Mitochondrial control of apoptosis. Immunol. Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- MAZEN A., MENISSIER-DE MURCIA J. , MOLINETE M., SIMONIN F., GRADWOHL G., POIRIER G., DE MURCIA G. Poly (ADP-ribose) polymerase: a novel finger protein. Nucleic Acids Res. 1989;17:4689–4698. doi: 10.1093/nar/17.12.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENISSIER-DE MURCIA J., MOLINETE M., GRADWOHL G., SIMONIN F., DE MURCIA G. Zinc-binding domain of poly(ADP-ribose)polymerase participates in the recognition of single strand breaks on DNA. J. Mol. Biol. 1989;210:229–233. doi: 10.1016/0022-2836(89)90302-1. [DOI] [PubMed] [Google Scholar]
- MCCABE M.J., JR, JIANG S.A., ORRENIUS S. Chelation of intracellular zinc triggers apoptosis in mature thymocytes. Lab. Invest. 1993;69:101–110. [PubMed] [Google Scholar]
- MICHIKAWA M., LIM K.T., MCLARNON J.G., KIM S.U. Oxygen radical-induced neurotoxicity in spinal cord neuron cultures. J. Neurosci. Res. 1994;37:62–70. doi: 10.1002/jnr.490370109. [DOI] [PubMed] [Google Scholar]
- MOON J.O., PARK S.K., NAGANO T. Hepatoprotective effect of Fe-TPEN on carbon tetrachloride induced liver injury in rats. Biol. Pharm. Bull. 1998;21:284–288. doi: 10.1248/bpb.21.284. [DOI] [PubMed] [Google Scholar]
- MUSICKI B., BEHRMAN H.R. Metal chelators reverse the action of hydrogen peroxide in rat luteal cells. Mol. Cell. Endocrinol. 1993;92:215–220. doi: 10.1016/0303-7207(93)90011-8. [DOI] [PubMed] [Google Scholar]
- PARAT M.O., RICHARD M.J., POLLET S., HADJUR C., FAVIER A., BEANI J.C. Zinc and DNA fragmentation in keratinocyte apoptosis: its inhibitory effect in UVB irradiated cells. J. Photochem. Photobiol. B. 1997;37:101–106. doi: 10.1016/s1011-1344(96)07334-4. [DOI] [PubMed] [Google Scholar]
- PERRY D.K. , SMYTH M.J., STENNICKE H.R., SALVESEN G.S., GURIEZ P., POIRIER G.G., HANNUN Y.A. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- ROTHE M., PAN M.G., HENZEL W.J., AYRES T.M., GOEDDEL D.V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- ROY N. , DEVERAUX Q.L., TAKAHASHI R., SALVESEN G.S., REED J.C. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 1997;16:6914–6925. doi: 10.1093/emboj/16.23.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SABRI A., BYRON K.L., SAMAREL A.M., BELL J., LUCCHESI P.A. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ. Res. 1998;82:1053–1062. doi: 10.1161/01.res.82.10.1053. [DOI] [PubMed] [Google Scholar]
- SZABÓ C. The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock. 1996;6:79–88. doi: 10.1097/00024382-199608000-00001. [DOI] [PubMed] [Google Scholar]
- SZABÓ C. Role of poly (ADP-ribose) synthetase in inflammation. Eur. J. Pharmacol. 1998;350:1–19. doi: 10.1016/s0014-2999(98)00249-0. [DOI] [PubMed] [Google Scholar]
- SZABÓ C., CUZZOCREA S., ZINGARELLI B., O'CONNOR M., SALZMAN L.A. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J. Clin. Invest. 1997a;100:723–735. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABÓ C., DAWSON V.L. Role of poly (ADP-ribose) synthetase activation in inflammation and reperfusion injury. Trends Pharmacol. Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- SZABÓ C., FERRER-SUETA G., ZINGARELLI B., SOUTHAN G.J., SALZMAN A.L., RADI R. Mercaptoethylguanidine and related guanidine nitric oxide synthase inhibitors react with peroxynitrite and protect against peroxynitrite-induced oxidative damage. J. Biol. Chem. 1997b;272:9030–9036. doi: 10.1074/jbc.272.14.9030. [DOI] [PubMed] [Google Scholar]
- SZABÓ C., LIM L.H., CUZZOCREA S., GETTING S.J., ZINGARELLI B., FLOWER R.J., SALZMAN A.L., PERRETTI M. Inhibition of poly (ADP-ribose) synthetase exerts anti-inflammatory effects and inhibits neutrophil recruitment. J. Exp. Med. 1997c;186:1041–1049. doi: 10.1084/jem.186.7.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABÓ C., SALZMAN A.L., ISCHIROPOULOS H. Endotoxin triggers the expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in the rat aorta in vivo. FEBS Letters. 1995;363:235–238. doi: 10.1016/0014-5793(95)00322-z. [DOI] [PubMed] [Google Scholar]
