

Short abstract
Computational methods have been used to characterize the genomic sequences adjacent to L1 retrotransposons, shedding light on the mechanisms of integration and the frequency of L1-mediated transduction.
Abstract
Background
A large fraction of the human genome is attributable to L1 retrotransposon sequences. Not only do L1s themselves make up a significant portion of the genome, but L1-encoded proteins are thought to be responsible for the transposition of other repetitive elements and processed pseudogenes. In addition, L1s can mobilize non-L1, 3'-flanking DNA in a process called 3' transduction. Using computational methods, we collected DNA sequences from the human genome for which we have high confidence of their mobilization through L1-mediated 3' transduction.
Results
The precursors of L1s with transduced sequence can often be identified, allowing us to reconstruct L1 element families in which a single parent L1 element begot many progeny L1s. Of the L1s exhibiting a sequence structure consistent with 3' transduction (L1 with transduction-derived sequence, L1-TD), the vast majority were located in duplicated regions of the genome and thus did not necessarily represent unique insertion events. Of the remaining L1-TDs, some lack a clear polyadenylation signal, but the alignment between the parent-progeny sequences nevertheless ends in an A-rich tract of DNA.
Conclusions
Sequence data suggest that during the integration into the genome of RNA representing an L1-TD, reverse transcription may be primed internally at A-rich sequences that lie downstream of the L1 3' untranslated region. The occurrence of L1-mediated transduction in the human genome may be less frequent than previously thought, and an accurate estimate is confounded by the frequent occurrence of segmental genomic duplications.
Background
Analysis of the initial draft of the human genome revealed that 45% of the sequence is transposable elements [1]. The expansion of the human genome that resulted from the mobilization of these transposable elements suggests they hold secrets of our evolution and increase the plasticity and variation in our genome. In some cases, transposable elements may have been domesticated by their host to serve clear functional roles [2-10]. Most human transposable elements are retrotransposons.
Among the retrotransposons in the human genome is the LINE-1 (L1) element. A full-length L1 insertion in the genome is approximately 6,000 nucleotides long and consists of a 5' untranslated region (UTR), two open reading frames (ORFs), and a 3' UTR terminating in a poly(A) tail [11]. The second ORF of L1 encodes three domains critical for L1 propagation: endonuclease (EN) [12], reverse transcriptase (RT) [13,14], and a 3' terminal zinc-finger-like domain [15]. The EN and RT nick a target site in DNA and reverse transcribe L1 RNA, respectively, to integrate into a new genomic locus [12,16-18]; this process is known as target-site-primed reverse transcription (TPRT). It is believed that the tendency of EN to nick target DNA at the consensus 3'-AA-TTTT-5' exposes a T-rich sequence, to which the poly(A) tail of an L1 transcripts can anneal thereby priming reverse transcription [16,19-21]. A new L1 insertion is usually flanked by short direct repeats derived from the target DNA locus upon L1 integration [22,23]; these repeats are referred to as target-site duplications (TSDs).
The role of L1 in shaping the human genome is unmistakable. Not only does L1 sequence itself contribute at least 462 megabases (Mb) to our genome (17% of the total length) [1], but copies of the Alu and SVA transposable elements and processed pseudogenes are also believed to have inserted into the genome by borrowing the EN and RT proteins encoded by L1 [13,20,21,24-27]. In addition to self-mobilization and mobilization of other transposable elements, L1s can also move unique flanking DNA sequence to another locus in the genome in a process known as 3' transduction. This occurs when an L1 transcript reads into a portion of the downstream flanking sequence. This 3' sequence becomes transduced, along with the L1 sequence, to a new genomic locus; a hypothesized cause of the imprecision of the 3' end of the L1 transcript is the weak polyadenylation signal in the L1 element [28]. Clear indications of 3' transduction have been documented in cases where an L1 inserted into the dystrophin gene [29], APC [30] and CYBB [31]. All these disease-producing L1 insertions, the boundaries of which were defined by flanking TSDs, contained novel sequences downstream of the L1 sequence itself. In addition, it has been suggested that the multiple copies of exon 9 of the cystic fibrosis transmembrane conductance regulator (CFTR) gene found in the human genome may have proliferated via L1-mediated transduction [32]. In most of these cases, the progenitor L1 element could be identified on the basis of the sequence of the 3'-transduced DNA segment.
A proposed consequence of L1 3' transduction is exon shuffling [28,33,34]. That is, an exon downstream of an L1 may be co-mobilized with that same L1 and inserted at a new locus such that the exon is integrated into another gene. Moran et al. demonstrated this experimentally in cultured cells by cloning a reporter gene containing a splice acceptor downstream from the polyadenylation signal of an intact L1 element [28]. This engineered L1 retrotransposed into transcriptionally active genomic loci, allowing the co-mobilized reporter to be expressed after being spliced into a transcript expressed in these cells, effectively creating a chimeric mRNA [28].
We have previously found that nearly 9% of recent L1 insertions in the human genome have TSDs that are consistent with 3' transduction [23]. That is, the 3' TSD of these L1s with transduction-derived sequence (L1-TDs) is preceded by a poly(A) tail and located up to several hundred nucleotides downstream from the end of the L1 3' UTR [35,36]. On the other hand, standard L1 insertions have TSDs that follow a poly(A) tail immediately flanking the L1 sequence. For L1 elements that have 3'-transduced sequence, sibling, progenitor, and/or descendant L1s can be identified by comparing the transduced sequence to the sequence downstream of other L1 elements in the genome.
Using a recently developed algorithm, TSDfinder [23,37,38], we have precisely identified L1 insertions in the human genome whose sequence signature suggests an L1-TD. We then determined which of these transduced sequences shared high similarity with one or more other genomic loci that were also located immediately downstream of an L1. In this way, we built families of L1s potentially derived from the same progenitor element. We found that many potential family members of L1-TDs were merely duplications. Bona fide transduced sequences were analyzed for functional annotation, such as coding regions of genes, in the human genome. In studying the architecture of the 3'-transduced sequences, we found that only a fraction had a recognizable polyadenylation signal. For some of the other transduced sequences, in lieu of a polyadenylation signal, the pairwise alignment between the presumed progenitor element plus its downstream sequence and a descendant L1-TD ended in poly(A) or a related A-rich sequence. These sequence structures may indicate internal priming of the L1 RT at A-rich tracts of a transcript during the process of TPRT.
