

Abstract
Hypercholesterolaemia often occurs in patients with type 2 diabetes, who therefore encounter administration of HMG-CoA reductase inhibitors. Alteration of pancreatic β-cell function leading to an impaired insulin secretory response to glucose plays a crucial role in the pathogenesis of type 2 diabetes. Therefore, it is important to examine the effects of HMG-CoA reductase inhibitors on β-cell function.
Cytosolic Ca2+ concentration ([Ca2+]i) plays a central role in the regulation of β-cell function. The present study examined the effects of HMG-CoA reductase inhibitors on the glucose-induced [Ca2+]i signalling and insulin secretion in rat islet β-cells.
Simvastatin, a lipophilic HMG-CoA reductase inhibitor, at 0.1–3 μg ml−1 concentration-dependently inhibited the first phase increase and oscillation of [Ca2+]i induced by 8.3 mM glucose in single β-cells. The less lipophilic inhibitor, simvastatin-acid, inhibited the first phase [Ca2+]i increase but was two orders of magnitude less potent. The hydrophilic inhibitor, pravastatin (100 μg ml−1), was without effect on [Ca2+]i.
Simvastatin (0.3 μg ml−1), more potently than simvastatin-acid (30 μg ml−1), inhibited glucose-induced insulin secretion from islets, whereas pravastatin (100 μg ml−1) had no effect.
Whole-cell patch clamp recordings demonstrated a reversible inhibition of the β-cell L-type Ca2+ channels by simvastatin, but not by pravastatin. Simvastatin also inhibited the [Ca2+]i increases by L-arginine and KCl, agents that act via opening of L-type Ca2+ channels.
In conclusion, lipophilic HMG-CoA reductase inhibitors can inhibit glucose-induced [Ca2+]i signalling and insulin secretion by blocking L-type Ca2+ channels in β-cells, and their inhibitory potencies parallel their lipophilicities. Precaution should be paid to these findings when HMG-CoA reductase inhibitors are used clinically, particularly in patients with type 2 diabetes.
Keywords: HMG-CoA reductase inhibitor, simvastatin, pravastatin, lipophilicity, islet, β-cell, cytosolic Ca2+, L-type Ca2+ channel, insulin secretion, diabetes
Introduction
The 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors are specific, competitive inhibitors of the rate-limiting enzyme in cholesterol biosynthesis (Alberts, 1988). Extended clinical studies on the long-term (2–5.3 years) administration of HMG-CoA reductase inhibitors have demonstrated that these drugs were effective in lowering plasma cholesterol levels and reducing the incidence of cardiovascular disease (Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995; Byington et al., 1995; Sacks et al., 1996). Consequently, HMG-CoA reductase inhibitors have been widely used for the treatment of patients with hypercholesterolaemia (Hoeg, 1990). Hypercholesterolaemia often occurs in diabetic patients and atherosclerotic cardiovascular disease is one of the major causes of death in diabetic patients. Therefore, diabetic patients are often administered HMG-CoA reductase inhibitors. Alteration of pancreatic β-cell function leading to an impaired insulin secretory response to glucose is thought to play a crucial role in the pathogenesis and progression of type 2 diabetes (non-insulin-dependent diabetes mellitus) (Perley & Kipnis, 1967; Kosaka et al., 1977; Pfeifer et al., 1981; O'Meary & Polonsky, 1994; Porte & Kahn, 1995). Therefore, it is of great importance to investigate the possible inhibitory effects of HMG-CoA reductase inhibitors on β-cell functions. It is well established that the cytosolic Ca2+ concentration ([Ca2+]i) plays a central role in the regulation of β-cell functions, including insulin secretion (Wollheim & Sharp, 1981; Gilon et al., 1993; Ammala et al., 1993; Yada et al., 1994; Henquin, 1994). It has recently been shown that glucose-induced oscillations in [Ca2+]i in a β-cell line, MIN6 cells, are linked to a long-lasting increase in mitochondrial Ca2+ (Nakazaki et al., 1998), a factor which is considered to be involved in the maintenance of the β-cell metabolism (Nakazaki et al., 1998; Kennedy et al., 1996). Regulation of gene expression by [Ca2+]i oscillations has recently been demonstrated (Dolmetsch et al., 1998; Li et al., 1998). Therefore, the present study examined the effects of HMG-CoA reductase inhibitors on the glucose-induced [Ca2+]i signalling, including oscillations, and insulin secretion in islet β-cells.
