

Abstract
The α1-adrenoceptor agonist methoxamine acted independently of receptor activation to reduce Ito and the sustained outward current in rat ventricular myocytes, and hKv 1.5 and Kv 4.2 cloned K+ channel currents. Two hundred μM methoxamine reduced Ito by 36% in the presence of 2 μM prazosin, and by 37 and 38% after preincubation of myocytes with either N-ethylmaleimide or phenoxybenzamine (n=6). The EC50 values at +60 mV for direct reduction of Ito, hKv 1.5, and Kv 4.2 by methoxamine were 239, 276, and 363 μM, respectively, with Hill coefficients of 0.87–1.5.
Methoxamine accelerated Ito and Kv 4.2 current inactivation in a concentration- and voltage-dependent manner. Apparent rate constants for methoxamine binding and unbinding gave Kd values in agreement with EC50 values measured from dose-response relations. The voltage-dependence of block supported charged methoxamine binding to a putative intracellular site that sensed ∼20% of the transmembrane electrical field.
In the presence of methoxamine, deactivating Kv 4.2 tail currents displayed a distinct rising phase, and were slowed relative to control, such that tail current crossover was observed. These observations support a dominant mechanism of open channel block, although closed channel block could not be ruled out.
Single-channel data from hKv 1.5 patches revealed increased closed times with blank sweeps and decreased burst duration in the presence of drug, and a reduction of mean channel open time from 1.8 ms in control to 0.4 ms in 500 μM methoxamine. For this channel, therefore, both open and closed channel block appeared to be important mechanisms for the action of methoxamine.
Keywords: Methoxamine, transient outward current, hKv 1.5, Kv 4.2, channel block
Introduction
Inhibition of repolarizing K+ currents in cardiac muscle in response to α-adrenoceptor stimulation by hormones, neurotransmitters, and drugs is one mechanism of positive inotropy in the heart, which becomes increasingly important pathophysiologically when α1-adrenoceptors are increased in proportion to other adrenoceptor subtypes, such as during heart failure (Hwang et al., 1996). Many previous studies of α1-adrenoceptor-mediated responses in cardiac tissues have used the α-adrenoceptor-specific agonist methoxamine (Fedida et al., 1990), since methoxamine lacks activity at β-adrenoceptors. Structurally, methoxamine is related to phenylephrine, but has greater specificity for the α1-adrenoceptor subtypes. Several studies, however, have demonstrated that methoxamine, especially at high concentrations, can produce pharmacological responses that differ from those observed in response to phenylephrine. In sinoatrial nodal cells, for example, methoxamine reduced the slow inward (Isi) and the hyperpolarization-activated (Ih) currents, whereas phenylephrine, in the presence of pindolol, increased both of these currents (Satoh & Hashimoto, 1988). The authors speculated that methoxamine may have had non-specific actions in addition to its activity at α1-adrenoceptors. Here we have investigated prazosin-insensitive actions of methoxamine on Ito in isolated rat ventricular cardiac myocytes, and in further experiments we have studied methoxamine block of current carried by two cloned potassium channels, hKv 1.5 and Kv 4.2. The results suggest that methoxamine can inhibit voltage-gated K+ currents independently of α1-adrenoceptor activation.
Methods
Myocyte isolation and cell culture
Hearts were rapidly removed from male rats weighing between 150 and 250 g previously anaesthetized with sodium pentobarbitone (80 mg kg−1, i.p.), and ventricular cells were isolated as previously described for rabbit myocytes (Braun et al., 1990). hKv 1.5 or Kv 4.2 were stably or transiently transfected into human embryonic kidney 293 (HEK) cells using the vector pCDNA3 (Invitrogen, San Diego, CA, U.S.A.). Transfected cells were plated on glass coverslips in 25 mm Petri dishes, and maintained in Modified Eagle's Medium (MEM) at 37°C in an air/5% CO2 incubator until use. Cells transiently expressing hKv 1.5 or Kv 4.2 were detected by co-transfection with pHook-1 (Invitrogen), as previously described (Zhang et al., 1997).
Electrophysiological solutions
For whole-cell recording from myocytes, the pipette filling solution contained (in mM): KCl, 20; K-aspartate, 120; EGTA, 1; MgCl2, 1; HEPES, 5; Na2ATP, 4; GTP, 0.1; pH 7.2 with KOH. For HEK cells, K-aspartate was substituted by KCl. The bath solution for both cell types contained (in mM): NaCl, 135; KCl, 5; sodium acetate, 2.8; MgCl1, 1; HEPES, 10; CaCl2, 1; pH 7.4 with NaOH. For cell-attached recordings, the pipette was filled with this bath solution and the membrane potential was zeroed with a solution that contained (in mM): KCl, 135; MgCl2, 1; HEPES, 10; CaCl2, 1; dextrose, 10; pH 7.4 with KOH. For myocyte experiments, bath solutions were continuously bubbled with 100% O2, and contained 5 μM propranolol to block β-adrenergic stimulation, and 300 μM CdCl2 to block calcium and calcium-activated currents. Methoxamine (Mox), phenylephrine (PE), phenoxybenzamine (PhBz), and N-ethylmaleimide (NEM) were prepared in water just prior to use, at appropriate stock concentrations. Prazosin (Pz) was dissolved in ethanol at 2 mM, and protected from light. The concentration of ethanol in the bath solution never exceeded 0.1%. When cells were exposed to methoxamine, effects on membrane currents usually reached a steady-state within 1–2 min so that drug exposure times rarely exceeded 5 min. All chemicals were from Sigma Chemical Co. (St. Louis, MO, U.S.A.).
Electrophysiological procedures and analysis
All experiments were carried out at 22–23°C. Whole-cell recordings were made using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, U.S.A.). Patch electrodes were pulled from thin-walled borosilicate glass (TW150, World Precision Instruments, FL, U.S.A.) and had resistances of 1.5–3.0 MΩ. Capacity compensation and 75–85% series resistance compensation were used in all measurements. In some experiments involving HEK cells, leak subtraction was applied to data. Data were filtered at 5–10 kHz before digitization and stored on a microcomputer for later analysis using pClamp6 software (Axon Instruments). The transient outward current (Ito) amplitude from rat ventricular myocytes was taken to represent the peak outward current measured during depolarizing voltage steps, as described previously (Apkon & Nerbonne, 1988). The dose-response curves (Figures 2B, 4D, and 5B) for current inhibition produced by methoxamine were computer-fitted to a Hill equation:
where f is the percentage current block (f=[1–Idrug/Icontrol]×100%) at drug concentration [D], max is the maximum fitted level of block, EC50 is the concentration producing half-maximal inhibition, and nH is the Hill coefficient.
Figure 2.
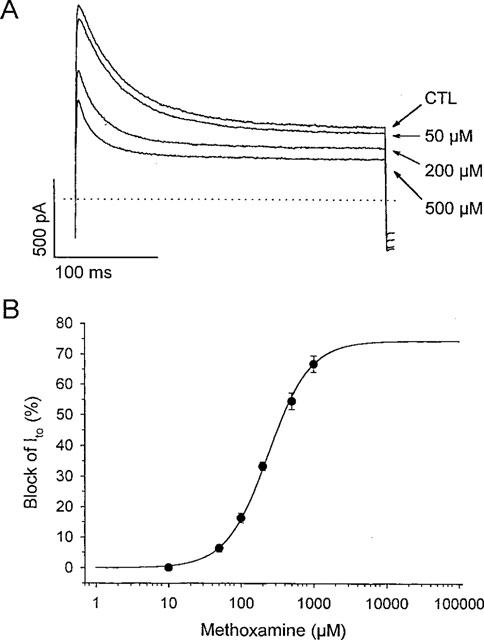
Concentration-dependence of non-specific action of methoxamine on Ito in rat ventricular myocytes. (A) currents recorded during steps to +60 mV from −80 mV in the presence of 2 μM prazosin (CTL), and in response to 50, 200, and 500 μM methoxamine in the presence of prazosin, as indicated. (B) dose-response curve for the non-specific action of methoxamine. Data are means±s.e.mean from 6–14 cells at each concentration. Solid line was fit to data using equation [1]. The EC50 value was 239 μM and the Hill coefficient, nH, was 1.5, with a maximum expected block of 75%.