Results
Finding L1-TDs
We used the RepeatMasker [39] and TSDfinder [23,37,38] programs to identify 6,178 L1 elements that had a sequence structure consistent with 3' transduction in a recent build of the human genome. These L1s had TSDs at least nine nucleotides long preceded by poly(A); they were classified as L1-TDs on the basis of having at least 20 nucleotides of sequence between the end of the 3' UTR and the start of the poly(A) tail that immediately preceded the 3' TSD. These L1-TDs represented 38% of all L1s for which we were able to identify TSDs [23].
Identifying related L1s
For these 3'-transduced sequences to be legitimate, they had to be located downstream of another L1 elsewhere in the genome. Otherwise, the mechanism for their mobilization or duplication in the genome might not be L1-dependent. To test for this, we collected 3 kilobases (kb) of sequence downstream from each 3' intact L1 that we found in the genome [23], formatted this collection of 3'-flanking sequences as a BLAST database [40], and queried each putative transduced sequence against it (see Materials and methods).
When a putative transduced sequence was found to be very similar to the downstream sequence of another L1 in the genome, certain criteria had to be met in order to merit further analysis. First, the two L1s could not be on the same chromosome and adjacent, otherwise the match was likely to be trivial and due to shared sequence lying downstream of both of the L1s (Figure 1a). Furthermore, the downstream sequences had to be equal to or greater than 90% identical (Figure 1b), the length of the alignment had to be equal to or greater than 30% of the putative transduced sequence length (Figure 1c), and the orientation of the matching downstream sequences with respect to the upstream L1 had to be the same (Figure 1a,1d). The start positions for both downstream sequences in their pairwise alignment were required to be within 20 nucleotides of each other (Figure 1e). Finally, if a putative 3'-transduced sequence passed all these tests, we checked to ensure that it was not part of a segmental duplication in the genome (Figure 1f) (see Materials and methods for details). This step was necessary because L1s can be, and often are, part of larger segmental duplications in the genome; in this case, identity between the downstream sequences of two such L1s cannot be attributed to 3' transduction without significant analysis by hand, and the sequence identity will generally continue well beyond the 3' TSD. An inordinate number of our putative L1-TDs were within genomic duplications located on the Y chromosome, whereas only two such occurrences were found on the gene-rich chromosome 19 (data not shown). Generally, the frequency of duplications found on each chromosome was in agreement with a previous study of segmental duplications in the human genome sequence [41]. Although exceptions to some of the above criteria could be envisaged such that a match to a putative transduced sequence could be legitimate, we settled on these conservative criteria to winnow the results. As outlined in Figure 1, this analysis greatly reduced the number of robust L1-TDs; these remaining L1-TDs were considered bona fide L1-TDs for the purposes of this study.
Figure 1.
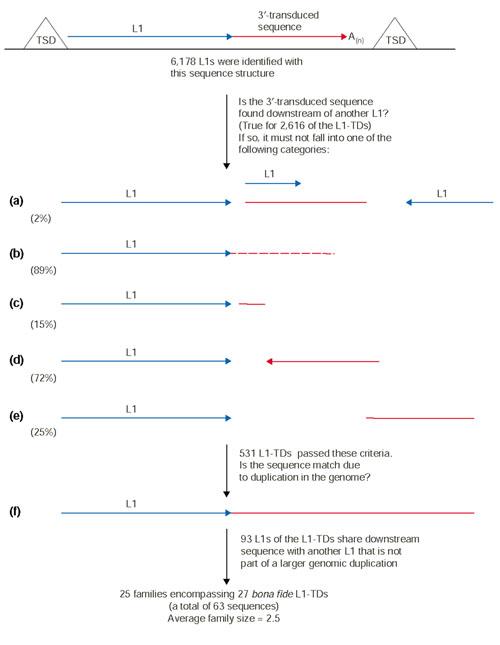
Criteria for rejecting related sequence downstream from another L1 in the genome. At the top of the figure is a schematic of an L1-TD. The red segment represents a 3'-transduced sequence; in (a-f), the red segment represents a possible BLAST hit with a sequence downstream of another L1 element. When searching for the master element that gave rise to the transduced sequence, the following scenarios must not be true: (a) two different but nearly adjacent L1s share the same 3 kb flanking sequence; (b) the sequence shares less than 90% identity with the transduced sequence; (c) the length of the match is less than 30% of the transduced sequence; (d) the match is in the opposite orientation with respect to the L1 sequence; (e) the position of the match with respect to the end of the L1 differs by more than 20 nucleotides; (f) the two genomic segments are duplications and the alignment extends past the TSD. The percentages shown below criteria (a-e) indicate the frequency of finding the depicted structure in our entire collection of L1-TDs (and thus rejecting a candidate on that basis).
For our final analysis, 28 families remained. In the case of families containing the L1s 12_173 and 2_22677, both of these L1-TDs 'found' the other as a family member; therefore the two families were consolidated into a single family (Table 1, family id 4). Furthermore, we observed some overlap in the families representing the L1-TDs 5_7396, 7_12643, and X_11447. We realized that 5_7396 and 7_12643 were duplicates and pooled them and their family members into the X_11447 family (family id 25). In the end, we found a total of 25 families made up of 63 L1 elements (Table 1).
Table 1.
