

Abstract
The effect of the administration of pertussis toxin (PTX) as well as modulators of different subtypes of K+ channels on the antinociception induced by clonidine and guanabenz was evaluated in the mouse hot plate test.
Pretreatment with pertussis toxin (0.25 μg per mouse i.c.v.) 7 days before the hot-plate test, prevented the antinociception induced by both clonidine (0.08–0.2 mg kg−1, s.c.) and guanabenz (0.1–0.5 mg kg−1, s.c.).
The administration of the KATP channel openers minoxidil (10 μg per mouse, i.c.v.), pinacidil (25 μg per mouse, i.c.v.) and diazoxide (100 mg kg−1, p.o.) potentiated the antinociception produced by clonidine and guanabenz whereas the KATP channel blocker gliquidone (6 μg per mouse, i.c.v.) prevented the α2 adrenoceptor agonist-induced analgesia.
Pretreatment with an antisense oligonucleotide (aODN) to mKv1.1, a voltage-gated K+ channel, at the dose of 2.0 nmol per single i.c.v. injection, prevented the antinociception induced by both clonidine and guanabenz in comparison with degenerate oligonucleotide (dODN)-treated mice.
The administration of the Ca2+-gated K+ channel blocker apamin (0.5–2.0 ng per mouse, i.c.v.) never modified clonidine and guanabenz analgesia.
At the highest effective doses, none of the drugs used modified animals' gross behaviour nor impaired motor coordination, as revealed by the rota-rod test.
The present data demonstrate that both KATP and mKv1.1 K+ channels represent an important step in the transduction mechanism underlying central antinociception induced by activation of α2 adrenoceptors.
Keywords: K+ channel, antinociception, clonidine, guanabenz, minoxidil, pinacidil, diazoxide, gliquidone, mKv1.1, apamin
Introduction
Agonists of α2-adrenoceptors produce a wide variety of effects. These include an antihypertensive action, alleviation of opiate-withdrawal syndrome, antinociception and sedation (Fielding & Lal, 1981; McDonald et al., 1997). Clonidine, an agonist of α2-adrenoceptors, is extremely potent as an antinociceptive agent showing equal or greater potency than morphine in rodents (Fielding et al., 1978). Clonidine has been shown to exhibit antinociceptive activity against a wide variety of noxious stimuli such as chemical irritants, heat, pressure and electrical stimuli (Fielding & Lal, 1981; Skingle et al., 1982).
Clonidine and other agonists of α2-adrenoceptors hyperpolarize neuronal membranes by opening K+ channels (Morita & North, 1981; Andrade & Aghajanian, 1985; Tatsumi et al., 1990). It has also been reported that clonidine antinociception is antagonized by blockers of ATP-dependent K+ channels (Ocana & Bayens, 1993; Raffa & Martinez, 1995). A possible relationship between the effects on KATP conductance and enhancement of the pain threshold produced by activation of α2-adrenoceptors has, therefore, been postulated.
Since several kinds of K+ channels with different electrophysiological characteristics and pharmacological sensitivities have been described in neurones (Halliwell, 1990; Aronson, 1992), in the present work we thought it worthwhile to employ different K+ channel modulators to elucidate better the role of K+ channels in the antinociception induced by α2-adrenoceptor agonists. Apamin has been reported to block specifically currents through the Ca2+-activated K+ channels (Cook, 1988). Sulphonylureas such as gliquidone block KATP channels in neurones whereas minoxidil, pinacidil and diazoxide open the same type (KATP) of K+ channel (Edwards & Weston, 1993). So far, the blockers of neuronal voltage-dependent K+ channels are not selective for any channels in particular (Cook & Quast, 1990; Halliwell, 1990). An antisense oligonucleotide (aODN) was, therefore, used to selectively block the expression of the gene coding for mKv1.1, a voltage-gated K+ channel (Wang et al., 1994).
To this purpose we have evaluated the effects produced by the K+ channel blockers apamin and gliquidone, the K+ channel openers minoxidil, pinacidil and diazoxide, as well as an aODN to mKv1.1 in the antinociception induced by α2-adrenoceptor agonists in the mouse hot-plate test.
Clonidine analgesia has been reported to be prevented by intracerebroventricular administration of pertussis toxin (PTX) (Sanchez-Blasquez & Garzon, 1991). We, therefore, investigated whether also guanabenz analgesia was sensitive to pertussis toxin pretreatment taking into account that α2-adrenoceptors are coupled to pertussis toxin-sensitive G-proteins which in turn can regulate ionic conductance (Aghajanian & Wang, 1986).
Methods
Animals
Male Swiss albino mice (23–30 g) from the Morini (San Polo d'Enza, Italy) breeding farm were used. Fifteen mice were housed per cage. The cages were placed in the experimental room 24 h before the test for acclimatization. The animals were fed a standard laboratory diet and tap water ad libitum and kept at 23±1°C with a 12 h light/dark cycle, light on at 07.00 h. Animals were used a single time with the exception of the aODN experiments. Experiments were conducted 72 h and 7 days after the end of the aODN treatment. Each animal was, therefore, used twice (at 72 h and at day 7). All experiments were carried out according to the guidelines of the European Community Council for experimental animal care.
Intracerebroventricular injection technique
Intracerebroventricular (i.c.v.) administration was performed under ether anaesthesia, according to the method described by Haley & McCormick (1957). Briefly, during anaesthesia, mice were grasped firmly by the loose skin behind the head. A 0.4 mm external diameter hypodermic needle attached to a 10 μl syringe was inserted perpendicularly through the skull and no more than 2 mm into the brain of the mouse, where 5 μl were then administered. The injection site was 1 mm to the right or left from the midpoint on a line drawn through to the anterior base of the ears. Injections were performed into the right or left ventricle randomly. To ascertain that the drugs were administered exactly into the cerebral ventricle, some mice (20%) were injected with 5 μl of diluted 1 : 10 India ink and their brains examined macroscopically after sectioning. The accuracy of the injection technique was evaluated and the percentage of correct injections was 95.