Figure 4.
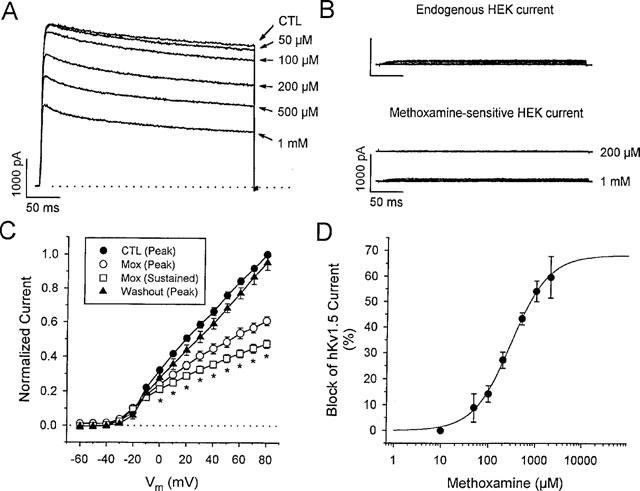
Reduction of hKv 1.5 current by methoxamine. (A) transfected HEK cell hKv 1.5 currents at +60 mV in control (CTL) and during exposure to a range of methoxamine concentrations as indicated. 2 μM prazosin was present in the bath at all times. The dotted line indicates zero current level. (B) endogenous HEK current in untransfected cells. The top panel shows typical endogenous currents in response to depolarizations over the range of −80 to +80 mV, from a holding potential of −80 mV. The lower panel illustrates the small methoxamine-sensitive endogenous currents obtained by subtracting currents in the presence of the indicated concentration of methoxamine from the corresponding currents in the absence of drug. The voltage-step protocol is the same as that described above. (C) normalized I-V relationships for peak control (n=5), peak (n=5) and sustained (n=5) hKv 1.5 currents in the presence of 500 μM methoxamine and peak current after washout (n=4), as indicated. Peak and sustained (at 300 ms) hKv 1.5 currents were individually normalized to peak and sustained current, respectively, measured at +80 mV under control conditions. *denotes statistically significant difference of both peak and sustained current amplitudes from control, P<0.05. (D) dose-response for the block of sustained hKv 1.5 current at +60 mV by methoxamine. The data are means±s.e.mean (n=5). The solid line represents a fit of the data to equation [1]. At +60 mV, the EC50 was 276 μM with nH=1.2, and a maximum expected block of 65.5%.
Figure 5.
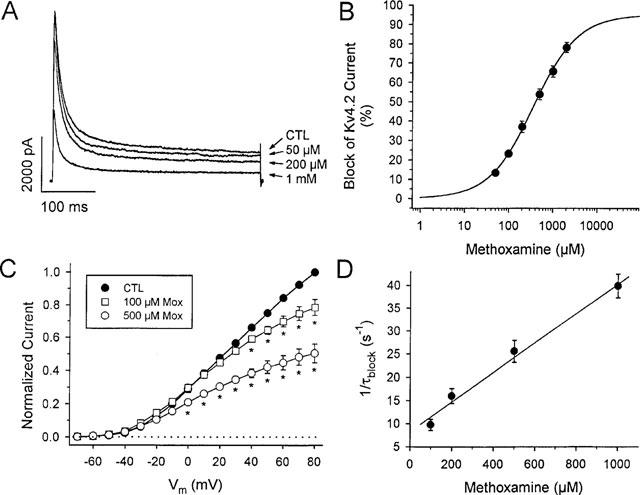
Reduction of Kv 4.2 current by methoxamine. (A) Kv 4.2 transfected HEK cell currents at +60 mV in control (CTL), and in the presence of 50, 200 μM, and 1 mM methoxamine, as indicated. (B) dose-response relation for Kv 4.2 current block by methoxamine. Current was measured at 300 ms and data points are means±s.e.mean (n=6 at each concentration). The solid line was fit to data using equation [1]. The EC50 was 363 μM with nH=0.87, and a maximum expected block of 95%. (C) normalized mean I-V relations for Kv 4.2 current in control (n=6), 100 μM (n=5), and 500 μM (n=6) methoxamine, as indicated. Currents were normalized to control currents at +80 mV, measured at 300 ms. *denotes statistically significant difference from control current amplitude, P<0.05. (D) 1/τblock values plotted vs methoxamine concentration. τblock values were extracted as described in the Methods from fits to the rapid phase of Kv 4.2 decay in the presence of methoxamine. From equation [3a] the best least-squares fit to these data, as indicated by the solid line in Figure 7D, gave an apparent k+1 of 3.2×104 M−1 s−1, and k−1=8.3 s−1. From equation [3b], the calculated Kd was 388 μM. Data are means±s.e.mean (n=5–9 at each concentration).
In rat ventricular myocytes, outward currents evoked upon depolarization contain at least two kinetic components, the first of which activates and inactivates rapidly (Ito), while the kinetics of the second are at least 10 fold slower, as previously described (Apkon & Nerbonne, 1991). Thus, peak outward current was taken to represent predominantly Ito, with only a negligible contribution from the second, slowly-activating sustained component of the outward current. Ito and the sustained current component also differ in their voltage-dependence of steady-state inactivation, and this can be used to separate them (Apkon & Nerbonne, 1991). After a 1 s conditioning step to −100 mV, outward currents evoked at a test potential of +60 mV exhibited both transient and sustained components (Figure 3A). After a conditioning step to −60 mV the sustained current was preferentially inactivated with only minimal effects on the peak outward current, whereas a prepulse to 0 mV inactivated both currents, and left only a small residual current representing leak and other non-inactivating currents. Ito was isolated by subtracting current traces recorded using a conditioning potential of 0 mV from those recorded using a conditioning potential of −60 mV. Single-exponential fits to this difference current, measured under control conditions, were taken to represent the time constant of inactivation (τinact) of Ito. For Kv 4.2, control current traces were better fit to a second order exponential function, and the fast inactivation time constant in control was denoted τinact. In the presence of drug, Ito and Kv 4.2 currents were fit to second-order exponentials, and the fast time constants of current decay in the presence of each concentration of drug were denoted τdecay, since they represented both inactivation and block of current by drug. We extracted time constants of methoxamine block (τblock) as an approximation of the drug-channel interaction kinetics as previously described (Slawsky & Castle, 1994; Snyders & Yeola, 1995), using:
and values of τinact measured as indicated above. In addition we calculated first order rate constants for block, using:
and
in which k+1 and k−1 are the apparent rate constants of binding and unbinding for the drug, respectively, and Kd is the equilibrium dissociation constant. For single channel analysis, blank sweeps were averaged and subtracted from all current traces to compensate for leak and capacity currents. Data were filtered at 1 kHz for presentation and analysis. Experimental values are given as means±s.e.mean. For the data in Figures 1E, 4C and 5C, repeated measures ANOVA was used to determine significance. In all other cases, paired or unpaired t-tests were used, as appropriate. A value of P<0.05 was considered statistically significant.