Overview of L1-TD based families
| Family id | L1 id (Chrom_number) | Gi | L1 5' start in Gi* | L1 end or end of transduced sequence in Gi* | Length of transduced sequence (nucleotides) | L1 length (nucleotides) | Parent element† | TSD | 5' inverted‡ |
| 1 | 2_4643 | 15294467 | 307978 | 311125 | 2,584 | 567 | aaaaataaa | ||
| Un_953 | 15306878 | 184575 | 184856 | NA | 285 | ||||
| Un_964 | 15306878 | 268932 | 269213 | NA | 285 | ||||
| 2 | 2_7114 | 15294791 | 255899 | 256304 | 265 | 141 | taaaatgcagaaactag | ||
| 10_7718 | 15298903 | 2767966 | 2773393 | NA | 5,451 | ||||
| 1_8235 | 15295669 | 348450 | 342868 | 160 | 5,449 | aaataaatg | |||
| 1_8446 | 15295734 | 237379 | 231775 | 176 | 5,449 | aaataaatg | |||
| 4_17987 | 15296429 | 154870 | 149448 | NA | 5,449 | gattcagtgtag | |||
| 3 | 2_17349 | 15296775 | 3661224 | 3663417 | 1,056 | 1,149 | gaaaacccccatt | Y | |
| 8_15027 | 15300852 | 2440737 | 2440139 | NA | 602 | Y | |||
| 4 | 2_22677 | 15297650 | 495140 | 494449 | 237 | 440 | gaaagtctcag | ||
| 12_173 | 13650683 | 155075 | 155558 | 244 | 241 | aaaagtgtat | |||
| 5 | 2_25220 | 15321353 | 1827382 | 1820496 | 869 | 6,028 | aaagctgctatgc | ||
| X_13522 | 15310267 | 360475 | 354463 | NA | 6,027 | Y | gaaaatctataacctt | ||
| 6 | 2_25509 | 15321375 | 1909492 | 1915693 | 181 | 6,029 | aagagttcaagaccag | ||
| 7_87 | 12732805 | 21100 | 27115 | NA | 6,029 | Y | aaagaagtttacggat | ||
| 7 | 3_1865 | 13643340 | 90808 | 92969 | 324 | 1,877 | agaac [a/t]tctggtttctc | Y | |
| 15_6107 | 15301004 | 875067 | 868748 | 308 | 6,028 | Y | gaaagttttctc | ||
| 1_1921 | 15294432 | 1613642 | 1609714 | NA | 3,940 | ||||
| 8 | 3_9516 | 15294771 | 235402 | 237115 | 901 | 853 | aaagaaaaatgcat | Y | |
| 3_8906 | 15294621 | 1207468 | 1206594 | NA | 888 | ||||
| 9 | 3_21623 | 15297547 | 436478 | 433456 | 1,346 | 1,666 | aaaagaaaagaaaa | ||
| 3_21596 | 15297547 | 226036 | 226431 | NA | 385 | ||||
| 10 | 5_11657 | 15296062 | 51646 | 48476 | 290 | 2,898 | aaaaagaaa | ||
| 7_871 | 14750593 | 827200 | 821606 | NA | 5,554 | ||||
| 11 | 6_4059 | 15299959 | 5480739 | 5478164 | 416 | 2,167 | aaagaatgtgttttccc | ||
| 2_23164 | 15297755 | 259721 | 265735 | NA | 6,029 | Y | aagaaaaggtggcacat | ||
| 9_9971 | 15298967 | 4929487 | 4935489 | NA | 6,028 | Y | aagaaaatg | ||
| 12 | 6_5918 | 15300319 | 1335113 | 1337115 | 216 | 1,786 | aaagatatagta | Y | |
| 4_13249 | 15295387 | 360002 | 353916 | 74 | 6,029 | Y | aaaagcaatcttgc | ||
| 13 | 9_6571 | 15298230 | 1090696 | 1086281 | 869 | 3,582 | aaat [g/t]acccatcatta | ||
| 7_6559 | 15299368 | 436642 | 437412 | NA | 791 | ||||
| 14 | 9_10645 | 15299021 | 1612561 | 1606351 | 193 | 6,028 | aaaagtttcag | ||
| 11_9728 | 15310185 | 12924346 | 12918249 | 80 | 6,028 | Y | aagaaggcatttcag | ||
| 15 | 12_12007 | 15307682 | 13065 | 12422 | 501 | 142 | aaaaaaatcccctt | ||
| 4_15129 | 15295977 | 549041 | 548937 | NA | 105 | ||||
| 16 | 13_482 | 14757668 | 240258 | 238846 | 47 | 1,353 | aagtaggta | ||
| X_17433 | 15311159 | 890742 | 891945 | NA | 1,190 | ||||
| 17 | 13_5911 | 15302172 | 527379 | 521229 | 125 | 6,029 | agaaaataattaacaa | ||
| 6_6642 | 15300443 | 3829157 | 3826437 | NA | 2,733 | Y | |||
| 18 | 13_10012 | 15303229 | 716846 | 718738 | 1,557 | 328 | aaaaagaaaa | ||
| 22_2286 | 15319019 | 17887232 | 17888051 | NA | 810 | aattggcctttga | |||
| Y_1832 | 15284289 | 141365 | 142172 | NA | 806 | aattggcctttga | |||
| 22_2857 | 15319360 | 187476 | 187124 | NA | 347 | ||||
| 19 | 14_785 | 15299776 | 2701347 | 2702113 | 398 | 373 | aaaaaaaaa | ||
| 13_6443 | 15302172 | 4825675 | 4827164 | NA | 1,507 | ||||
| 20 | 14_2523 | 15299963 | 1505451 | 1509166 | 176 | 3,539 | aagagtaaa | ||
| 8_9380 | 15299954 | 317860 | 323975 | 99 | 6,029 | Y | gaaaggacaaaaagg | ||
| 21 | 15_2689 | 15300281 | 2523261 | 2517133 | 133 | 6,029 | aaaaagaaataag | ||
| 2_25800 | 15321383 | 1783087 | 1782231 | 113 | 745 | tttaaaaaa | Y | ||
| 18_6857 | 15306195 | 246156 | 240742 | NA | 5,438 | ||||
| 18_6870 | 15306195 | 347799 | 353795 | NA | 6,027 | Y | |||
| 22 | 16_1147 | 15315724 | 78409 | 84467 | 50 | 6,028 | aaga [g/c]ggttattctg | ||
| 5_5158 | 15294637 | 18076 | 12135 | NA | 5,964 | ||||
| 23 | 19_4956 | 15321727 | 603330 | 598565 | 2,883 | 1,910 | aataaataaataaataa | ||
| 19_4990 | 15321727 | 762962 | 769106 | NA | 6,132 | Y | |||
| 24 | 20_1331 | 15304545 | 16832755 | 16832512 | 112 | 132 | aagagacag | ||
| Y_3590 | 15284296 | 451303 | 453712 | NA | 2,382 | Y | |||
| 25 | X_11447 | 15310114 | 1438124 | 1444236 | 94 | 6,029 | aaagaacacctggg | ||
| 5_7396§ | 15295056 | 6999 | 483 | 500 | 6,029 | Y | aaaaatttactgtcta | ||
| 7_12643§ | 15300287 | 781397 | 774887 | 494 | 6,029 | Y | aaaaatttactgtcta | ||
| 18_6119 | 15305941 | 1578214 | 1576721 | NA | 1,494 | ||||
| 6_17369 | 15302692 | 2484984 | 2485139 | NA | 156 |
*An end coordinate less than the start coordinate indicates that the L1 is on the complementary strand. †Y indicates that the L1 element may be the parent element. ‡Y indicates that the L1 element is 5' inverted. §These L1-TDs are duplicates.
Families of L1 elements
The average size of the final high-confidence L1 families was 2.5 members. The length distribution of these bona fide transduced sequences is shown in Figure 2. The vast majority of the transduced sequences were less than 500 nucleotides, and the median length was 290 nucleotides.
Figure 2.