Hot-plate test
The method adopted was described by O'Callaghan & Holtzman (1975). Mice were placed inside a stainless steel container, which was set thermostatically at 52.5±0.1°C in a precision water-bath from KW Mechanical Workshop, Siena, Italy. Reaction times (s), were measured with a stopwatch before and 15, 30, 45 and 60 min after clonidine treatment or 15, 30, 45, 60, 75, 90, 120 and 180 min after guanabenz administration. The endpoint used was the licking of the fore or hind paws. Those mice scoring less than 12 and more than 18 s in the pretest were rejected (30%). An arbitrary cut-off time of 45 s was adopted.
The licking latency values reported in all figures were evaluated in relation to the maximum analgesic effect of clonidine and guanabenz which were respectively 30 and 75 min after administration.
Rota-rod test
The apparatus consisted of a base platform and a rotating rod of 3 cm diameter with a non-slippery surface. The rod was placed at a height of 15 cm from the base. The rod, 30 cm in length, was divided into five equal sections by six disks. Thus, up to five mice were tested simultaneously on the apparatus, with a rod-rotating speed of 16 r.p.m. The integrity of motor coordination was assessed on the basis of the number of falls from the rod in 30 s according to Vaught et al. (1985). Those mice scoring less than 3 and more than 6 falls in the pretest were rejected (20%). The performance time was measured before and 15, 30, 45 and 60 min after treatment with the exception of animals treated with guanabenz which were tested up to 120 min after injection.
Antisense oligonucleotides
The 24mer phosphodiester oligonucleotides were capped by a terminal phosphorothioate double substitution and purified by chromatography (Genosys, The Woodlands, U.S.A.). The antisense ODN (5′-CGA CAT CAC CGT CAT GAT GAA AGC-3′) was designed to target the 5′ portion of the murine Kv1.1 (mKv1.1) mRNA, residues 575–598 of the published cDNA sequence (Chandy et al., 1990). A fully degenerate 24mer ODN (dODN) was used as control.
Mice were randomly assigned to antisense oligodeoxyribonucleotide (mKv1.1 aODN), degenerate oligodeoxyribonucleotide (mKv1.1 dODN) or vector groups. 400 μM ODNs were preincubated at 37°C for 30 min with 13 μM DOTAP (N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammoniummethyl sulphate), used as vector. Each group received a single intracerebroventricular (i.c.v.) injection on days 1, 4 and 7. The experimental conditions for the antisense studies (time, dose and decreases in mRNA levels) have been chosen on the basis of previous studies (Galeotti et al., 1997).
Drugs
The following drugs were used: clonidine hydrochloride, guanabenz acetate, minoxidil, pinacidil, apamin, diazoxide, pertussis toxin (RBI); gliquidone (Boehringer Ingelheim).
Drugs were dissolved in isotonic (NaCl 0.9%) saline solution, with the exception of pinacidil, that was dissolved in a water and dimethyl sulphoxide (DMSO) (3 : 1) vehicle, and diazoxide, which was dispersed in a 1% solution of sodium carboxymethyl cellulose, immediately before use. Drug concentrations were prepared in such a way that the necessary dose could be administered in a volume of 5 μl per mouse by i.c.v. injection and 10 ml kg−1 by esophageal (p.o.) and subcutaneous (s.c.) injection.
Apamin, gliquidone, minoxidil and pinacidil were injected i.c.v. whereas diazoxide was administered p.o.; all drugs were injected 15 min before the test. The drug administration schedule was chosen on the basis of preliminary experiments in which the time-course and the dose-response curves in the hot-plate test for every compound were determined.
Concerning pertussis toxin treatment, mice were randomly assigned to a vehicle (water solution containing 0.01 M sodium phosphate buffer, pH=7.0, with 0.05 M sodium chloride) or a pertussis toxin group (0.25 μg per mouse) which received a single i.c.v. injection on day 0. The hot-plate test was performed 7 days after pretreatment.
Following the pretreatment schedule with saline, vehicle, aODN or dODN mentioned in the above section, the antinociceptive effect of clonidine and guanabenz was tested 72 h and 7 days after the last i.c.v. injection.
Statistical analysis
All experimental results are given as the mean±s.e.mean. Analysis of variance (ANOVA), followed by Fisher's Protected Least Significant Difference (PLSD) procedure for post hoc comparison, and overall ANOVA were used to verify significance between two means. Data were analysed with the StatView software for the Macintosh (1992). P values of less than 0.05 were considered significant.
Results
Effect of pertussis toxin on clonidine and guanabenz antinociception
Both clonidine (0.08–0.20 mg kg−1, s.c.) and guanabenz (0.05–0.50 mg kg−1, s.c.) produced a dose-dependent antinociception in the mouse hot-plate test (Figure 1). Pertussis toxin, administered at the dose of 0.25 μg per mouse i.c.v. 7 days prior to the test, led to a prevention of the antinociceptive effect of clonidine (Figure 1a) and guanabenz (Figure 1b).
Figure 1.