Figure 3.
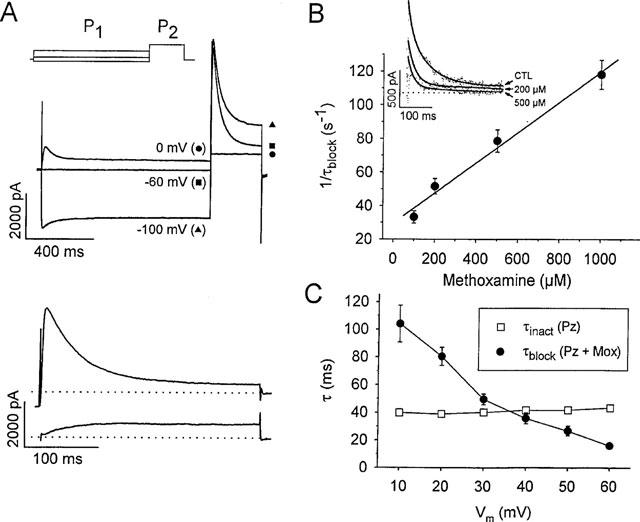
Concentration- and voltage-dependent kinetics of Ito block due to the non-specific action of methoxamine. (A) outward currents evoked from rat ventricular myocytes during a step to +60 mV (P2), preceded by a 1 s conditioning prepulse (P1) to −100 mV, −60 mV, or 0 mV, as indicated in the top panel. The lower panel shows difference currents isolating Ito (above) and the sustained outward current (below), derived as described in the Methods. Dotted lines indicate zero current level. (B) inset shows isolated Ito currents at +60 mV in control (CTL), and in the presence of 200 and 500 μM methoxamine, as indicated. Control currents were fit to a single exponential function, and currents in the presence of drug were fit to a double exponential (solid lines). The time constant for drug-channel interaction (τblock) was extracted as described in the Methods. In the main panel, 1/τblock values have been plotted vs methoxamine concentration. The solid lines fit to data using equation [3a] gave apparent association (k+1) and dissociation (k−1) rate constants of 8.9×104 M−1 s−1 and 32.9 s−1 respectively. The Kd value from equation [3b] was 307 μM. Data are means±s.e.mean from 5–10 cells at each concentration of methoxamine, and 2 μM prazosin was continuously present. (C) voltage-dependent acceleration of Ito inactivation by methoxamine in rat ventricular myocytes. Inactivation time constants were obtained from fits to the relaxation phase of Ito (see Methods) during 300 ms voltage steps from −80 mV. Control current inactivation was fit to a single exponential function, yielding τinact. In 2 μM prazosin and 200 μM methoxamine, the current decay was accelerated and τblock was extracted as described in the Methods. τblock values between +10 to +60 mV were accelerated, but τinact were not voltage dependent. Data are means±s.e.mean (n=5–10 at each voltage).
Figure 1.
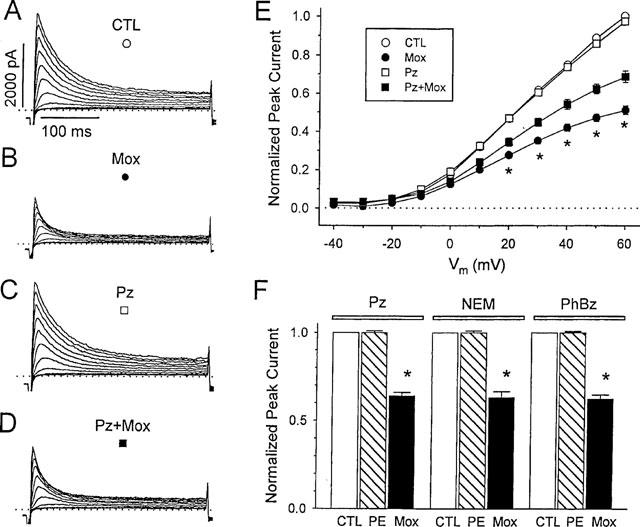
Methoxamine directly blocks outward currents in rat ventricular myocytes. A–D, currents from a representative myocyte during depolarizations from −80 mV to between −20 and +60 mV, in 10 mVsteps at a pulse frequency of 0.2 Hz. Dotted lines denote zero current level. Control (CTL, A) and with 200 μM methoxamine (Mox, B) after 5 min exposure. After 15 min of wash in control solution the cell was exposed to 2 μM prazosin (Pz, C) and then 200 μM methoxamine plus 2 μM prazosin (Pz+Mox, D). (E) mean peak Ito current-voltage relations in control and during exposure to prazosin or methoxamine, or both (n=6). For each cell, Ito was normalized to peak current in control solution measured at +60 mV. Symbols are as marked in panels A–D. * Indicates significant differences between current amplitudes in methoxamine alone compared with both methoxamine and prazosin, P<0.05. (F) block of signal transduction does not prevent the action of methoxamine. Effects of 100 μM phenylephrine (PE) and 200 μM methoxamine (Mox) on Ito amplitude in the presence of 2 μM prazosin (Pz), or after 30 min pretreatment with 50 μM N-ethylmaleimide (NEM) or 1 μM phenoxybenzamine (PhBz), as indicated. Currents were measured at +60 mV, and were normalized to Ito amplitude in the absence of α-agonist, but in the presence of Pz, NEM, or PhBz. Data are means±s.e.mean (n=6). * indicates a significant difference from control, P<0.05.
Results
Methoxamine can reduce outward currents independently of α1-adrenoceptor activation
In isolated rat ventricular myocytes, methoxamine reduced the amplitude of the outward currents, and this effect had both prazosin-sensitive and prazosin-insensitive components, as illustrated by the data in Figure 1ABCD. Addition of 200 μM methoxamine (B) reduced the amplitude of transient and sustained outward currents compared with control (A) across the entire potential range where currents could be measured. After wash, 2 μM prazosin was added to the bath solution (C), which has been shown to inhibit α-adrenergic responses in isolated cardiac myocytes (Fedida et al., 1991; Li et al., 1996). Even in the presence of prazosin, however, 200 μM methoxamine was still able to reduce current amplitude (D), although this effect was not as great as that observed with methoxamine alone. Specifically, at +60 mV, 200 μM methoxamine in the presence of prazosin was able to reduce peak outward current (Ito, see Methods) to 69.5±0.4% (n=6) of control. After washout, some cells were exposed for a second time to the same concentration of methoxamine and prazosin, which resulted in a reduction of Ito to 69.2±0.7% of control (n=5), which was not significantly different from that produced by the first exposure. Phenylephrine is approximately ten times more potent than methoxamine in stimulating α1-adrenoceptors (Yang & Endoh, 1994), and addition of 100 μM phenylephrine resulted in a significant decrease in Ito amplitude that was completely blocked by 2 μM prazosin (n=5).
Mean current-voltage relations for peak current reduction in response to methoxamine are shown in Figure 1E. Both the prazosin-sensitive and -insensitive actions of methoxamine affected current at all positive potentials. However, while the prazosin-sensitive action of methoxamine was relatively potential-independent, the prazosin-insensitive inhibition of Ito increased at the more positive potentials studied, greater than +20 mV. These data suggest that methoxamine reduced Ito in a manner that was both dependent upon, and independent of α-adrenoceptor activation. The increasing block of Ito at more positive potentials caused by methoxamine in the presence of prazosin was fitted with a Woodhull equation (Woodhull, 1973) which allowed calculation of the fraction of the electric field (δ) sensed by a singly charged drug molecule at its receptor site. From data in Figure 1E, a value of δ=0.18 was obtained.