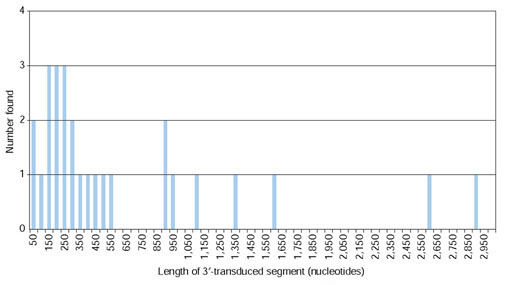
Lengths of 3'-transduced sequences. Length was calculated as the distance from the end of the L1 3' UTR to the start of poly(A) tail that precedes the 3' TSD. Lengths were placed into bins representing intervals of 50 nucleotides. Only the lengths of the 27 3'-transduced sequences that pass all the criteria shown in Figure 1 are considered.
For each family of L1 elements, we tried to determine the relationship between the family members. That is, an L1-TD is either a sibling of the other family members found, or it is the child of a progenitor element. In addition, an L1-TD could be the progenitor of subsequent L1-TDs, giving rise to composite transduction events. For an L1 to be a bona fide progenitor element of a L1-TD, it must be longer than, or of the same length as, the L1 element of the L1-TD. Furthermore, in order to have been transcribed and transposed to give rise to progeny elements, the progenitor L1 must in principle be long enough to include the internal promoter in the 5' UTR. The majority of L1s in the genome are 5' truncated [42], and over time, a full-length L1 may be disrupted by mutation, insertion of another transposable element, or other DNA rearrangements. Consequently, in our set of 25 families, only 10 have a nearly full-length candidate for the progenitor element; their family ids are: 5, 6, 7, 11, 12, 14, 20, 21, 23, and 25 (Table 1). For the remainder of the families, although downstream sequences were similar and TSDs marked the end of aligned sequences, the L1s in a given family were either shorter than the L1 for which a transduced sequence was found, or too short to have been transcribed from the internal L1 promoter. This would imply that, in many of our cases, L1s in a family are siblings that arose from the same progenitor L1.
Four of the L1-TDs in our final set appeared to be composite transpositions. That is, we identified an L1 with downstream sequence that matched the proximal part of the transduced sequence, but we did not find any sequence downstream from an L1 that matched the distal end of the transduced sequence.
Functional annotation of the transduced sequences
We next studied the location of all the transduced sequences in Table 1 on the set of annotated 'NT_' contigs assembled at the National Center for Biotechnology Information (NCBI). In particular, we were interested to see if any transduced sequences were annotated as an exon, lending direct support to the mechanism of L1-mediated exon shuffling [28,33,34]. None of the transduced sequences downstream of any of the L1 family members in Table 1 was annotated as an exonic sequence. Of the 63 sequences that make up these families 12 were annotated as intronic sequences. One of the sequences was within 1,650 nucleotides of the start of an mRNA annotation predicted by automated computational analysis (L1 id 13_10012 and gene LOC92404 that is similar to a putative protein-tyrosine phosphatase). Thus, it is possible that this transduced sequence contributes important regulatory elements to the promoter region, influencing the expression of this gene. However, it is important to note that because our studies are confined to relatively young elements with TSDs, our failure to identify such examples by no means rules out exon shuffling by 3' transduction as a potentially important evolutionary mechanism. For example, one such event appears to have occurred 7-10 million years ago (Mya) with exon 9 of CFTR, the caveat being that there is no L1 element upstream of this particular exon in CFTR itself [32]. In our analysis, we found three transduced sequences with similarity to CFTR exon 9; however, two of them were part of a segmental duplication, and the third had a nearby sequencing gap, precluding assessment of its duplication status.
Polyadenylation signals
To understand the mechanism by which our set of transduced sequences were mobilized by an L1, we examined them for a polyadenylation signal upstream of the 3'-terminal poly(A) tail. We manually inspected these sequences for the presence of either AATAAA or ATTAAA polyadenylation signals no more than 100 nucleotides upstream of the poly(A) tail that preceded the 3' TSD [43]. We were able to identify a polyadenylation signal in 11 of our 25 examples of 3' transduction events (Table 2 and Figure 3a).
Table 2.
Poly(A) tails of L1s
| Family id | L1 id (Chrom_ number) | Polyadenylation signal* | Alignment ends in A-rich stretch*,† | Tail |
| 1 | 2_4643 | x | x | AAAAAAATAAATAAA |
| Un_953 | ||||
| Un_964 | ||||
| 2 | 2_7114 | x | x | AAAAAAAAAAATAAATAAATAAATAAAA |
| 10_7718 | ||||
| 1_8235 | AAAATAAAAAAATAAA | |||
| 1_8446 | AAAAAAATAAATAAATAAAAAATAAATAAA | |||
| 4_17987 | AAAAAAAAAAA | |||
| 3 | 2_17349 | aataaa | x | AAAAAAAAAAAAAAAAGAAAAA |
| 8_15027 | ||||
| 4 | 2_22677 | aataaa | x | ACAAAAAAGAAAAAAA |
| 12_173 | AAAAAATAAATGAATAAA | |||
| 5 | 2_25220 | attaaa | x | AAAATAATTAAAACATAAAAAAAAAAA |
| X_13522 | AAAAAAAAAATTAAAAAAAAAAAA | |||
| 6 | 2_25509 | x | x | AAAAAAAAAAAAAAAAAA |
| 7_87 | AAAAAAAAAAAAAA | |||
| 7 | 3_1865 | attaaa | A(19) | AAAAAAAAACAAAGAACAAAAAAAAA |
| 15_6107 | AAAAAAAAAAAAAAAAAAA | |||
| 1_1921 | ||||
| 8 | 3_9516 | x | short A(8) | AAAAAAAGAAAAAGA |
| 3_8906 | ||||
| 9 | 3_21623 | x | A(39) | AATAAAAATAAAAAAAGAAAA |
| 3_21596 | ||||
| 10 | 5_11657 | x | x | AAAAAAATGA |
| 7_871 | ||||
| 11 | 6_4059 | x | A(10) | AAAGTATAAAAAAA |
| 2_23164 | ATAATAAATTAAAAAAAAAGAAA | |||
| 9_9971 | ATAATAAATTAAAAAAAA | |||