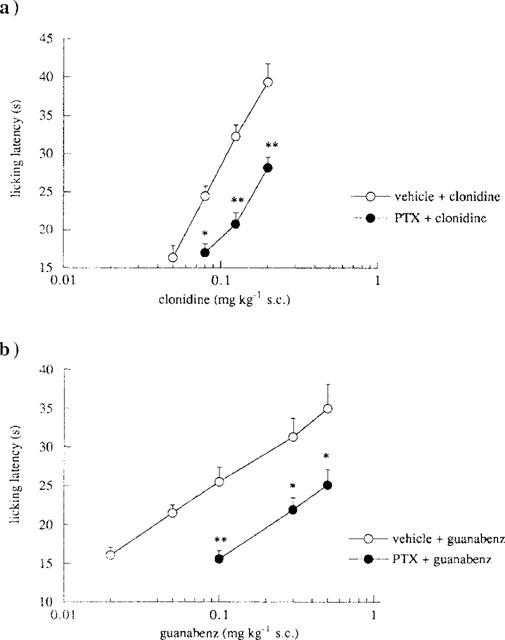
Effect of pertussis toxin (PTX) pretreatment on clonidine- and guanabenz-induced antinociception in the mouse hot-plate test. (a) Prevention by PTX (0.25 μg per mouse, i.c.v.) of clonidine (0.05–0.20 mg kg−1, s.c.) antinociception. The test was performed 7 days after a single i.c.v. injection of vehicle or PTX. Vertical lines represent s.e.mean; between 11 and 16 mice were tested. *P<0.05, **P<0.01 in comparison with vehicle-clonidine treated mice. (b) Prevention by PTX (0.25 μg per mouse, i.c.v.) of guanabenz (0.02–0.50 mg kg−1, s.c.) antinociception. The test was performed 7 days after a single i.c.v. injection of vehicle or PTX. Vertical lines represent s.e.mean; between 12 and 19 mice were tested. *P<0.05, **P<0.01 in comparison with vehicle-guanabenz treated mice.
Effect of KATP channel modulators on clonidine and guanabenz antinociception
The effect produced by the KATP channel openers diazoxide (100 mg kg−1, p.o.), minoxidil (10 μg per mouse, i.c.v.) and pinacidil (25 μg per mouse, i.c.v.) on the antinociception induced by clonidine (0.125 mg kg−1, s.c.) and guanabenz (0.30 mg kg−1, s.c.) was investigated in the mouse hot-plate test (Figure 2). All K+ channel openers potentiated the antinociceptive activity of both α2-adrenoceptor agonists investigated (Figure 2). Furthermore, pinacidil (25 μg per mouse i.c.v.), taken as an example, exerted its potentiating effect at various doses of clonidine (0.05–0.20 mg kg−1, s.c.; Figure 3a) and guanabenz (0.05–0.30 mg kg−1, s.c.; Figure 3b), shifting to the left their dose-response curves.
Figure 2.
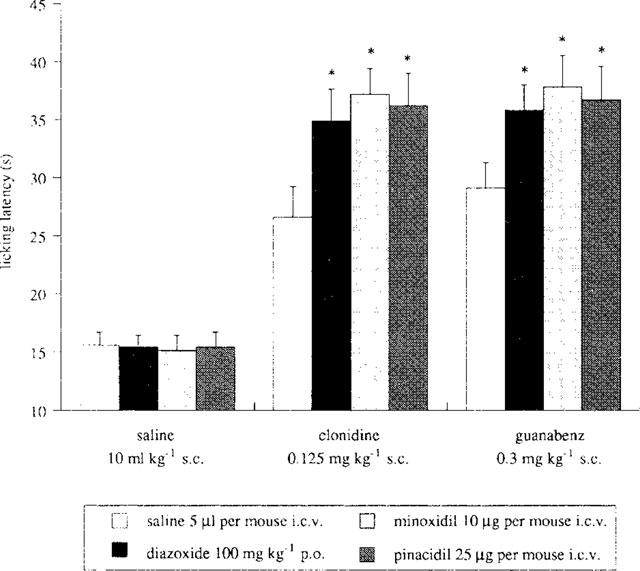
Enhancement of clonidine (0.125 mg kg−1, s.c.) and guanabenz (0.30 mg kg−1, s.c.) antinociception by the KATP openers diazoxide (100 mg kg−1, p.o.), minoxidil (10 μg per mouse i.c.v.) and pinacidil (25 μg per mouse, i.c.v.) in the mouse hot plate test. Vertical lines represent s.e.mean; between 12 and 16 mice were tested. *P<0.05 in comparison with corresponding analgesic drug-treated mice.
Figure 3.
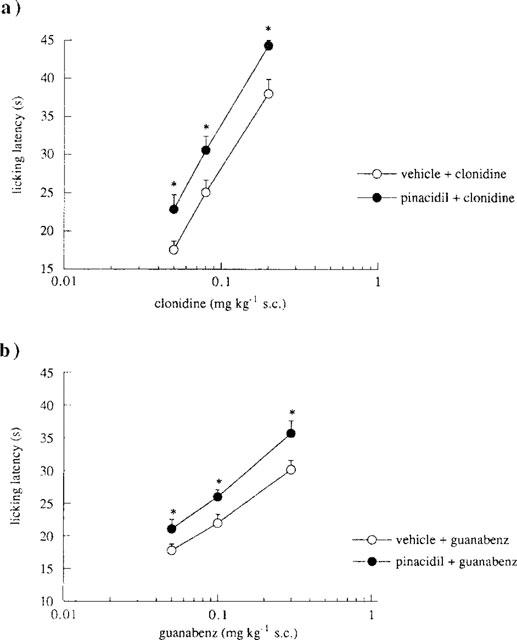
Effect of pinacidil on different doses of clonidine and guanabenz in the mouse hot-plate test. (a) Enhancement by pinacidil (25 μg per mouse, i.c.v.) of clonidine (0.05–0.20 mg kg−1, s.c.) antinociception. Vertical lines represent s.e.mean; between 14 and 16 mice were tested. *P<0.05 in comparison with clonidine-treated mice. (b) Enhancement by pinacidil (25 μg per mouse, i.c.v.) of guanabenz (0.05–0.30 mg kg−1, s.c.) antinociception. Vertical lines represent s.e.mean; between 15 and 18 mice were tested. *P<0.05 in comparison with guanabenz-treated mice.