Prazosin is a competitive antagonist of α-adrenoceptors. To explore the possibility that methoxamine was merely overcoming prazosin block, cells were exposed to the alkylating agents NEM or PhBz (Figure 1F). NEM, which alkylates sulphhydryl groups, has been shown to inhibit GTP-binding proteins, including G-proteins (Ueda et al., 1990). There is substantial molecular homogeneity between cardiac α1-adrenoceptors and the primary sequences of other members of the G-protein-binding superfamily (Harrison et al., 1991), and it appears that in mammalian cardiac cells, the effects of α1-adrenoceptor stimulation are primarily transduced via G-proteins (Im & Graham, 1990; Wu et al., 1992; Nakaoka et al., 1994), and it has been demonstrated that NEM can inhibit α1-adrenoceptor-mediated responses (Liebau et al., 1989). PhBz irreversibly alkylates α-adrenoceptors, with relative specificity for the α1 subtypes, and its use as an α-adrenoceptor antagonist has been well described (Minneman et al., 1994).
Preincubation of isolated cardiac myocytes for 30 min with either 50 μM NEM or 1 μM PhBz failed to prevent the reduction of peak outward current observed in response to methoxamine (Figure 1F). The mean reduction of Ito in response to 200 μM methoxamine in NEM- or PhBz-pretreated cells was 37.0±3.5% (n=6, mean±s.e.mean), and 37.8±2.4% (n=6) respectively, as shown by the solid bars in Figure 1F. These results are comparable with, and not significantly different from, the mean reduction observed in response to methoxamine in the presence of prazosin, which was 36.0±1.9% (n=6). In contrast to methoxamine, all three agents antagonized the action of 100 μM phenylephrine. These data also suggest that methoxamine resulted in an inhibition of Ito that was not mediated through α1-adrenergic receptor pathways.
Inhibition of Ito by methoxamine is concentration dependent
The non-specific action of methoxamine was concentration-dependent, as illustrated in Figure 2. Increasing concentrations of methoxamine, in the presence of 2 μM prazosin, decreased both the peak and steady-state levels of the outward currents recorded on depolarization (Figure 2A). As described previously for adult rat ventricular myocytes, depolarization-activated outward currents are composed of at least two kinetically distinct components: one that activates and inactivates rapidly (Ito), and one that activates and inactivates slowly (Apkon & Nerbonne, 1991). Since the more rapid of these components activates approximately 10 fold faster and inactivates approximately 30 fold faster than the slowly-activating component, the peak outward current predominantly reflects Ito, whereas the plateau current remaining after 100 ms should largely reflect the contribution of the second slower component. For purposes of analysis, therefore, we have considered only the effects of methoxamine on the peak transient current as a reflection of drug effects on Ito. In the dose-response relation in Figure 2B, data points correspond to percentage current block by methoxamine of the peak current in response to a depolarizing voltage step to +60 mV from a holding potential of −80 mV. The solid line was fitted to the data using equation [1]. The EC50 at +60 mV was 239 μM, and nH was 1.5. The threshold for non-specific inhibition of Ito by methoxamine was between 10 and 50 μM, and the peak amplitude of Ito was then decreased by methoxamine in a concentration-dependent manner. The concentration of methoxamine that resulted in a maximum reduction of Ito was difficult to determine, as concentrations greater than 1 mM were often toxic to the myocytes. Based on the results of the fit to the Hill equation, it is likely that a maximum reduction of Ito of about 75% could be achieved at concentrations of methoxamine greater than 2–5 mM.
Methoxamine accelerates Ito inactivation in a concentration and voltage-dependent manner
In addition to decreasing Ito amplitude, increasing concentrations of methoxamine also apparently accelerated the rate of Ito decay. However, methoxamine reduced not only Ito, but also the sustained outward current component as well. Therefore, in order to measure the rate of Ito decay alone, we separated these two current components using a voltage protocol based on the differences in voltage-dependence of steady-state inactivation of Ito and the sustained current component. Outward currents were measured at +60 mV after 1 s conditioning pulses to −100, −60, or 0 mV, as shown in Figure 3A. After a −100 mV prepulse, outward current comprised both transient and sustained components, but after a conditioning prepulse to −60 mV, the sustained component was largely inactivated with little effect on peak current. Both components were inactivated by a 0 mV prepulse. Ito and the sustained current component were isolated by subtraction as described in the Methods, and are illustrated in the lower panel of Figure 3A. The current traces in the inset to Figure 3B show Ito under control conditions and in the presence of 200 and 500 μM methoxamine, as indicated. Control currents were fitted with a single exponential function, whereas in the presence of drug, currents have been fitted to a double exponential decay (solid lines). There is a clear concentration-dependent acceleration of current decay in the presence of the drug. In controls, during 250 ms depolarizations, Ito inactivation at +60 mV had a time constant, τinact of 34.6±1.2 ms (n=9). In the presence of methoxamine and prazosin the rapid time constant of current decay, τdecay, accelerated with increasing concentrations of methoxamine. From these data we extracted the concentration-dependence of (1/τblock) as described in the Methods and shown in Figure 3B. 1/τblock increased linearly over a wide range of drug concentrations, and τblock was taken as an approximation of the time course of drug-channel interaction (Slawsky & Castle, 1994). From equation [3a] the best least-squares fit to 1/τblock, as indicated by the solid line in Figure 3B, resulted in an apparent association rate constant, k+1, of 8.9×104 M−1 s−1, and an apparent dissociation rate constant, k−1, of 32.9 s−1. Using equation [3b], the calculated Kd was 307 μM, which is in agreement with the EC50 value of 239 μM obtained from the dose-response curve (Figure 2B).
τblock also decreased in a voltage-dependent manner, while time constants obtained from monoexponential fits to control current inactivation (τinact) showed little voltage-dependence over the same potential range (Figure 3C, □). On the other hand, τdecay, measured in the presence of 200 μM methoxamine, decreased from a value of 28.8±1.9 ms at +10 mV to a value of 11.6±1.0 ms at +60 mV. From these measurements we extracted values for τblock (Figure 3C, •), which decreased from 103±13.9 ms at +10 mV to 15.9±1.9 ms at +60 mV. The values indicate that an increase in the electrical gradient accelerated block, which supports the idea that τblock reflects the time course of channel block by a positively charged form of methoxamine. In addition, the data support the observations described in Figure 1, where the degree of block increased at the more positive potentials studied.