| 12 | 6_5918 | aataaa | x | AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 4_13249 | AGAAGCAAAAAAAAAAAAAAAAAAAAAAAA | |||
| 13 | 9_6571 | x | short A(8) | AAAAATAAATAAATAAACAGATAAATAAATAAATAAATAA |
| 7_6559 | ||||
| 14 | 9_10645 | aataaa | x | ACAAATAAAAAACAAACAAACAAAAA |
| 11_9728 | AAAAAAAAAAAAAAAAAAAAA | |||
| 15 | 12_12007 | aataaa | A(6) | AATAAATTAAAAAACTAAAAAAAAAAAAAAAAAGA |
| 4_15129 | ||||
| 16 | 13_482 | x | x | AAAGAAAACAAAAAA |
| X_17433 | ||||
| 17 | 13_5911 | aataaa | A(12) | AAAAAAAAAA |
| 6_6642 | ||||
| 18 | 13_10012 | x | short | AGAAAAAAAAAAAAGA |
| 22_2286 | AAAAAAAACAA | |||
| Y_1832 | AAAAAAAACAA | |||
| 22_2857 | ||||
| 19 | 14_785 | x | A(21) | AAAAAAATAAATAAATAAGAAAAACATTA |
| 13_6443 | ||||
| 20 | 14_2523 | attaaa | short | AAAATGAAAAAATACTAAAAAAAAAA |
| 8_9380 | AACAGAGAAAAAAAAAAAAAA | |||
| 21 | 15_2689 | aataaa | A(35) | AAAAAATAAAAAATAAAAAATAAAAAATAATTAAAAAAAAGAAAATTAAAAAAAAAAGAAAA |
| 2_25800 | AAAAAATAAAATAAAATAAAAATAAATAAAAATAAATTAAAAAAA | |||
| 18_6857 | ||||
| 18_6870 | ||||
| 22 | 16_1147 | aataaa | A(10) | ACAGGAAAAAAA |
| 5_5158 | ||||
| 23 | 19_4956 | x | A(11) | AAAAATAAATAAATAAA |
| 19_4990 | ||||
| 24 | 20_1331 | x | A(23) | AAAAAAAAAAAAAAAAAAAAAAAA |
| Y_3590 | ||||
| 25 | X_11447 | x | x | AAAAAAAAAAA |
| 5_7396‡ | AACCAGAAAAAAAAAAAA | |||
| 7_12643‡ | AACCAGAAAAAAAAAA | |||
| 18_6119 | ||||
| 6_17369 |
*'x' indicates that a polyadenylation signal was not found or the alignment did not end in an A-rich stretch. †'short' indicates that the putative 3'-transduced sequence may be a composite transduction event; no L1 family member was found with downstream sequence that aligned with the distal part of the transduced sequence. If the proximal sequence alignment ended at an A-rich sequence, the number of As is indicated in parentheses. ‡These L1-TDs are duplicates.
Figure 3.

Alignments of 3' flanks of L1 family members. Two examples of parent-child pairs are shown. The TSDs of the L1-TDs are highlighted in blue, and the TSDs of other family members are highlighted in yellow. If L1s were 5' inversion events, the reverse complement of the 5' segment was used to view the alignment. The intensity of the background sequence shading is a function of the level of sequence conservation among all sequences. Numbers above the L1.3 consensus sequence indicate the sequence position. In calculating the percent identity, indels were counted as mismatches. In the case of family 12 depicted in (a), it appears that a cellular polyadenylation signal was used to generate a de novo poly(A) tail on the readthrough RNA. The polyadenylation signal is highlighted in pink. In the case of family 11 depicted in (b) no such signal is present; thus, this class of events may have arisen by an internal priming mechanism at an A-rich sequence.
For five of the transduced sequences lacking a clear polyadenylation signal, the alignment between the 3'-transduced sequence and family members ended at an A-rich sequence; the 3' TSD of the 3'-transduction event was found immediately downstream of this A-rich sequence in the DNA (Figure 3b). A similar sequence structure was reported by Ovchinnikov et al. for a 3'-transduction event [44]. Several possible explanations for this sequence structure are addressed in the Discussion. One explanation given by Ovchinnikov et al. [44] is that the transcripts may have been internally primed at an A-rich sequence (see Additional data file and [44]); a different type of internal priming is also required by a current model for 5' inversion of L1s [45].
If internal priming is a mechanism by which standard L1 transcripts could be copied into the genome, we would expect to find 3'-truncated L1 elements in the genome whose 3' ends coincide with internal A-rich regions of the L1 sequence. To investigate this possibility, we analyzed a set of 332,587 L1 insertions in the human genome [23]. The 3' end positions of these L1s were pooled into bins of 50 nucleotides along the L1 sequence, and their distribution is shown in Figure 4. The proportion of As in each 50-nucleotide bin of the L1.3 reference sequence is also shown. The great majority of L1 sequences had an intact 3' end, and we observed no clear trend of premature 3' ends that correspond to A-rich tracts in the L1 sequence.
Figure 4.
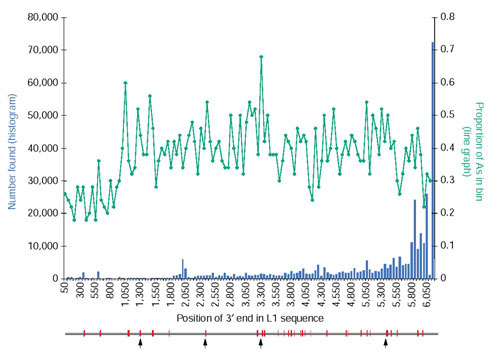
Distribution of L1 3' ends and L1 sequence A content. The histogram shows the annotated 3' endpoints of 332,587 young L1s that were collected from the human genome using RepeatMasker; the L1.3 sequence (GenBank accession number L19088.1) was used as the query library [11]. The L1 sequence was separated into bins of 50 nucleotides and each 3' end was placed into the appropriate bin. The line graph shows the proportion of A nucleotides in each 50-nucleotide bin of the L1.3 element. Below the graph is a schematic of the L1.3 element, and the red regions indicate locations that are at least 5 nucleotides long and with 85% or more A content. The arrows indicate the four A(7) regions.
It is possible that the L1 sequence has evolved to avoid long internal tracts of As in order to prevent internal priming of the transcript. The L1 consensus sequence has a 40% A content, not including the 5' UTR. We calculated the probability of finding an eight-nucleotide-long tract of As in a DNA sequence with such a sequence composition is 87%. However, the longest stretch of As found in the L1.3 reference sequence is seven (probability of 99%), and there are four such tracts.