The administration of the KATP channel blocker gliquidone (6 μg per mouse, i.c.v.) prevented the antinociception induced by clonidine (0.125 mg kg−1, s.c.) and guanabenz (0.30 mg kg−1, s.c.) (Figure 4). A lower dose of gliquidone (3 μg per mouse, i.c.v.) was ineffective (Figure 4).
Figure 4.
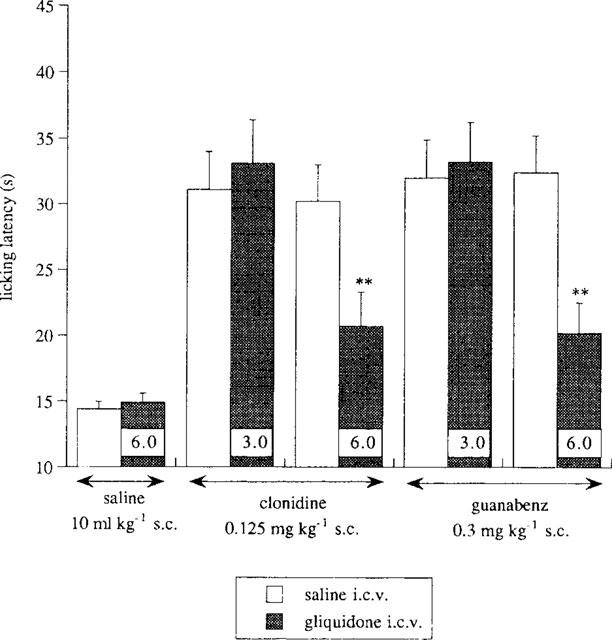
Prevention by the KATP blocker gliquidone (3.0–6.0 μg per mouse, i.c.v.) of clonidine (0.125 mg kg−1, s.c.) and guanabenz (0.30 mg kg−1, s.c.) antinociception in the mouse hot-plate test. Each column shows the dose of gliquidone administered. Vertical lines represent s.e.mean; between 10 and 15 mice were tested. **P<0.01 in comparison with the corresponding analgesic-treated mice.
In the same experimental conditions, neither diazoxide, minoxidil, pinacidil (Figure 2) nor gliquidone (Figure 4) modified the licking latency values of mice when given alone.
Effect of an aODN to mKv1.1 on clonidine and guanabenz antinociception
The effect produced by the antisense oligonucleotide (aODN) to the mKv1.1 gene on clonidine (0.05–0.20 mg kg−1, s.c.) and guanabenz (0.05–0.50 mg kg−1, s.c.) analgesia was evaluated by using the mouse hot-plate test.
Repeated administration of aODN (2 nmol per i.c.v. injection) prevented the antinociception induced by increasing concentrations of clonidine (0.08–0.20 mg kg−1, s.c.; Figure 5a) and guanabenz (0.10–0.50 mg kg−1, s.c.; Figure 5b) in comparison with the mice pretreated with the degenerate oligonucleotide (dODN). The aODN prevented the enhancement induced by the two α2-adrenoceptor agonists displacing to the right the clonidine (Figure 5a) and guanabenz (Figure 5b) dose-response line in a parallel fashion. This antagonistic effect was detected 72 h after the end of the aODN administration. The pretreatment with the dODN never modified clonidine and guanabenz antinociception in comparison with vector i.c.v.-injected mice (data not shown).
Figure 5.
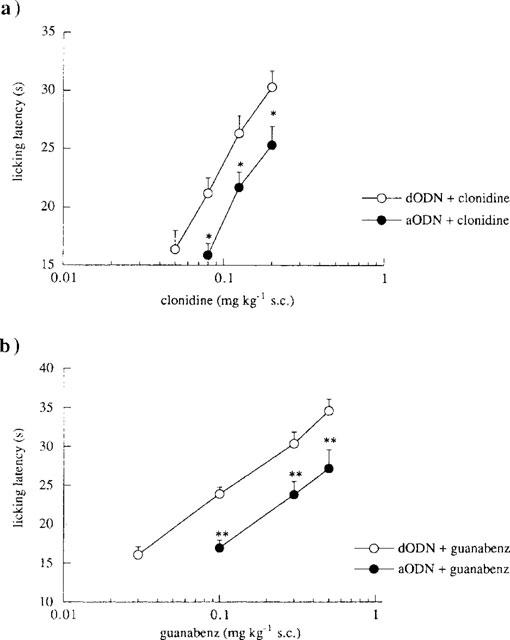
Effect of pretreatment with an antisense oligonucleotide (aODN) to mKv1.1 gene on clonidine- and guanabenz-induced antinociception in the mouse hot-plate test. (a) Prevention by aODN (2.0 nmol per single i.c.v. injection) of clonidine (0.05–0.20 mg kg−1, s.c.) antinociception. The test was performed 72 h after the last i.c.v. injection of degenerate ODN (dODN; 2.0 nmol per single i.c.v. injection) or aODN. Vertical lines represent s.e.mean; between 12 and 16 mice were tested. *P<0.05 in comparison with dODN+clonidine group. (b) Prevention by aODN (2.0 nmol per single i.c.v. injection) of guanabenz (0.02–0.50 mg kg−1, s.c.) antinociception. The test was 72 h after the last i.c.v. injection of degenerate ODN (dODN; 2.0 nmol per single i.c.v. injection) or aODN. Vertical lines represent s.e.mean; between 13 and 19 mice were tested. *P<0.05; **P<0.01 in comparison with dODN+guanabenz group.