There are two forms of HMG-CoA reductase inhibitors: a hydrophilic inhibitor, pravastatin, and lipophilic inhibitors such as simvastatin and lovastatin. Simvastatin is lipophilic because of its lactone ring. The open acid form of simvastatin (simvastatin-acid), an in vivo active metabolite, is partially hydrophilic (Serajuddin et al., 1991). Pravastatin, occurring in the acid form, is hydrophilic (Serajuddin et al., 1991). Pravastatin and both forms of simvastatin are equally active as HMG-CoA reductase inhibitors for lowering serum cholesterol levels. HMG-CoA reductase inhibitors have been reported to cause myotonia and acute rhabdomyolysis clinically and experimentally, though the frequency is very low (Tobert, 1988; 1995; Hurminghake, 1992; Nakahara et al., 1992). The action of HMG-CoA reductase inhibitors to induce rapid cell damage in L6 myocytes has been shown to be closely related to the lipophilicity of these drugs (Nakahara et al., 1994). In the present study, therefore, the comparative effects of simvastatin, simvastatin-acid, and pravastatin on glucose-stimulated β-cells were examined.
Methods
Preparation of islets and single β-cells and selection of β-cells
Islets and single β-cells were prepared as previously reported (Yada et al., 1992; 1997). Briefly, islets of Langerhans were isolated from Wistar rats aged 8–12 weeks by collagenase digestion. Animals were anaesthetized with an intraperitoneal injection of pentobarbitone at 80 mg kg−1. The abdomen was opened, and collagenase (3 mg ml−1) dissolved in 5 mM Ca2+-containing Krebs-Ringer bicarbonate buffer (KRB) was injected into the common bile duct at the distal end after ligation of the duct proximal to the pancreas. The rats were killed by cervical dislocation. The pancreas was dissected out and incubated at 37°C for 17 min. Islets were hand-collected and either used for insulin release experiments or dispersed into single cells in Ca2+-free KRB. The single cells were plated on coverslips and maintained in short-term culture for up to 3 days in Eagle's minimal essential medium containing 5.6 mM glucose supplemented with 10% foetal bovine serum (FBS), 100 μg ml−1 streptomycin and 100 μU ml−1 penicillin at 37°C in a 95% air and 5% CO2 atmosphere. The cells during this culture period responded to the test agents in a consistent manner. β-cells were selected according to their morphological and physiological criteria as reported previously (Yada et al., 1995).
Solutions
KRB was composed of (in mM): NaCl 121.7, KCl 4.4, KH2PO4 1.2, CaCl2 2.0, MgSO4 1.2, NaHCO3 5.0 and 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES; at pH 7.4) 10 supplemented with 0.1% bovine serum albumin. The standard pipette solution for whole-cell patch clamp recording of Ca2+ channel current was composed of (in mM): aspartate 75, tetraethylammonium (TEA) 30, HEPES 11, EGTA 11, MgCl2 3, CaCl2 1, ATP-Na2 3 (Boehringer Mannheim, Indianapolis, IN) and GTP 0.1 (at pH 7.2) (Boehringer). External solution used for superfusion to establish whole-cell clamp was composed of (in mM): Tris-HCl 100, TEA-Cl 30, HEPES 10 and CaCl2 (at pH 7.3) 2.
Measurements of [Ca2+]i
[Ca2+]i was measured by dual-wavelength fura-2 microfluorometry combined with imaging as previously reported (Yada et al., 1992; 1994). Briefly, cells on coverslips were incubated with 1 μM fura-2 acetoxymethylester for 30 min at 37°C in KRB containing 2.8 mM glucose. Cells were then mounted in a chamber and superfused with KRB at a rate of 1 ml min−1 at 37°C. Cells were excited at 340 and 380 nm alternately every 2.5 s, emission signals at 510 nm were detected with an intensified charge-coupled device (ICDD) camera, and ratio images were produced by an Argus-50 system (Hamamatsu Photonics, Hamamatsu, Japan). Ratio values were converted to [Ca2+]i according to calibration curves (Yada et al., 1992).