Finally, we observed that for two of the L1-TDs that do not have a polyadenylation signal, the sequence that the TSDfinder program defined as the poly(A) tail is 'patterned'; that is, the tail is composed of A-rich simple repeats (for example, AAATAAAT...) [23]. Since these poly(A) tails themselves contain polyadenylation signals, it is possible that their assignments as poly(A) tails are false positives, and the real tail formed by polyadenylation has shortened over time and is not detectable [44,46,47]. Four of the remaining L1-TDs for which no polyadenylation signal was found appeared to result from multiple sequential transduction events; we did not have family members that align with the 3' end of the transduced sequence to determine whether the poly(A) stretch could have been encoded in DNA.
Discussion
In this study, we used computational methods to analyze human L1s whose sequence structure was consistent with 3' transduction. The vast majority of these putative L1-TDs could not be thoroughly evaluated for family members because of gaps in the genome sequence, excessive repetitive DNA content, sequence duplications and other practical limitations. Therefore, we are unable to calculate the overall rate of transduction events. Of the putative 3' transduced sequences, 58% (3,562) lacked detectable BLAST hits in our database of sequences downstream from L1s. Possible reasons for this are: first, that the sequence is unique in the human genome and was never 3' transduced (false positive); second, that the sequence has a counterpart in the unsequenced portion of the human genome; third, the loss of the full-length progenitor L1 due to recombination [48], or fourth, that the progenitor L1 sequence suffered from extensive mutations that precluded its detection as a 3' intact L1, and therefore, the downstream sequence was not included in our BLAST database.
It is surprising how few of the examples of putative L1-TDs can be directly verified by finding either progenitor-progeny or sibling pairs. This can be understood by considering the facts that the human population is highly outbred, and L1 elements are preferentially cis-acting [24,27]. Progenitor elements for L1-TDs must be transpositionally competent and thus are likely to be relatively young. Their youth means that they are likely to be present in the human population in an 'unfixed' (heterozygous) state and in a relatively small population. Once such an element spawns a progeny element, the progenitor element (as well as the progeny element) has a high likelihood of extinction due to genetic drift; the donor and the progeny elements will be separated from each other by outcrossing (see [48] for further discussion). Alternatively, Boissinot et al. have hypothesized that full-length L1 elements may be selectively removed from the genome by recombination and thus not be found [48].
We were unable to build families around a high proportion of our initial L1-TDs because of their residence in duplicated genomic regions. This finding is consistent with data showing an abundance of both interchromosomal and intrachromosomal duplications in the human genome [41]. It is unclear how many of these duplications are due to errors in genome assembly and how many represent authentic segmental duplications; correctly assembling duplications as a genuine landscape features of the genome sequence is a formidable informatics challenge [41]. In the future, as better, more accurate, genome builds become available, particularly with regard to the presence of duplications and other rearrangements and the annotation of genes and their promoter regions becomes more thorough and correct, it will be important to repeat this study for the whole genome sequence.
Although in a relatively recent genome build we did not find any clear examples of transduced exons, we did find one transduced sequence located less than 2 kb upstream from a putative gene. This particular transduced sequence could contribute to the regulatory regions of that gene. To find examples of exons shuffled via L1 retrotransposition, an analysis similar to the method we used in this study may have to be performed with older subfamilies of L1s. According to RepeatMasker annotation, all the L1-TDs in Table 1 are of a primate-specific lineage, and it is believed that the ancestral primate genome, the structure of which is thought to be very similar to the modern human genome, existed 60-70 Mya [49]. Thus, at the earliest point in time that we can reliably detect intact L1 elements, most human genes may have been largely established. That is, older L1s and older families of LINEs may have had more influence on the exon composition of genes which themselves are generally rather old. Nevertheless, until more complete genome sequences are available for comparative genomic analyses, we can only speculate on the mechanism by which genes have been altered, sometimes contributing to the formation of a new species. Furthermore, although GC- and gene-rich regions generally lack L1s [1], it has been hypothesized that at any given time during the evolution of the human genome, L1s have inserted randomly, but over time, they have been selected against in GC-rich regions [44]. If this is true, it may be difficult to find evidence in our genome of L1-mediated exon shuffling.
Some L1s with 3'-transduced sequence lack a polyadenylation signal but a common stretch of As delineate the end of an alignment between family members (Figure 3b). One explanation for this sequence structure is that the transcripts were polyadenylated at the same location, and the polyadenylation signal mutated beyond recognition after the insertion event occurred; interestingly, Ovchinnikov et al. reported that polyadenylation signals tend to degrade rapidly after L1 insertion [44]. A second explanation is that the poly(A) sequence is encoded in DNA and either the transcript was degraded up to the position of the A-rich sequence or RNA polymerase II failed to elongate the message past the A-rich sequence; the resulting transcript would then resemble one that had been polyadenylated. Alternatively, there may be a propensity for the L1 transcript to break at A-rich sequences, resulting in an A-rich 3' end that would mimic a 3' polyadenylated transcript.
Interestingly, a recently proposed mechanism of 5' inversion of L1 elements requires that first-strand cDNA synthesis be primed internally on the L1 transcript [45]. Therefore, it is possible that the cDNA synthesis of a 3'-transducing transcript may also be primed at an internal A-rich site as suggested by Ovchinnikov et al. [44] (see Additional data file). Since the L1 EN consensus site is 3'-AA-TTTT-5', the first cut would expose a T-rich sequence on the target sequence from which an A-rich template could be primed for reverse transcription. Indeed, the TSDs for the 3'-transduction events that lack a polyadenylation signal are A-rich at their 5' ends (Table 1), indicating that the target site at which priming occurred would have been T-rich.
We did not find evidence of internal priming for standard L1 insertions predicted to produce 3' truncated L1 elements with well defined endpoints. Rather, L1 elements that are 3' truncated were mostly interrupted by insertions of another transposable element. Lack of internal priming on a standard L1 transcript may be due to interference by the L1 ORF1 protein that has been reported to bind specifically to L1 RNA [50]. Moreover, it is possible that the L1 transcript has a secondary structure that would inhibit an A-rich region from being available as a template for reverse transcription, whereas a 3' tail on the L1 transcript, especially if derived from transduced sequence, may be much more accessible. Finally, in the L1 sequence minus the 5' UTR, the length of the longest internal A-tract (seven nucleotides) is shorter than would be expected by random chance. Thus L1 may have evolved multiple mechanisms to avoid internal priming on A-rich tracts, which would generate defective elements, while allowing it to occur in the flanking sequences, where polyadenylation signals might or might not be found.