Methoxamine inhibits hKv 1.5 and Kv 4.2 currents
Data presented in Figure 1 suggested that methoxamine reduced both peak (Ito) and sustained outward currents recorded from myocytes, and it was therefore of interest to examine individual cloned channel currents to find out which, if any, were preferentially blocked by the non-specific action of methoxamine. Both Kv 4.2 and hKv 1.5 are thought to be important components of outward current in cardiac tissue. Kv 4.2 and Kv 4.3 are thought to underlie Ito in rat and human ventricular myocytes (Barry et al., 1995; Dixon et al., 1996). hKv 1.5, cloned from human heart (Tamkun et al., 1991; Fedida et al., 1993) is thought to be a component of sustained current (Fedida et al., 1993; Wang et al., 1993), perhaps IKur in human myocytes (Feng et al., 1997). hKv 1.5 and Kv 4.2 were heterologously expressed in HEK cells, which lack α1-adrenoceptors (Theroux et al., 1996), and currents expressed in these cells failed to show any response to phenylephrine (n=6). Figure 4A illustrates current traces obtained from an HEK cell transfected with hKv 1.5, in response to depolarizing voltage steps to +60 mV, during exposure to a range of methoxamine concentrations. hKv 1.5 currents are relatively slowly inactivating and the effect of methoxamine was to reduce both peak and sustained hKv 1.5 current amplitude with less of an acceleration of the rate of current decay than seen for Ito in myocytes. Current traces obtained in the presence of methoxamine activated initially as in control (see Figure 4A), but reached a lower peak and subsequently decayed more quickly. Untransfected HEK cells are known to contain a small endogenous voltage-activated delayed rectifier current. The average magnitude of this endogenous current, measured in untransfected HEK cells at +60 mV and at 300 ms, was 274±32 pA (n=10), and current tracings from a typical cell, obtained in response to a series of depolarizations over the range of −80 to +80 mV, are shown in the top panel of Figure 4B. The lower panel of Figure 4B illustrates typical drug-sensitive endogenous currents, in the presence of 200 μM and 1 mM methoxamine. This endogenous current was insensitive to concentrations of methoxamine <200 μM (n=6), and partial inhibition was noted at concentrations >500 μM. This suggests that the reduction of sustained currents observed in hKv 1.5-transfected cells with <500 μM methoxamine must represent solely block of hKv 1.5 current. Even with 1 mM methoxamine, the average magnitude of the methoxamine-sensitive current, measured at +80 mV, was 89 pA. Nonetheless, to minimize the contributions of this endogenous current, our analyses of expressed hKv 1.5 cloned channel currents included only those cells that contained relatively large currents, such that the methoxamine-sensitive endogenous current would be expected to contribute less than 3% of the total measured methoxamine-sensitive hKv 1.5 current. Mean peak (○) and sustained (□) current-voltage relations for cells exposed to 500 μM methoxamine are shown in Figure 4C, and illustrate the large effects of methoxamine on peak hKv 1.5 current, as well as reversibility of the drug action on washout (▴). Block of hKv 1.5 current by methoxamine was voltage- (Figure 4C) and concentration-dependent (Figure 4D). Fit to current-voltage data positive to +20 mV gave a value for δ=0.18, similar to that determined for Ito with methoxamine and prazosin (see text to Figure 1). Data points in Figure 4D were well fit by equation [1], which yielded an EC50 value, at +60 mV, of 276 μM, and nH=1.2, with a maximum level of block of 65.5% predicted from the fit.
The response of Kv 4.2 channel currents to methoxamine was similar to that observed for Ito. Threshold drug actions were observed at 50 μM and consisted predominantly of an acceleration of the rate of current decay. Higher concentrations of methoxamine not only accelerated the rate of Kv 4.2 current decay, but also reduced peak current, with little effect of methoxamine on the initial activation of the currents relative to control (Figure 5A). As for hKv 1.5 data, at drug concentrations greater than 500 μM, the methoxamine-sensitive endogenous current was small, and would be expected to compose less than 5% of the total methoxamine-sensitive current in all Kv 4.2-transfected cells included in analyses. The EC50 for steady-state Kv 4.2 current reduction was 363 μM, with nH=0.87, with a maximum expected block of 95% predicted from the fit of the data in Figure 5B. I-V relationships for cells in control solution (Figure 5C, •, n=10) and in the presence of 100 μM (□, n=6) and 500 μM (○, n=7) methoxamine again illustrate a voltage-dependence of block, and a fit of the current-voltage data positive to +20 mV in the presence of 500 μM methoxamine yielded a value of δ=0.21. For Kv 4.2 the control current inactivation was bi-exponential with fast time constants that were relatively voltage-independent in control (data not shown). The time constant for drug-channel interaction (Figure 5D; 1/τblock, •) was again concentration-dependent as for Ito. From equation [3a] the best least-squares fit to these data, as indicated by the solid line in Figure 5D, gave an apparent k+1 of 3.2×104 M−1s−1, and k−1=8.3 s−1. From equation [3b], the calculated Kd was 388 μM, which is in close agreement with the EC50 value of 363 μM obtained from the dose-response curve (Figure 5B).
Methoxamine modifies Kv 4.2 tail currents
Under control conditions, at −40 mV, Kv 4.2 current deactivated with a time constant (τdeactivation) of 24.4±1.6 ms (n=6), which represents the closing of channels from the open state. In the presence of methoxamine, the peak tail current amplitude was reduced relative to control, but also displayed an initial rising phase that was not observed in the absence of drug (n=8). This rising phase was more pronounced with higher doses of methoxamine, and represents unblock of open channels previously inhibited by methoxamine. Figure 6A shows representative tail currents obtained at −40 mV, after 100 ms depolarizations in control conditions, and in the presence of 100, 500 μM or 1 mM methoxamine. Not only was the peak more reduced and the rising phase more pronounced at higher methoxamine concentrations, but subsequent decay of the tail currents was significantly slowed in the presence of methoxamine, and this effect was dose-dependent (Figure 6B). This had the result that tail currents recorded at the higher concentrations of methoxamine crossed over the control tail current (Figure 6A, inset). These data indicate that upon repolarization, methoxamine dissociated from blocked channels, allowing these channels to become conducting and resulting in the rising phase of tail current. Subsequently, a dynamic equilibrium was reached, wherein a fraction of these open, unblocked channels became blocked again, delaying the irreversible closing of these channels and resulting in the slowed rate of tail current decay relative to control.
Figure 6.
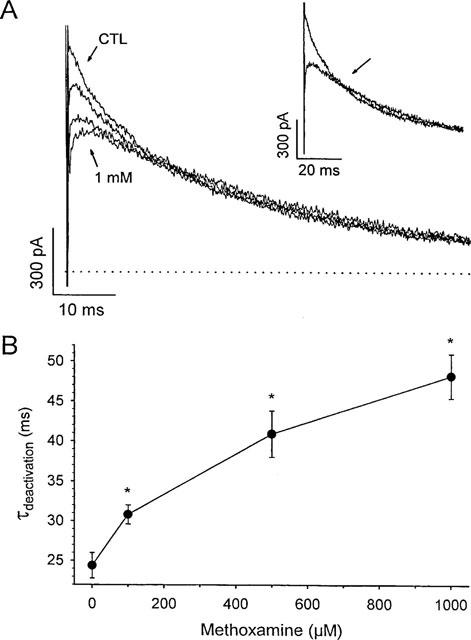
Methoxamine modifies Kv 4.2 deactivating tail currents. (A) typical deactivating tail currents in control (CTL), 100 μM, 500 μM, and 1 mM methoxamine. Peak tail current amplitudes decreased with increasing dose. The cell was held at −80 mV, and tails were recorded at −40 mV after a 100 ms depolarization to +50 mV. Dotted line indicates zero current level. Inset panel shows crossover of tail in control and 1 mM methoxamine and crossover point is arrowed. (B) slowing of rate of decay of tail currents with increasing doses of methoxamine. Tail currents in control and in the presence of drug were obtained as in (A) and were computer-fitted to a single exponential function to obtain the deactivation time constant, τdeactivation, at each concentration of drug. Data are means±s.e.mean (n=6). *denotes significant difference from control (i.e. 0 μM methoxamine), P<0.05.