Electrophysiological recordings
Voltage-dependent Ca2+ channel currents were recorded in single β-cells, under superfusion conditions at 1 ml min−1 at 30°C, using the standard whole-cell configuration of the patch-clamp method (Hamill et al., 1981). Pipettes were pulled from borosilicate glass (Sutter Instruments Co., Novata, CA, U.S.A.), coated with Sylgard 184 (Dow Corning, Midland, MI, U.S.A.) near their tips and fire polished. Their resistance was 2–7 MΩ when filled with the standard pipette solution. Membrane currents were measured using an Axopatch-200B amplifier and the software, pCLAMP 6 (Axon Instruments, Inc., Foster City, CA, U.S.A.). The potential was held at −70 mV and depolarizing voltage pulses to 0 mV with a duration of 100 ms were applied at a frequency of 0.2 Hz. The data were filtered at 5 kHz, digitized at 10 kHz, stored in a computer (IBM; Tokyo, Japan), and analysed with the Clampfit program. The reference potential was the zero-current potential of the pipette obtained immediately after correction for the junction potential and before establishment of the seal.
Measurement of insulin secretion
Measurements of insulin release were carried out as previously described (Yada et al., 1994; 1997). Briefly, groups of five islets, isolated from Wistar rats 8–12 weeks of age, were first incubated for 30 min in KRB containing 2.8 mM glucose for stabilization. Islets were then incubated at 37°C for 30 min in 1 ml of KRB. Insulin concentration was determined by enzyme immunoassay using a kit (Morinaga, Yokohama, Japan).
Materials
Simvastatin, simvastatin-Na (simvastatin-acid) and pravastatin were synthesized and kindly provided by Sankyo Co. (Tokyo, Japan). Simvastatin was dissolved in 100% ethanol, while simvastatin-acid and pravastatin were in distilled water. For experiments, small aliquots of the stock solutions of HMG-CoA reductase inhibitors were added to KRB. The final concentration of ethanol was less than 0.1%, at which it had no effect on [Ca2+]i in β-cells. Fura-2 and fura-2 acetoxymethylester were obtained from Dojin Chemical (Kumamoto, Japan). FBS was obtained from Life Technologies Inc. (New York, NY, U.S.A.). Mevalonic acid lactone was obtained from Nacalai-Tesque Chemical Co. (Kyoto, Japan). All other chemicals were from Sigma Chemical Co. (St. Louis, MO, U.S.A.) or Nacalai-Tesque.
Statistical analysis
The calculated values were expressed as the mean±s.e.mean (n=number of observations). The statistical analysis was carried out with the Student's t-test. Differences were considered statistically significant when P<0.05.
Results
Temporal profile of the glucose-induced increase in [Ca2+]i in single rat pancreatic β-cells
[Ca2+]i in single β-cells at a basal glucose concentration of 2.8 mM ranged between 30 and 150 nM (76.8±3.7 nM for 58 cells). An elevation of the glucose concentration from 2.8 mM to 8.3 or 16.7 mM increased [Ca2+]i in β-cells in a biphasic manner: an initial peak at around 400–500 nM (first phase) (Figure 1) followed by a moderate elevation at around 100–200 nM which was occasionally superimposed with an oscillation of [Ca2+]i (second phase), confirming previous reports (Yada et al., 1992; 1994).
Figure 1.
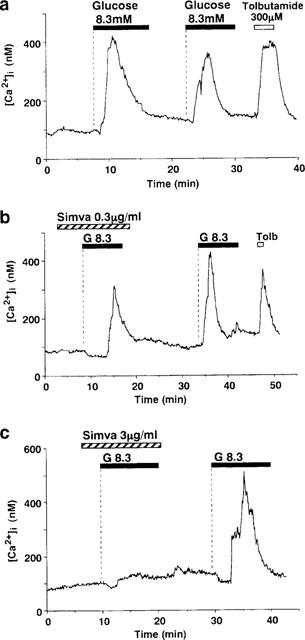
Glucose-induced first phase [Ca2+]i increases and their inhibition by simvastatin in single β-cells. (a) A rise in glucose concentration from 2.8 to 8.3 M, under superfusion conditions, evoked the first phase [Ca2+]i increase in single rat pancreatic β-cells. The majority of β-cells responded to two sequential challenges by 8.3 mM glucose (G8.3) with increases in [Ca2+]i with a similar pattern. The amplitude of the first phase [Ca2+]i increase in response to the first challenge was either larger than or comparable to that in response to the second challenge. In the presence of simvastatin (Simva) at 0.3 μg ml−1 (b) and 3 μg ml−1 (c), the first phase [Ca2+]i increase was reduced and abolished, respectively. After washing out simvastatin, the first phase [Ca2+]i increase was restored in response to the second challenge with 8.3 mM glucose. Tolbutamide at 300 μM (Tolb) induced rapid increases in [Ca2+]i as seen in (a) and (b). The bars above the tracing indicate the periods of exposure to the agents specified. Dotted lines indicate beginning of exposure. The basal glucose concentration was 2.8 mM throughout measurements when not indicated otherwise. The results shown are representative of 14 cells in (a), six in (b) and ten in (c).