The prevention of clonidine (0.125 mg kg−1, s.c.) and guanabenz (0.3 mg kg−1, s.c.) analgesia produced by aODN (2.0 nmol per i.c.v. injection) disappeared 7 days after the end of the aODN pretreatment (clonidine: 27.3±1.8; cloni-dine+aODN: 27.8±1.6; guanabenz: 28.5±1.3; guanabenz +aODN: 28.3±0.9).
The aODN pretreatment did not reduce the pain threshold in mice, having no hyperalgesic effect at both 72 h and 7 days after the last i.c.v. injection (data not shown).
Effect of the Ca2+-activated K+ channel blocker apamin on clonidine and guanabenz antinociception
The administration of apamin (0.5–2.0 ng per mouse, i.c.v.), at all doses used, never modified the enhancement of the pain threshold induced by clonidine (0.125 mg kg−1, s.c.) as illustrated in Figure 6. Furthermore, apamin, at the dose of 1.0 ng per mouse i.c.v., was not able to produce any effect on guanabenz (0.3 mg kg−1, s.c.) antinociception (Figure 6). Apamin, when given alone, showed neither hyperalgesic nor analgesic effect (Figure 6).
Figure 6.
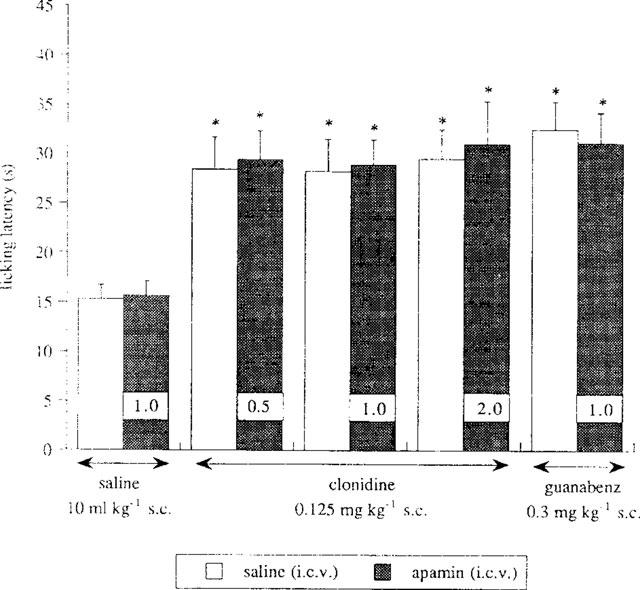
Lack of effect by the Ca2+-gated and K+ channel blocker apamin (0.5–2.0 ng per mouse, i.c.v.) on clonidine (0.125 mg kg−1, s.c.) and guanabenz (0.30 mg kg−1, s.c.) antinociception in the mouse hot-plate test. Each column shows the dose of apamin administered. Vertical lines give s.e.mean; there were 15 mice per group. *P<0.01 in comparison with saline-treated mice.
Higher doses of apamin were not investigated since, at 3.0 ng per mouse i.c.v., this compound produced convulsions in mice.
Effect of clonidine and guanabenz on mouse rota-rod test
It should be noted that both clonidine and guanabenz, at the doses used in the present work, elicited their antinociceptive effect without changing either gross animal behaviour or motor coordination as revealed by the rota-rod test. The rota-rod performance, evaluated as number of falls in 30 s, of mice treated with clonidine at the doses of 0.125 (4.0±0.6) and 0.20 mg kg−1 s.c. (3.8±0.3), and guanabenz at the doses of 0.30 (1.7±0.8) and 0.50 mg kg−1 s.c. (3.9±0.8) was not impaired in comparison with saline-treated mice (2.9±0.6). On the contrary, clonidine and guanabenz administered at higher doses (0.50 and 1.0 mg kg−1, s.c., respectively), produced a significant impairment of the rota-rod performance by increasing the number of falls from the rotating rod (5.8±0.4 and 5.9±0.7 respectively) in comparison with saline-treated mice and with pretest values (5.0±0.6 and 5.1±0.7 respectively).
The number of falls were recorded in correspondence, with the maximum analgesic effect of clonidine and guanabenz which occurred respectively 30 and 75 min after administration.
Discussion
The present results indicate that neuronal K+ channels play an important role in the mechanism of analgesic action of the α2-adrenoceptor agonists clonidine and guanabenz.
The administration of gliquidone, a potent blocker of ATP-dependent K+ channels (KATP) (Amoroso et al., 1990), prevented the antinociception induced by both α2-adreno-ceptor agonists investigated. Pretreatment with minoxidil, pinacidil and diazoxide, openers of neuronal KATP channels (Longman & Hamilton, 1992), potentiated the enhancement of the pain threshold produced by clonidine and guanabenz. This potentiation may occur because minoxidil, pinacidil and diazoxide facilitate the opening of KATP channels induced by the α2-adrenoceptor agonists. The functionality of KATP channels appears, therefore, fundamental in the antinociception produced by α2-adrenoceptor agonists. These observations are in agreement with previous studies on the activity of clonidine in which an antagonism of clonidine K+ conductance by Ce2+, Ba2+ and Rb+, blockers of several types of K+ channels including KATP channels (Quayle et al., 1988; Criddle & Moura, 1995) was reported (Cherubini & North, 1985; Williams & North, 1985; Williams et al., 1988). According to these electrophysiological data, behavioural studies showed that different sulphonylureas, blockers of KATP channels, antagonized clonidine antinociception (Ocaña & Bayens, 1993; Raffa & Martinez, 1995) whereas cromakalim, a KATP channel opener in neurones (Edwards & Weston, 1993), enhanced clonidine-induced antinociception in the mouse tail flick test (Ocaña et al., 1996).