Materials and methods
Identifying L1-TDs
The human genomic DNA sequence records used were the set of contigs assembled at the NCBI [51] as of 29 August 2001 (build 25). L1 elements in the contigs were annotated using RepeatMasker [39] with a custom library containing only the L1.3 element (GenBank accession number L19088.1); in this way, we dealt primarily with young L1 elements [23]. The TSDfinder program [23,37,38] was run on the RepeatMasker *.out output files in order to find the TSDs of the L1 elements, thereby refining the L1 boundaries. Each time a 3' intact L1 insertion was found (no more than 30 nucleotides missing from the 3' end of the 3' UTR), the 3 kb of sequence downstream from the L1 was collected. A total of 72,582 such 3-kb sequences were collected. These sequences were placed into a file and formatted as a BLAST database (formatdb parameters: -n ThrPrSeqDB -p F -V T -o T).
During the same run of the TSDfinder program, L1-TDs were identified. To be classified as an L1-TD, the distance between the end of the L1 3' UTR and the start of the 3' TSD had to be greater than 20 nucleotides (not counting the length of the poly(A) tail preceding the TSD nor the number of nucleotides missing from the end of the L1 3' UTR according to the RepeatMasker annotation of the L1 element). The classification of an L1 insertion as an L1-TD was only allowed when a 3' TSD closer to the L1, indicating a standard insertion event, could not be found (see [23,37,38]). The coordinates of the candidate transduced regions (the gi record and the begin and end coordinates) were stored for later analysis.
Analyzing putative 3'-transduced sequences
Transduced sequences were masked to avoid multiple ambiguous matches. The masking was accomplished using the default settings of RepeatMasker [39] (parameter -xsmall). For 2,085 (33%) of the 6,178 L1-TDs, the putative transduced sequence was nearly completely masked. The blastn program [40] was run for the putative transduced sequences against ThrPrSeqDB (parameters: -d ThrPrSeqDB -e 0.05 -J T -U T -F 'm D;R' -Z 150). By doing so, we ensured that any significant match in the genome was also downstream of a potential progenitor L1 for any particular transduced sequence.
A series of Perl scripts was used to examine the BLAST results (see rationale in Figure 1). To test for duplications, the 3 kb of downstream sequence for each putative L1-TD and the potentially related L1(s) identified by BLAST were collected. These sequences were input into bl2seq [52] (parameters: -g T -F 'm D;R' -S 1 -e 10 -X 100 -q -1). If the alignment between the sequences extended beyond the end of the putative transduced sequence, the sequence was labeled as a duplication and was not analyzed any further. Some sequences were removed from analysis because of nearby sequencing gaps that precluded a conclusion regarding the duplication status. Finally, for some alignments produced by bl2seq, the alignment was less than 90% identical or was considered too poor an alignment to continue further analysis with that particular set of L1s.
The duplication status could not be properly assessed for 154 of the initial putative transduced sequences, and they were consequently removed from consideration. One reason for ambiguity of the duplication status was gaps in the genome sequence; if either the query or the subject L1 had a stretch of more than 50 Ns in the 3 kb of downstream flanking sequence, indicating a gap in genome sequence, these were excluded from the analysis because they tended to interfere with the assessment of duplication and confound the automatic analysis. No proof of mapping to a genomic duplication was detected for 93 of the 6,178 initial 3' transduction candidates and their respective family members. These 93 families were made up of 652 total members. The DNA sequence of each member of these families was collected and multiple alignments among the family members were performed using the clustalx and GeneDoc software [53-55]. Manual inspection of the alignment of the family member sequences revealed that 12 of these families had more than 10 members, and it was immediately clear that the matches with the putative transduced sequences in these families were based solely on patchy alignments of largely low-complexity sequence. For the remaining 81 families, 43 were eliminated because of low-complexity matches only (largely poly(A) sequence) or previously missed duplications in the 3' flank. One of the families was eliminated because both L1 elements were full length, yet one of them had a 131-nucleotide insertion in its 5' UTR and the other did not, indicating that these L1s were not directly related [23,56]. Finally, for nine families, although alignment of 3'-transduced sequence with a family member was clearly delineated by the 3' TSD, we found sequence duplication in the 5' flank of the L1s. These families with L1s exhibiting identity in the 5' flank were eliminated from further analysis, as the L1s may represent the same insertion event that was part of a segmental duplication in the genome whose endpoint happened to coincide with the 3' TSD.
GenBank headers of the appropriate gi record (NT_* contigs) were checked for whether the final, bona fide transduced sequences were included in the annotation of any mRNA or CDS.
Adenine content of L1s
The L1s used to generate the data in Figure 4 represent all L1s that were found in the human genome using RepeatMasker [39] with the L1.3 sequence as a custom library (see [23]). To calculate the probability that the length of a stretch of pure A nucleotides in the L1 sequence was less than y, we used the formula:
P(A tract < y) = e^(-nqpy)
where n is the length of the L1 sequence, p is the probability of finding an A in the L1 sequence, and q is (1 - p) [57].
Additional data files
A figure showing how transcripts may have been internally primed at an A-rich sequence is available as a PowerPoint file (Additional data file 1). The model is adapted from [44].
Supplementary Material
A figure showing how transcripts may have been internally primed at an A-rich sequence
Acknowledgments
Acknowledgements
We are grateful to members of the Boeke lab and Landsman group for helpful discussions during the preparation of this manuscript, especially Liora Strichman-Almashanu, John S.J. Anderson, Wataru Fujibuchi and David Symer. This work was supported by NIH grant CA16519 to J.D.B.