In the present study, the effects of HMG-CoA reductase inhibitors on the first phase [Ca2+]i increase were studied unless otherwise indicated. To evaluate the effects of the inhibitors at the single cell level, glucose stimulation was carried out twice: first in the presence and second in the absence of inhibitors. In control experiments in which β-cells were stimulated twice with 8.3 mM glucose, 24 out of 29 cells responded to the first stimulation and 20 cells to the second stimulation with increases in [Ca2+]i. In 22 out of these 24 responding cells (92%), the response to the first stimulation was either larger than or comparable to the response to the second stimulation (Figure 1a) with regard to the amplitude of the first phase [Ca2+]i increase. Therefore, if the experiments showed that the response to the first glucose stimulation obtained in the presence of HMG-CoA reductase inhibitors was consistently smaller than the response to the second stimulation obtained after washing out the inhibitors, the inhibition was evident at the single cell level. At the end of experiments, it was confirmed that the cells responded to 300 μM tolbutamide with large increases in [Ca2+]i, the property characteristic of normal β-cells (Fujitani et al., 1997).
Effects of simvastatin, simvastatin-acid and pravastatin on the glucose-induced first phase [Ca2+]i increase in single β-cells
In the presence of 0.3 μg ml−1 simvastatin, the peak of the first phase [Ca2+]i increase in response to 8.3 mM glucose was reduced (Figure 1b). Simvastatin at a higher concentration of 3 μg ml−1 strongly inhibited or even abolished the first phase [Ca2+]i increase (Figure 1c). After washing out simvastatin, the first phase [Ca2+]i increase in response to the second stimulation with 8.3 mM glucose was restored, indicating that the inhibition was reversible (Figure 1b and c).
The mean amplitude of the first phase [Ca2+]i increase, averaged for the single β-cells examined, was also reduced by simvastatin at 0.1–3 μg ml−1 (P<0.05 for 0.1 μg ml−1, P<0.02 for 0.3 μg ml−1, P<0.001 for 1 μg ml−1, and P<0.0001 for 3 μg ml−1 simvastatin) (Figure 2). Thus, simvastatin inhibits the first phase [Ca2+]i response to glucose in a concentration-dependent manner.
Figure 2.
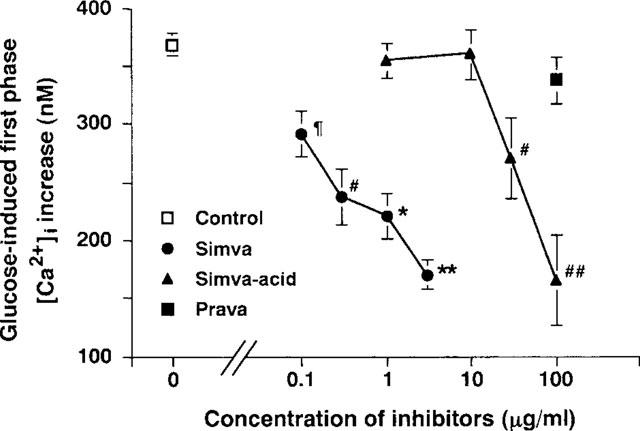
Concentration-response relationship for the inhibition of glucose-induced first phase [Ca2+]i increases by HMG-CoA reductase inhibitors in single β-cells. The mean amplitude of the first phase [Ca2+]i increase in response to 8.3 mM glucose was averaged and expressed as the mean±s.e.mean. The number of single β-cells examined was 127 for the control and 18–50 for each experimental condition. The mean amplitude of the first phase [Ca2+]i increase was reduced by simvastatin at 0.1–3 μg ml−1 and simvastatin-acid at 30–100 μg ml−1. }P<0.05 for 0.1 μg ml−1 simvastatin, #P<0.02 for 0.3 μg ml−1 simvastatin and 30 μg ml−1 simvastatin-acid, ##P<0.002 for 100 μg ml−1 simvastatin-acid, *P<0.001 for 1 μg ml−1 simvastatin, and **P<0.0001 for 3 μg ml−1 simvastatin vs the control.