The present study also provides evidence for the involvement of voltage-gated K+ channel in central antinociception induced by α2-adrenoceptor agonists. By using an antisense ODN (aODN) to the mKv1.1 gene coding for the mouse Shaker-like Kv1.1, the importance of the integrity and functionality of the above-mentioned K+ channel has been evidenced. mKv1.1 is a K+ channel of the Shaker-like subfamily that, when expressed in Xenopus oocytes, gives rise to a fast activating, slowly inactivating K+ current (Hopkins & Tempel, 1992). The investigation into the involvement of mKv1.1 in central analgesia was carried out on the basis of its wide distribution in the mammalian brain, including areas involved in the modulation of the pain threshold (Wang et al., 1994). Repeated i.c.v. administration of aODN to mKv1.1 prevented clondine and guanabenz antinociception. The possibility that the antagonism exerted by aODN could be caused by sequence-independent effects on cerebral structures was ruled out since results obtained from a quantitative RT–PCR indicated a lowering of mKv1.1 mRNA brain levels specifically in the anti-mKv1.1 aODN-treated mice (Galeotti et al., 1997). The inhibition of clonidine- and guanabenz-induced enhancement of the pain threshold disappeared 7 days after the last i.c.v. injection of the aODN indicating a lack of damage or toxicity associated with aODN treatment.
The intracellular mechanism of analgesic action of clonidine and guanabenz involves the activation of a pertussis toxin (PTX)-sensitive G-protein, since not only clonidine, in agreement with previous data (Sanchez-Blasquez & Garzon, 1991), but also guanabenz analgesia is prevented by the i.c.v. administration of PTX. PTX-sensitive G-proteins represent the most widespread modulatory signaling pathway in neurones (Holz et al., 1986) and are responsible for modulation of ionic conductance through a direct interaction with the ion channel and/or by lowering intracellular cyclic AMP levels (Hille, 1994). The KATP channels can be opened through a mechanism mediated by G-proteins. It has been reported that the interaction between the α-GTP subunit and the KATP channel produces a conformational change that stimulates the opening of the channel (Edwards & Weston, 1993). In the presence of GTPγS, a non hydrolysable GTP analogue, an irreversible activation of KATP is obtained. Furthermore, the α subunits involved in the modulation of KATP channel function have been identified as belonging to the αi and αo subtypes (Edwards & Weston, 1993). Also voltage-gated K+ channels can be modulated by neurotransmitters and hormones in many tissues. Indeed, protein kinase A modulation of delayed rectifier-type K+ currents has been shown in the heart and in lymphocytes (Giles et al., 1989; Soliven & Nelson, 1990). More recently an increase in mKv1.1 K+ channel expression at the levels of RNA, protein and current density in Chinese hamster ovary cells was found as a result of reduced basal protein kinase A activity (Bosma et al., 1993). PTX-sensitive G-proteins can, therefore, modulate not only KATP but also mKv1.1 channel function through a different mechanism: a direct interaction (KATP) or a modulation of protein kinase A levels (mKv1.1). From this evidence it is plausible to suppose that both K+ channel subtypes are involved in α2-adrenoceptor agonist-induced analgesia as an intracellular effector underlying the activation of a Gi/o protein.
The i.c.v. administration of the bee venom apamin, a blocker of small (low) conductance Ca2+-gated K+ channels (Rudy, 1988), did not modify the antinociception induced by both clonidine and guanabenz, indicating the lack of involvement of Ca2+-gated K+ channels in the modulation of the pain threshold produced by activation of α2-adrenoceptors. These results are in agreement with electrophysiological studies in which apamin was unable to block the inhibitory effect produced by clonidine on peripheral sensory nerves (Stretton et al., 1992; Miura et al., 1992). Doses of apamin higher than 2.0 ng per mouse i.c.v. were not investigated since they induced convulsions in mice. However, we can exclude that the lack of prevention exerted by apamin could be due to the employment of inadequate concentrations since, in the same range of doses used in the present study, apamin was able to modulate different neuronal functions such as learning and memory (Ghelardini et al., 1998) and food consumption (Ghelardini et al., 1997).
In these experimental conditions, neither the K+ channel blocker gliquidone and the aODN to mKv1.1, nor the K+ channel openers used modified the licking latency values of mice in comparison with control groups. The lack of effect of both gliquidone and aODN to mKv1.1 agrees with results of studies in which these compounds did not modify the nociceptive threshold against thermal noxious stimuli (Welch & Dunlow, 1993; Raffa & Martinez, 1995; Galeotti et al., 1997). The administration of the K+ channel openers diazoxide and minoxidil has been reported to induce antinociception in the tail flick test, but this effect was detectable only at doses higher than those employed in the present study (Welch & Dunlow, 1993). We can, therefore, exclude that the prevention or the enhancement of clonidine and guanabenz antinociception is due respectively to a hyperalgesic or analgesic effect of the K+ channel modulators used.
Agonists of α2-adrenoceptors induce not only analgesia but also numerous other pharmacological effects such as sedation and hypotension (McDonald et al., 1997) whose appearance could lead to an alteration of the licking latency values observed in the hot-plate test. It has been, therefore, necessary to choose a range of doses of clonidine and guanabenz at which these compounds showed antinociceptive properties without any behavioural side effects. Doses of clonidine and guanabenz higher than 0.2 and 0.5 mg kg−1, s.c. respectively were not used because they impaired the mice's rota-rod performance. Similarly, the K+ channel modulators and the aODN to mKv1.1 were used at doses which did not modify the animals' behaviour as revealed by the rota-rod and hole board tests (Galeotti et al., 1997; Ghelardini et al., 1997; 1998).