Methoxamine block of single Kv channels
For single-channel recording, HEK cells were depolarized in high K+ solution containing 2 μM prazosin. Pipettes contained 5 mM K+ solution and patches containing one or a few hKv 1.5 channels were held at a transmembrane potential of −80 mV and depolarized to +60 mV, at 0.5 Hz. Representative control sweeps are presented in Figure 7A, and tracings recorded with 500 μM methoxamine in the bath solution are illustrated in Figure 7B. Under control conditions, hKv 1.5 channels opened in multiple bursts that were characterized by frequent brief closings. These kinetics for hKv 1.5 have been reported previously from our laboratory (Chen & Fedida, 1998) and are quite different from the maintained openings with few closings that characterize the endogenous current in HEK cells (data not shown). In the presence of methoxamine, burst duration was greatly reduced, rapid closings within bursts became even more frequent, and the channel rarely opened to its full amplitude. The mean single channel current in control solution, as determined by a Gaussian fit to an all-points amplitude histogram (data not shown) was 1.1±0.01 pA, which corresponds to a single channel conductance of 9 pS, consistent with that reported previously for hKv 1.5 (Grissmer et al., 1994). In the presence of methoxamine, however, the mean single channel current amplitude was significantly reduced to 0.3±0.01 pA. These two actions of 500 μM methoxamine had the effect of reducing the ensemble-average current by 73% (Figure 7D) relative to control (Figure 7C). This may be compared with the 43% reduction of hKv 1.5 current observed in the whole cell configuration (Figure 4C). In addition, methoxamine significantly decreased the mean channel open time of hKv 1.5. Fits to open channel dwell time distributions for a cell in control solution (Figure 7E) and following addition of methoxamine (Figure 7F) yielded time constants (τopen), corresponding to mean channel open times, of 1.8±0.01 ms and 0.4±0.06 ms, respectively. Similar results were seen in four other cells.
Figure 7.
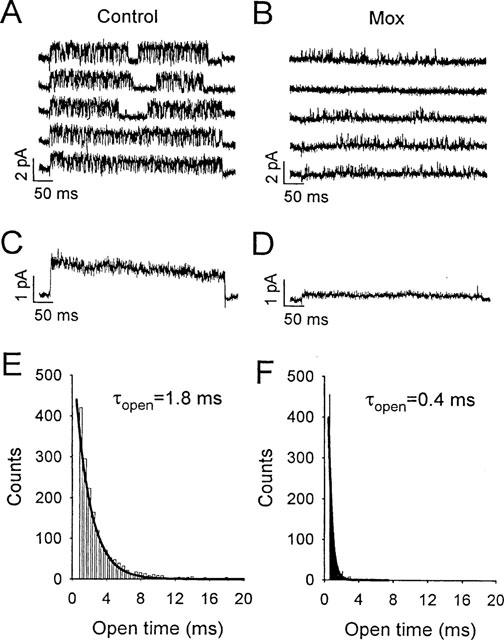
Effects of methoxamine on hKv 1.5 single channels in cell-attached patches with 140 mM K+ in the bath (see Methods). (A and B) cells were held at −80 mV and single-channel currents obtained at +60 mV, in control and in 500 μM methoxamine. Each sweep was recorded during 500 ms depolarizations. (C and D) ensemble averages (n=20) in control and in 500 μM methoxamine. (E and F) open-channel dwell time distributions in control and 500 μM methoxamine. Bin widths were 0.5 ms in E and 0.1 ms in F. Solid lines are single-exponential fits to data, with τopen=1.8±0.01 ms in control, and 0.4±0.06 ms in methoxamine.
Discussion
Methoxamine has both α1-adrenoceptor-dependent and α1-adrenoceptor-independent actions
Methoxamine and phenylephrine have both been widely used in studies of α-adrenergic effects on cardiac ionic currents. In our studies, both phenylephrine and methoxamine reduced peak Ito amplitude in rat ventricular myocytes, but the response to phenylephrine was prevented by prazosin, whereas prazosin could reduce, but not abolish, the response to methoxamine (Figure 1). Similarly, preincubation of ventricular cells with prazosin, NEM, or PhBz prevented the action of phenylephrine, but failed to prevent the reduction in Ito observed during exposure to 200 μM methoxamine (see Figure 1F). This suggests that although prazosin, NEM, and PhBz act to disrupt different stages of the α-adrenergic signal transduction pathway, the non-specific action of methoxamine was not dependent on receptor activation, or on signal transduction by heterotrimeric G-proteins.
Methoxamine reduced both peak (Ito) and sustained outward currents in rat ventricular myocytes, independently of α1-adrenoceptor stimulation, so we examined the action of methoxamine on two classes of Kv channels. These were Kv 4.2, a rapidly inactivating channel thought to underlie Ito (Barry et al., 1995; Dixon et al., 1996) and hKv 1.5, a slowly-inactivating delayed rectifier-type of channel thought to contribute to sustained outward current (Van Wagoner et al., 1996; Feng et al., 1997). Our data demonstrated that methoxamine could reduce the whole-cell currents recorded from hKv 1.5 and Kv 4.2 in HEK cells (Figures 4 and 5) which express no detectable endogenous α1-adrenoceptors (Theroux et al., 1996).
Concentration- and voltage-dependence of receptor-independent action of methoxamine
In the presence of prazosin, methoxamine decreased the amplitudes of Ito, hKv 1.5, and Kv 4.2 in a concentration- and voltage-dependent manner. The EC50s for observed fractional block were similar for all three currents, and Hill coefficients were also similar, suggesting a single binding site model in each case. In myocytes, methoxamine characteristically increased the rate of decay of Ito (Figure 3). This was similar to data obtained for Kv 4.2 (Figure 5), but appeared to be much less important an effect for hKv 1.5 (Figure 4). We extracted a methoxamine-dependent time constant for block, τblock (Figures 3 and 5), which decreased with increasing drug concentrations. The calculated Kd values, at +60 mV, were in good agreement with EC50 values obtained from dose-response relations (Figures 2B and 5B).
The observed voltage-dependence of Ito (Figure 1E), hKv 1.5 (Figure 4C) and Kv 4.2 (Figure 5C) inhibition, increasing with potential, is expected if the positively charged form of methoxamine (at pH 7.4, with pKa=9.2) blocks the channel and therefore responds to changes in membrane potential. Uncharged methoxamine could cross the plasma membrane and become protonated and charged within the cell, and then reach a putative intracellular binding site. This is supported by single channel data in Figure 7 where methoxamine was added to the bath solution and the external face of the membrane at the recording site was shielded by the pipette. The fraction of the membrane electrical field (δ) sensed by the drug at its receptor site was determined to be 0.18 for Ito and for hKv 1.5, and 0.21 for Kv 4.2. These values are similar to those for quinidine (Fedida, 1997) and quaternary ammonium compounds (Choi et al., 1993), and suggests that positively charged agents may bind to a common intracellularly-accessible site (Snyders & Yeola, 1995).
Non-specific action of methoxamine is consistent with both open- and closed-channel block
Methoxamine accelerated the rate of decay of Ito and Kv 4.2 without markedly affecting the initial time course of activation (Figures 2A and 5A), which is generally suggestive of a mechanism of open-channel block (Slawsky & Castle, 1994). In the presence of higher concentrations of methoxamine, the reduction in peak Ito, hKv 1.5 and Kv 4.2 currents can be explained by rapid drug-channel interaction according to the first-order rate constant given by k+1[D]. In the presence of methoxamine, deactivating Kv 4.2 tail currents showed a distinct rising phase that was not observed under control conditions, and decay of the current tails was significantly slowed in the presence of drug (Figure 6). These observations are consistent with a simple open channel block scheme for methoxamine and Kv 4.2:
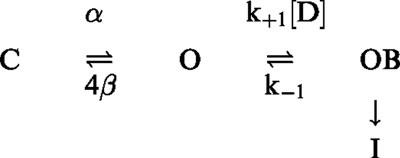 |
In this model, α and 4β refer to standard opening and closing rate constants, respectively, between the last closed (C) and open (O) states in a Hodgkin-Huxley four-subunit model, (I) is the inactivated state, and k+1 and k−1 are the apparent binding and unbinding rates for methoxamine and Kv 4.2, which were calculated to be 3.2×104 M−1 s−1 and 8.3 s−1, respectively (Figure 5D). Since the unbinding rate is relatively fast, upon repolarization tail currents in the presence of drug should exhibit a rising phase indicative of the OB→O transition. Subsequently, these unblocked open channels can reblock, which slows tail current decay according to the relative values of 4β and k+1[D]. If drug binding is sufficiently rapid relative to channel closing, a ‘crossover' of tail currents can be observed, as is the case for quinidine (Snyders et al., 1992; Fedida, 1997), and was demonstrated by data in Figure 6.