References
- International Human Genome Sequencing Consortium (IHGSC) Initial sequencing of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Britten RJ. Mobile elements inserted in the distant past have taken on important functions. Gene. 1997;205:177–182. doi: 10.1016/s0378-1119(97)00399-5. [DOI] [PubMed] [Google Scholar]
- Kidwell MG, Lisch D. Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci USA. 1997;94:7704–7711. doi: 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WJ, McDonald JF, Pinsker W. Molecular domestication of mobile elements. Genetica. 1997;100:261–270. [PubMed] [Google Scholar]
- Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Li WH. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001;17:619–621. doi: 10.1016/s0168-9525(01)02445-3. [DOI] [PubMed] [Google Scholar]
- Craig NL. Unity in transposition reactions. Science. 1995;270:253–254. doi: 10.1126/science.270.5234.253. [DOI] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M. DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Mizuuchi K, Gellert M. Similarities between initiation of V(D)J recombination and retroviral integration. Science. 1996;271:1592–1594. doi: 10.1126/science.271.5255.1592. [DOI] [PubMed] [Google Scholar]
- Schatz DG. Transposition mediated by RAG1 and RAG2 and the evolution of the adaptive immune system. Immunol Res. 1999;19:169–182. doi: 10.1007/BF02786485. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Mathias SL, Nanthakumar E, Scott AF, Kazazian HH., Jr Isolation of an active human transposable element. Science. 1991;254:1805–1808. doi: 10.1126/science.1662412. [DOI] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Feng Q, Mathias SL, Sassaman DM, Scott AF, Kazazian HH, Jr, Boeke JD. An in vivo assay for the reverse transcriptase of human retrotransposon L1 in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:4485–4492. doi: 10.1128/mcb.14.7.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias SL, Scott AF, Kazazian HH, Jr, Boeke JD, Gabriel A. Reverse transcriptase encoded by a human transposable element. Science. 1991;254:1808–1810. doi: 10.1126/science.1722352. [DOI] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- Feng Q, Schumann G, Boeke JD. Retrotransposon R1Bm endonuclease cleaves the target sequence. Proc Natl Acad Sci USA. 1998;95:2083–2088. doi: 10.1073/pnas.95.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Cost GJ, Feng Q, Jacquier A, Boeke JD. Human L1 element target-primed reverse transcription in vitro. EMBO J. 2002;21:5899–5910. doi: 10.1093/emboj/cdf592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cost GJ, Boeke JD. Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry. 1998;37:18081–18093. doi: 10.1021/bi981858s. [DOI] [PubMed] [Google Scholar]
- Boeke JD. LINEs and Alus - the polyA connection. Nat Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning TG, Singer MF. LINE-1: a mammalian transposable element. Biochim Biophys Acta. 1987;910:203–212. doi: 10.1016/0167-4781(87)90112-6. [DOI] [PubMed] [Google Scholar]
- Szak ST, Pickeral OK, Makalowski W, Boguski MS, Landsman D, Boeke JD. Molecular archeology of L1 insertions in the human genome. Genome Biol. 2002;3:research0052.1–0052.18. doi: 10.1186/gb-2002-3-10-research0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Dhellin O, Maestre J, Heidmann T. Functional differences between the human LINE retrotransposon and retroviral reverse transcriptases for in vivo mRNA reverse transcription. EMBO J. 1997;16:6590–6602. doi: 10.1093/emboj/16.21.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol. 2001;21:1429–1439. doi: 10.1128/MCB.21.4.1429-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, DeBerardinis RJ, Kazazian HH., Jr Exon shuffling by L1 retrotransposition. Science. 1999;283:1530–1534. doi: 10.1126/science.283.5407.1530. [DOI] [PubMed] [Google Scholar]
- Holmes SE, Dombroski BA, Krebs CM, Boehm CD, Kazazian HH., Jr A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet. 1994;7:143–148. doi: 10.1038/ng0694-143. [DOI] [PubMed] [Google Scholar]
- Miki Y, Nishisho I, Horii A, Miyoshi Y, Utsunomiya J, Kinzler KW, Vogelstein B, Nakamura Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992;52:643–645. [PubMed] [Google Scholar]
- Meischl C, Boer M, Ahlin A, Roos D. A new exon created by intronic insertion of a rearranged LINE-1 element as the cause of chronic granulomatous disease. Eur J Hum Genet. 2000;8:697–703. doi: 10.1038/sj.ejhg.5200523. [DOI] [PubMed] [Google Scholar]
- Rozmahel R, Heng HH, Duncan AM, Shi XM, Rommens JM, Tsui LC. Amplification of CFTR exon 9 sequences to multiple locations in the human genome. Genomics. 1997;45:554–561. doi: 10.1006/geno.1997.4968. [DOI] [PubMed] [Google Scholar]
- Eickbush T. Exon shuffling in retrospect. Science. 1999;283:1465–1467. doi: 10.1126/science.283.5407.1465. [DOI] [PubMed] [Google Scholar]
- Boeke JD, Pickeral OK. Retroshuffling the genomic deck. Nature. 1999;398:108–111. doi: 10.1038/18118. [DOI] [PubMed] [Google Scholar]
- Pickeral OK, Makalowski W, Boguski MS, Boeke JD. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000;10:411–415. doi: 10.1101/gr.10.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Ostertag EM, Kazazian HH. Transduction of 3'-flanking sequences is common in L1 retrotransposition. Hum Mol Genet. 2000;9:653–657. doi: 10.1093/hmg/9.4.653. [DOI] [PubMed] [Google Scholar]
- Pickeral OK. Bioinformatics of human retrotransposons. PhD dissertation Baltimore, MD: The Johns Hopkins University. 2000.
- TSDfinder code http://www.ncbi.nlm.nih.gov/CBBresearch/Landsman/TSDfinder
- RepeatMasker http://repeatmasker.genome.washington.edu
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AF, Schmeckpeper BJ, Abdelrazik M, Comey CT, O'Hara B, Rossiter JP, Cooley T, Heath P, Smith KD, Margolet L. Origin of the human L1 elements: proposed progenitor genes deduced from a consensus DNA sequence. Genomics. 1987;1:113–125. doi: 10.1016/0888-7543(87)90003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan DF, Manley JL. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov I, Troxel AB, Swergold GD. Genomic characterization of recent human LINE-1 insertions: evidence supporting random insertion. Genome Res. 2001;11:2050–2058. doi: 10.1101/gr.194701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001;11:2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human L1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- Gilbert N, Prigge SL, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Furano AV. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol. 2001;18:926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Stanyon R, O'Brien SJ. Evolution of mammalian genome organization inferred from comparative gene mapping. Genome Biol. 2001;2:research0005.1–0005.8. doi: 10.1186/gb-2001-2-6-reviews0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohjoh H, Singer MF. Sequence-specific single-strand RNA binding protein encoded by the human LINE-1 retrotransposon. EMBO J. 1997;16:6034–6043. doi: 10.1093/emboj/16.19.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI genome resources - H sapiens ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences [Erratum: FEMS Microbiol Lett 1999 Aug 1;177(1):187-8]. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- GeneDoc http://www.psc.edu/biomed/genedoc
- Nicholas KB, Nicholas HB, Jr, Deerfield DW., II GeneDoc: analysis and visualization of genetic variation. EMBNEW News. 1997;4:14. [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Hidaka S, Sakaki Y. Sequence analysis of a KpnI family member near the 3' end of human beta-globin gene. Nucleic Acids Res. 1985;13:7813–7827. doi: 10.1093/nar/13.21.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spouge JL. Finite-size corrections to Poisson approximations of rare events in renewal processes. J Appl Prob. 2001;38:554–569. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A figure showing how transcripts may have been internally primed at an A-rich sequence