In conclusion, the present data demonstrates that both KATP and mKv1.1 K+ channels are an important step in the transduction mechanism underlying central antinociception induced by activation of α2-adrenoceptors.
Acknowledgments
The authors wish to thank Mary Forrest for linguistic revision of the manuscript. This work was supported by grants from MURST.
Abbreviations
- aODN
antisense oligonucleotide
- dODN
degenerate oligonucleotide
- i.c.v.
intracerebroventricular
- KATP
ATP-dependent K+ channel
- PTX
pertussis toxin
References
- AGHAJANIAN G.K., WANG Y.-Y. Pertussis toxin blocks the outward currents evoked by opiate and α2-agonists in locus coeruleus neurones. Brain Res. 1986;371:390–394. doi: 10.1016/0006-8993(86)90382-3. [DOI] [PubMed] [Google Scholar]
- AMOROSO S., SCHMIDT-ANTOMARCHI H., FOSSET M., LAZDUNSKI M. Glucose, sulphonylureas, and neurotransmitter release: role of ATP-sensitive K+ channels. Science. 1990;247:852–854. doi: 10.1126/science.2305257. [DOI] [PubMed] [Google Scholar]
- ANDRADE R., AGHAJANIAN G.K. Opiate- and α-2-adrenoceptor-induced hyperpolarization of locus coeruleus neurones in brain slices: Reversal by cyclic adenosine 3′ : 5′-monophosphate analogues. J. Neurosci. 1985;5:2359–2364. doi: 10.1523/JNEUROSCI.05-09-02359.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARONSON J.K. Potassium channels in nervous tissue. Biochem. Pharmacol. 1992;43:11–14. doi: 10.1016/0006-2952(92)90653-z. [DOI] [PubMed] [Google Scholar]
- BOSMA M.M., ALLEN M.L., MARTIN T.M., TEMPEL B.L. PKA-dependent regulation of mKv1.1, a mouse Shaker-like potassium channel gene, when expressed in CHO cells. J. Neurosci. 1993;13:5242–5250. doi: 10.1523/JNEUROSCI.13-12-05242.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDY K.G., WILLIAMS C.B., SPENCER R.H., AGUILAR B.A., GHANSHANI S., TEMPEL B.L., GUTMAN G.A. A family of three mouse potassium channel genes with intronless coding regions. Science. 1990;247:973–975. doi: 10.1126/science.2305265. [DOI] [PubMed] [Google Scholar]
- CHERUBINI E., NORTH R.A. μ and κ opioids inhibit transmitter release by different mechanisms. Proc. Natl. Acad. Sci. U.S.A. 1985;82:1860–1863. doi: 10.1073/pnas.82.6.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK N.S. The pharmacology of potassium channels and their therapeutic potential. Trend Pharmacol. Sci. 1988;9:21–28. doi: 10.1016/0165-6147(88)90238-6. [DOI] [PubMed] [Google Scholar]
- COOK N.S., QUAST U.Potassium channel pharmacology Potassium Channels: Structure, Classification, Function and Therapeutic Potential 1990Chichester: Ellis Horwood Limited; 181–255.ed. Cook, N.S. pp [Google Scholar]
- CRIDDLE D.N., MOURA R.S. Inhibitory effect of Rb+ on the responses to levcromakalim and P1060 in the isolated human myometrium. Eur. J. Pharmacol. 1995;272:293–296. doi: 10.1016/0014-2999(94)00706-d. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. The pharmacology of ATP-sensitive potassium channels. Annu. Rev. Pharmacol. Toxicol. 1993;33:597–637. doi: 10.1146/annurev.pa.33.040193.003121. [DOI] [PubMed] [Google Scholar]
- FIELDING S., LAL H. Clonidine: new research in psychotropic drugs pharmacology. Med. Res. Rev. 1981;1:97–123. doi: 10.1002/med.2610010106. [DOI] [PubMed] [Google Scholar]
- FIELDING S., WILKER J., HYNES J., SZEWCZAK M., NOVIC W.J., LAL M. A comparison of clonidine with morphine for antinociceptive and antiwithdrawal actions. J. Pharmacol. Exp. Ther. 1978;270:899–905. [PubMed] [Google Scholar]
- GALEOTTI N., GHELARDINI C., PAPUCCI L., QUATTRONE A., CAPACCIOLI S., BARTOLINI A. An antisense oligonucleotide to the mouse Shaker-like potassium channel Kv1.1 gene prevents the antinociception induced by morphine and baclofen. J. Pharmacol. Exp. Ther. 1997;281:941–949. [PubMed] [Google Scholar]
- GHELARDINI C., GALEOTTI N., BARTOLINI A. Potassium channel modulators influence cognitive processes in mice. Br. J. Pharmacol. 1998;123:1079–1084. doi: 10.1038/sj.bjp.0701709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHELARDINI C., GALEOTTI N., PECORI VETTORI A., CAPACCI-OLI S., QUATTRONE A., BARTOLINI A. Effect of potassium channels modulation on mouse feeding behaviour. Eur. J. Pharmacol. 1997;329:1–8. doi: 10.1016/s0014-2999(97)10102-9. [DOI] [PubMed] [Google Scholar]
- GILES W., NAKAJIMA T., ONO K., SHIBATA E.F. Modulation of a delayed rectifier K+ current by isoprenaline in bull-frog atrial myocytes. J. Physiol. 1989;415:233–249. doi: 10.1113/jphysiol.1989.sp017720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALEY T.J., MCCORMICK W.G. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br. J. Pharmacol. Chemother. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALLIWELL J.V.K+ channels in the central nervous system Potassium Channels: Structure, Classification, Function and Therapeutic Potential 1990Chichester: Ellis Horwood Limited; 348–381.ed. Cook, N.S. pp [Google Scholar]