In addition to accelerating the rate of decay of whole cell current, methoxamine decreased the mean hKv 1.5 single channel open time, consistent with rapid association and dissociation of the drug and the channel, and supportive of open-channel block. However, mean single channel current was also reduced, which may reflect brief openings not visualized at our bandwidth (1 kHz), or alternatively, alteration of the channel conductance by methoxamine. The number of blank sweeps also increased during methoxamine, and this increase in channel closed times strongly suggests an additional interaction with resting or closed states of hKv 1.5.
Implications for previous studies
Methoxamine has been a widely-used agonist to study the role of α1-adrenergic modulation of cardiac function, and the concentrations of drug used in these studies vary widely over the range of 10−6–10−2 M. In rat ventricular myocytes a 25–50% reduction in the magnitude of the outward K+ currents was observed in response to 20 μM methoxamine (Apkon & Nerbonne, 1988). At this concentration of methoxamine, the non-specific component of Ito inhibition is negligible (Figure 2B) compared to the α1-adrenoceptor-mediated response. Many studies, however, have used higher concentrations (e.g. >100 μM methoxamine), at which there is likely to be a significant non-specific component to the observed action that cannot be overlooked. In the absence of prazosin, 100 μM methoxamine resulted in ∼35% reduction of Ito in rat ventricular cells (Fedida & Bouchard, 1992), at which concentration our results indicate that Ito would be reduced by ∼15% due to a non-specific action alone. Yang et al. (1996) routinely used 100 μM methoxamine to examine positive inotropy in isolated rabbit myocardium, and methoxamine concentrations up to 1 mM have been used to demonstrate modulation of Ito in hyperthyroid rabbit myocytes (Shimoni & Banno, 1993). Interestingly, in rabbit atrium, 200 μM methoxamine has also been shown to decrease the Ito mean single channel open time (Braun et al., 1990). This result is similar to our α-adrenoceptor-independent action on single hKv 1.5 channels, although we noted additional closed channel effects (Figure 7). All these results suggest that great caution must be exercised in the use of methoxamine as an α-agonist in systems which possess significant numbers of voltage-gated K+ channels or where ion channel modulation itself is being directly studied.
Interestingly, other compounds that interact with adrenoceptors have also been shown to have channel-blocking properties. The α-antagonist phentolamine can block ATP-sensitive K+ (KATP) channels in rabbit ventricle (Wilde et al., 1994) and pancreatic β-cells (Plant & Henquin, 1990), as can the specific α2-antagonist yohimbine. Dicentrene, an alkaloid α-antagonist, inhibits Ito, IK, and the sodium current (INa) in guinea-pig ventricular cells independently of adrenoceptor binding (Su et al., 1994). To our knowledge, however, this is the first report of an α-adrenoceptor agonist with K+ channel-blocking properties.
Acknowledgments
Supported by grants from the Heart and Stroke Foundation of Ontario and the Medical Research Council of Canada to David Fedida. The authors would like to thank Shunping Lin for valuable assistance in data collection.
Abbreviations
- CTL
control
- δ
fraction of transmembrane electric field sensed by a singly-charged drug molecule at its receptor site
- Mox
methoxamine
- NEM
N-ethylmaleimide
- PE
phenylephrine
- PhBz
phenoxybenzamine
- Pz
prazosin
- τblock
time constant of block of current by drug
- τdeactivation
time constant of deactivation of tail currents
- τdecay
time constant of current decay in the presence of drug
- τinact
time constant of control current inactivation
References
- APKON M., NERBONNE J.M. α1-adrenergic agonists selectively suppress voltage-dependent K+ currents in rat ventricular myocytes. Proc. Natl. Acad. Sci. U.S.A. 1988;85:8756–8760. doi: 10.1073/pnas.85.22.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APKON M., NERBONNE J.M. Characterization of two distinct depolarization-activated K+ currents in isolated adult rat ventricular myocytes. J. Gen. Physiol. 1991;97:973–1011. doi: 10.1085/jgp.97.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRY D.M., TRIMMER J.S., MERLIE J.P., NERBONNE J.M. Differential expression of voltage-gated K+ channel subunits in adult rat heart: Relation to functional K+ channels. Circ. Res. 1995;77:361–369. doi: 10.1161/01.res.77.2.361. [DOI] [PubMed] [Google Scholar]
- BRAUN A.P., FEDIDA D., CLARK R.B., GILES W.R. Intracellular mechanisms for α1-adrenergic regulation of the transient outward current in rabbit atrial myocytes. J. Physiol. (Lond.) 1990;431:689–712. doi: 10.1113/jphysiol.1990.sp018355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN F.S.P., FEDIDA D. On the mechanism by which 4-aminopyridine occludes quinidine block of the cardiac K+ channel, hKv 1.5. J. Gen. Physiol. 1998;111:539–554. doi: 10.1085/jgp.111.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI K.L., MOSSMAN C., AUBÉ J., YELLEN G. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- DIXON J.E., SHI W.M., WANG H.S., MCDONALD C., YU H., WYMORE R.S., COHEN I.S., MCKINNON D. Role of the Kv 4.3 K+ channel in ventricular muscle-A molecular correlate for the transient outward current. Circ. Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- FEDIDA D., SHIMONI Y., GILES W.R. α-Adrenergic modulation of the transient outward current in rabbit atrial myocytes. J. Physiol. (Lond.) 1990;423:257–277. doi: 10.1113/jphysiol.1990.sp018021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEDIDA D., BRAUN A.P., GILES W.R. α1-Adrenoceptors reduce background K+ current in rabbit ventricular myocytes. J. Physiol. (Lond.) 1991;441:673–684. doi: 10.1113/jphysiol.1991.sp018772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEDIDA D., WIBLE B., WANG Z., FERMINI B., FAUST F., NATTEL S., BROWN A.M. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ. Res. 1993;73:210–216. doi: 10.1161/01.res.73.1.210. [DOI] [PubMed] [Google Scholar]
- FEDIDA D. Gating charge and ionic currents associated with quinidine block of human Kv 1.5 delayed rectifier K+ channels. J. Physiol. (Lond.) 1997;499:661–675. doi: 10.1113/jphysiol.1997.sp021959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEDIDA D., BOUCHARD R.A. Mechanisms for the positive inotropic effect of α1-adrenoceptor stimulation in rat cardiac myocytes. Circ. Res. 1992;71:673–688. doi: 10.1161/01.res.71.3.673. [DOI] [PubMed] [Google Scholar]