- SZABÓ C., VIRÁG L., CUZZOCREA S., SCOTT G.J., HAKE P., O'CONNOR M.P., ZINGARELLI B., SALZMAN A.L., KUN E. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly (ADP-ribose) synthetase. Proc. Natl. Acad. Sci. U.S.A. 1998;95:3867–3872. doi: 10.1073/pnas.95.7.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABÓ C., ZINGARELLI B., O'CONNOR M., SALZMAN A.L. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc. Natl. Acad. Sci. U.S.A. 1996a;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABÓ C., ZINGARELLI B., SALZMAN A.L. Role of poly-ADP ribosyltransferase activation in the vascular contractile and energetic failure elicited by exogenous and endogenous nitric oxide and peroxynitrite. Circ. Res. 1996b;78:1051–1063. doi: 10.1161/01.res.78.6.1051. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI R., DEVERAUX Q., TAMM I., WELSH K., ASSA-MUNT N., SALVESEN G.S., REED J.S. A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 1998;273:7787–7790. doi: 10.1074/jbc.273.14.7787. [DOI] [PubMed] [Google Scholar]
- TREVES S., TRENTINI P.L., ASCANELLI M., BUCCI G., DI VIRGILIO F. Apoptosis is dependent on intracellular zinc and independent of intracellular calcium in lymphocytes. Exp. Cell Res. 1994;211:339–343. doi: 10.1006/excr.1994.1096. [DOI] [PubMed] [Google Scholar]
- TRUCCO C., OLIVER F.J., DE MURCIA G., MENISSIER-DE MURCIA J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998;26:2644–2649. doi: 10.1093/nar/26.11.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANAGS D.M., PÖRN-ARES M., COPPOLA S., BURGESS D.H., ORENIUS S. Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J. Biol. Chem. 1997;271:31075–31085. doi: 10.1074/jbc.271.49.31075. [DOI] [PubMed] [Google Scholar]
- VERMES I., HAANEN C., STEFFENS-NAKKEN H., REUTELINGSPERGER C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- VIRÁG L., SALZMAN A.L., SZABÓ C. Poly (ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J. Immunol. 1998a;161:3753–3759. [PubMed] [Google Scholar]
- VIRÁG L., SCOTT G., CUZZOCREA S., MARMER D., SALZMAN A.L., SZABÓ C. Peroxynitrite-induced thymocyte apoptosis: the role of caspases and poly-(ADP-ribose) synthetase (PARS) activation. Immunology. 1998b;94:345–355. doi: 10.1046/j.1365-2567.1998.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHISLER R.L., GOYETTE M.A., GRANTS I.S., NEWHOUSE Y.G. Sublethal levels of oxidant stress stimulate multiple serine/threonine kinases and suppress protein phosphatases in Jurkat T cells. Arch. Biochem. Biophys. 1995;319:23–25. doi: 10.1006/abbi.1995.1263. [DOI] [PubMed] [Google Scholar]
- ZAMZAMI N., MARCHETTI P., CASTEDO M., ZANIN C., VAYSSIERE J.L., PETIT P.X., KROEMER G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZINGARELLI B., CUZZOCREA S., ZSENGELLÉR Z., SALZMAN A.L., SZABÓ C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc. Res. 1997a;36:205–215. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., DAY B.J., CRAPO J.D., SALZMAN A.L., SZABÓ C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br. J. Pharmacol. 1997b;120:259–267. doi: 10.1038/sj.bjp.0700872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZINGARELLI B., O'CONNOR M., WONG H., SALZMAN A.L., SZABÓ C. Peroxynitrite-mediated DNA strand breakage activates poly-ADP ribosyl synthetase and causes cellular energy depletion in macrophages stimulated with bacterial lipopolysaccharide. J. Immunol. 1996;156:350–358. [PubMed] [Google Scholar]
- ZINGARELLI B., SALZMAN A.L., SZABÓ C. Genetic disruption of poly (ADP ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia-reperfusion injury. Circ. Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]