- HILLE B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- HOLZ G.G., RANE S.G., DUNLAP K. GTP-binding proteins mediate transmitter inhibition of voltage-dependent calcium channels. Nature. 1986;319:670–672. doi: 10.1038/319670a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOPKINS W.F., TEMPEL B.L. Members of a mouse subfamily of genes encoding voltage-gated potassium channel subunits form heteromultimers when coexpressed in Xenopus oocytes. Soc. Neurosci. Abstr. 1992;18:1093. [Google Scholar]
- LONGMAN S.D., HAMILTON T.C. Potassium channel activator drugs: mechanism of action, pharmacological properties, and therapeutic potential. Med. Res. Rev. 1992;12:73–148. doi: 10.1002/med.2610120202. [DOI] [PubMed] [Google Scholar]
- MCDONALD E., KOBILKA B.K., SCHEININ M. Gene targeting homing in on α2-adrenoceptor-subtype function. Trends Pharmacol. Sci. 1997;18:211–219. doi: 10.1016/s0165-6147(97)01063-8. [DOI] [PubMed] [Google Scholar]
- MIURA M., BELVISI M.G., STRETTON C.D., YACOUB M.H., BARNES P.J. Role of K+ channels in the modulation of cholinergic neural responses in guinea-pig and human airways. J. Physiol. 1992;455:1–15. doi: 10.1113/jphysiol.1992.sp019287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORITA K., NORTH R.A. Clonidine activates membrane potassium conductance in myenteric neurones. Br. J. Pharmacol. 1981;74:419–428. doi: 10.1111/j.1476-5381.1981.tb09987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'CALLAGHAN J.P., HOLTZMAN S.G. Quantification of the analgesic activity of narcotic antagonists by a modified hot-plate procedure. J. Pharmacol. Exp. Ther. 1975;192:497–505. [PubMed] [Google Scholar]
- OCAÑA M., BAEYENS J.M. Differential effect of K+ channel blockers on antinociception induced by α2-adrenoceptor, GABAB and κ-opioid receptor agonists. Br. J. Pharmacol. 1993;110:1049–1054. doi: 10.1111/j.1476-5381.1993.tb13919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OCAÑA M., BARRIOS M., BAEYENS J.M. Cromakalim differentially enhances antinociception induced by agonists of alpha2 adrenoceptors, γ-aminobutyric acidB, mu and kappa opioid receptors. J. Pharmacol. Exp. Ther. 1996;276:1136–1142. [PubMed] [Google Scholar]
- QUAYLE J.M., STANDEN N.B., STANFIELD P.R. The voltage-dependent block of ATP-sensitive potassium channels of frog skeletal muscle by caesium and barium ions. J. Physiol. 1988;405:677–697. doi: 10.1113/jphysiol.1988.sp017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAFFA R.B., MARTINEZ R.P. The glibenclamide shift of centrally-acting antinociceptive agents in mice. Brain Res. 1995;677:277–282. doi: 10.1016/0006-8993(95)00164-l. [DOI] [PubMed] [Google Scholar]
- RUDY B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-BLASQUEZ P., GARZON P. Cholera toxin and pertussis toxin on opioid- and α2-mediated supraspinal analgesia in mice. Life Sci. 1991;48:1721–1727. doi: 10.1016/0024-3205(91)90208-s. [DOI] [PubMed] [Google Scholar]
- SKINGLE M., HAYES A.G., TYERS M.B. Antinociceptive activity of clonidine in the mouse, rat and dog. Life Sci. 1982;31:1123–1132. doi: 10.1016/0024-3205(82)90086-8. [DOI] [PubMed] [Google Scholar]
- SOLIVEN B., NELSON D.J. Beta adrenergic modulation of K+ current in human T lymphocytes. J. Membr. Biol. 1990;117:263–274. doi: 10.1007/BF01868456. [DOI] [PubMed] [Google Scholar]
- STRETTON D., MIURA M., BELVISI M.G., BARNES P.J. Calcium-activated potassium channels mediate prejunctional inhibition of peripheral sensory nerves. Proc. Natl. Acad. Sci. U.S.A. 1992;89:1325–1329. doi: 10.1073/pnas.89.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TATSUMI H., COSTA M., SCHIMERLIK M., NORTH R.A. Potassium conductance increased by noradrenaline, opioids, somatostatin, and G-proteins: whole-cell recording from guinea-pig submucous neurons. J. Neurosci. 1990;10:1675–1682. doi: 10.1523/JNEUROSCI.10-05-01675.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAUGHT J., PELLEY K., COSTA L.G., SETHER P., ENNA S.J. A comparison of the antinociceptive responses to GABA-receptor agonists THIP and baclofen. Neuropharmacology. 1985;24:211–216. doi: 10.1016/0028-3908(85)90076-0. [DOI] [PubMed] [Google Scholar]
- WANG H., KUNKEL D.D., SCHWARTZKROIN P.A., TEMPEL B.L. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J. Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WELCH S.P., DUNLOW L.D. Antinociceptive activity of intrathecally administered potassium channel openers and opioid agonists: a common mechanism of action. J. Pharmacol. Exp. Ther. 1993;267:390–399. [PubMed] [Google Scholar]
- WILLIAMS J.T., NORTH R.A. Catecholamine inhibition of calcium action potentials in rat locus coeruleus neurones. Neuroscience. 1985;14:103–109. doi: 10.1016/0306-4522(85)90167-8. [DOI] [PubMed] [Google Scholar]
- WILLIAMS J.T., NORTH R.A., TOKIMASA T. Inward rectification of resting and opiate-activated potassium currents in rat locus coeruleus neurones. J. Neurosci. 1988;8:4299–4306. doi: 10.1523/JNEUROSCI.08-11-04299.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]