- FENG J.L., WIBLE B., LI G.R., WANG Z.G., NATTEL S. Antisense oligodeoxynucleotides directed against Kv 1.5 mRNA specifically inhibit ultrarapid delayed rectifier K+ current in cultured adult human atrial myocytes. Circ. Res. 1997;80:572–579. doi: 10.1161/01.res.80.4.572. [DOI] [PubMed] [Google Scholar]
- GRISSMER S., NGUYEN A.N., AIYAR J., HANSON D.C., MATHER R.J., GUTMAN G.A., KARMILOWICZ M.J., AUPERIN D.D., CHANDY K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv 1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- HARRISON J.K., PEARSON W.R., LYNCH K.R. Molecular characterization of α1- and α2-adrenoceptors. TIPS. 1991;12:62–67. doi: 10.1016/0165-6147(91)90499-i. [DOI] [PubMed] [Google Scholar]
- HWANG K.C., GRAY C.D., SWEET W.E., MORRAVEC C.S., IM M.-J. α1-adrenergic receptor coupling with Gh in the failing human heart. Circulation. 1996;94:718–726. doi: 10.1161/01.cir.94.4.718. [DOI] [PubMed] [Google Scholar]
- IM M.-J., GRAHAM R.M. A novel guanine nucleotide-binding protein coupled to the α1-adrenergic receptor. I. Identification by photolabeling of membrane and ternary complex preparations. J. Biol. Chem. 1990;265:18944–18951. [PubMed] [Google Scholar]
- LI G.R., FENG J.L., WANG Z.G., FERMINI B., NATTEL S. Adrenergic modulation of ultrarapid delayed rectifier K+ current in human atrial myocytes. Circ. Res. 1996;78:903–915. doi: 10.1161/01.res.78.5.903. [DOI] [PubMed] [Google Scholar]
- LIEBAU S., HOHLFELD J., FORSTERMANN U. The inhibition of α1-adrenoceptor-mediated contraction of rabbit pulmonary artery by Ca2+-withdrawal, pertussis toxin, and N-ethylmaleimide. Naun. Schmied. Arch. Pharmacol. 1989;339:496–502. doi: 10.1007/BF00167251. [DOI] [PubMed] [Google Scholar]
- MINNEMAN K.P., THEROUX T.L., HOLLINGER S., HAN C., ESBENSHADE T.A. Selectivity of agonists for cloned α1-adrenergic receptor subtypes. Mol. Pharmacol. 1994;46:929–936. [PubMed] [Google Scholar]
- NAKAOKA H., PEREZ D.M., BAEK K.J., DAS T., HUSAIN A., MISONO K., IM M.-J., GRAHAM R.M. Gh: a GTP-binding protein with transglutaminase activity and receptor signalling function. Science. 1994;264:1593–1595. doi: 10.1126/science.7911253. [DOI] [PubMed] [Google Scholar]
- PLANT T.D., HENQUIN J.C. Phentolamine and yohimbine inhibit ATP-sensitive K+ channels in mouse pancreatic β-cells. Br. J. Pharm. 1990;101:115–120. doi: 10.1111/j.1476-5381.1990.tb12099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATOH H., HASHIMOTO K. Effect of α1-adrenoceptor stimulation with methoxamine and phenylephrine on spontaneously beating rabbit sino-atrial node cells. Naun. Schmied. Arch. Pharmacol. 1988;337:415–422. doi: 10.1007/BF00169533. [DOI] [PubMed] [Google Scholar]
- SHIMONI Y., BANNO H. α-Adrenergic modulation of transient outward current in hyperthyroid rabbit myocytes. Am. J. Physiol. Heart Circ. Physiol. 1993;264:H74–H77. doi: 10.1152/ajpheart.1993.264.1.H74. [DOI] [PubMed] [Google Scholar]
- SLAWSKY M.T., CASTLE N.A. K+ channel blocking actions of flecainide compared with those of propafenone and quinidine in adult rat ventricular myocytes. J. Pharmacol. Exp. Ther. 1994;269:66–74. [PubMed] [Google Scholar]
- SNYDERS D.J., KNOTH K.M., ROBERDS S.L., TAMKUN M.M. Time-, voltage-, and state-dependent block by quinidine of a cloned human cardiac potassium channel. Mol. Pharmacol. 1992;41:322–330. [PubMed] [Google Scholar]
- SNYDERS D.J., YEOLA S.W. Determinants of antiarrhythmic drug action – Electrostatic and hydrophobic components of block of the human cardiac hKv 1.5 channel. Circ. Res. 1995;77:575–583. doi: 10.1161/01.res.77.3.575. [DOI] [PubMed] [Google Scholar]
- SU M.J., NIEH Y.C., HUANG H.W., CHEN C.C. Dicentrine, an α-adrenoceptor antagonist with sodium and potassium channel blocking activities. Naunyn-Schmiedebergs Arch. Pharm. 1994;349:42–49. doi: 10.1007/BF00178204. [DOI] [PubMed] [Google Scholar]
- TAMKUN M.M., KNOTH K.M., WALBRIDGE J.A., KROEMER H., RODEN D.M., GLOVER D.H. Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB J. 1991;5:331–337. doi: 10.1096/fasebj.5.3.2001794. [DOI] [PubMed] [Google Scholar]
- THEROUX T.L., ESBENSHADE T.A., PEAVY R.D., MINNEMAN K.P. Coupling efficiencies of human α1-adrenergic receptor subtypes: Titration of receptor density and responsiveness with inducible and repressible expression vectors. Mol. Pharmacol. 1996;50:1376–1387. [PubMed] [Google Scholar]
- UEDA H., MISAWA H., KATADA T., UI M., TAKAGI H., SATOH M. Functional reconstitution of purified Gi and Go with μ-opioid receptors in guinea-pig striatal membranes pretreated with micromolar concentrations of N-ethylmaleimide. J. Neurochem. 1990;54:841–848. doi: 10.1111/j.1471-4159.1990.tb02328.x. [DOI] [PubMed] [Google Scholar]
- VAN WAGONER D.R., KIRIAN M., LAMORGESE M. Phenylephrine suppresses outward K+ currents in rat atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 1996;271:H937–H946. doi: 10.1152/ajpheart.1996.271.3.H937. [DOI] [PubMed] [Google Scholar]
- WANG Z., FERMINI B., NATTEL S. Sustained depolarization-induced outward current in human atrial myocytes: Evidence for a novel delayed rectifer K+ current similar to Kv 1.5 cloned channel currents. Circ. Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- WILDE A.A., VELDKAMP M.W., VAN GINNEKEN A.C., OPTHOF T. Phentolamine blocks ATP sensitive potassium channels in cardiac ventricular cells. Cardiovasc. Res. 1994;28:847–850. doi: 10.1093/cvr/28.6.847. [DOI] [PubMed] [Google Scholar]
- WOODHULL A.M. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU D., KATZ A., LEE C.-H., SIMON M.I. Activation of phospholipase C by α1-adrenergic receptors is mediated by the α subunits of Gq family. J. Biol. Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- YANG H., ENDOH M. Dissociation of the positive inotropic effect of methoxamine from the hydrolysis of phosphoinositide in rabbit ventricular myocardium: a comparison with the effects of phenylephrine and the subtype of the α1 adrenoceptor involved. J. Pharm. Exp. Ther. 1994;269:732–742. [PubMed] [Google Scholar]
- YANG H.T., NOROTA I., ZHU Y., ENDOH M. Methoxamine-induced inhibition of the positive inotropic effect of endothelin via α1-adrenoceptors in the rabbit heart. Eur. J. Pharmacol. 1996;296:47–54. doi: 10.1016/0014-2999(95)00672-9. [DOI] [PubMed] [Google Scholar]
- ZHANG X., ANDERSON J.W., FEDIDA D. Characterization of nifedipine block of the human heart delayed rectifier, hKv 1.5. J. Pharmacol. Exp. Ther. 1997;281:1247–1256. [PubMed] [Google Scholar]