Simvastatin-acid at 1 and 10 μg ml−1, concentrations at which simvastatin had inhibitory effects, was without effect on the first phase [Ca2+]i increase in response to 8.3 mM glucose. However, simvastatin-acid at 30 μg ml−1 mildly (Figure 3a) and at 100 μg ml−1 strongly (Figure 3b) inhibited the first phase [Ca2+]i increase in a reversible manner. The mean amplitude of the first phase [Ca2+]i increase was significantly reduced by 30 and 100 μg ml−1 simvastatin-acid (P<0.01 for 30 μg ml−1 and P<0.002 for 100 μg ml−1) (Figure 2). Thus, simvastatin-acid inhibits the first phase [Ca2+]i response to glucose in a concentration-dependent manner, but the inhibitory action is about two orders of magnitude less potent than simvastatin (Figure 2).
Figure 3.
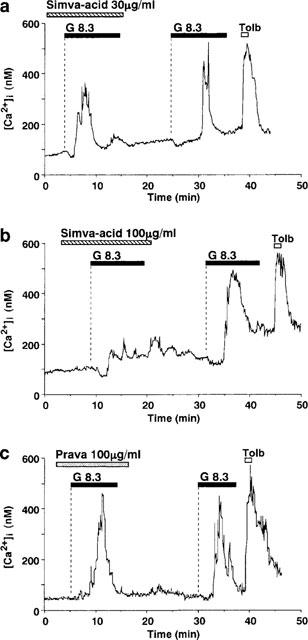
Simvastatin-acid, but not pravastatin, inhibits glucose-induced first phase [Ca2+]i increases in single β-cells. Simvastatin-acid (Simva-acid) at 30 μg ml−1 reduced (a) and at 100 μg ml−1 abolished (b) the first phase [Ca2+]i increase in response to 8.3 mM glucose (G8.3). After washing out simvastatin-acid, the first phase [Ca2+]i increase was restored in response to the second challenge with 8.3 mM glucose. (c) Pravastatin (Prava) at 100 μg ml−1 had no effects on the first phase [Ca2+]i increase in response to 8.3 mM glucose. The results shown are representative of eight cells in (a), six in (b) and 17 in (c).
Pravastatin in a concentration range up to 100 μg ml−1, when added to the superfusion solution, failed to affect the [Ca2+]i response to 8.3 mM glucose (Figures 2 and 3c). The results indicated that the hydrophilic HMG-CoA reductase inhibitor was without an acute effect on the β-cell response to glucose.
Effects of simvastatin and pravastatin on the glucose-induced [Ca2+]i oscillation in single β-cells
It has been suggested that [Ca2+]i oscillations in islet β-cells are causally related to the pulsatile secretion of insulin (Gilon et al., 1993; Hellman et al., 1992; Bergsten, 1995) which plays an important role in the physiological control of glucose metabolism (O'Meara & Polonsky, 1994; Matthews et al., 1983; O'Rahilly et al., 1988). Therefore, the effects of HMG-CoA reductase inhibitors on the [Ca2+]i oscillations were examined. Simvastatin at 0.3 μg ml−1 partially and at 3 μg ml−1 almost completely inhibited the [Ca2+]i oscillations induced by 8.3 M glucose, and they were either fully or partly reversible after washing out the inhibitor (Figure 4a). Simvastatin at 0.1 and 1 μg ml−1 also partially inhibited the [Ca2+]i oscillations (data not shown). In contrast, pravastatin in a concentration range up to 100 μg ml−1 was without any appreciable effect (Figure 4b). Among 14 cells that exhibited [Ca2+]i oscillations, only two cells (14%) continued to clearly oscillate during treatment with 1 μg ml−1 simvastatin for about 15 min, and 13 cells (93%) oscillated after washing out this drug, showing a strong and reversible inhibition (Figure 4c). In contrast, among 18 cells that exhibited [Ca2+]i oscillations, 17 (94%) and 15 cells (88%) exhibited oscillations during treatment with and after washing out 100 μg ml−1 pravastatin, respectively, in a manner similar to that before the drug treatment.
Figure 4.
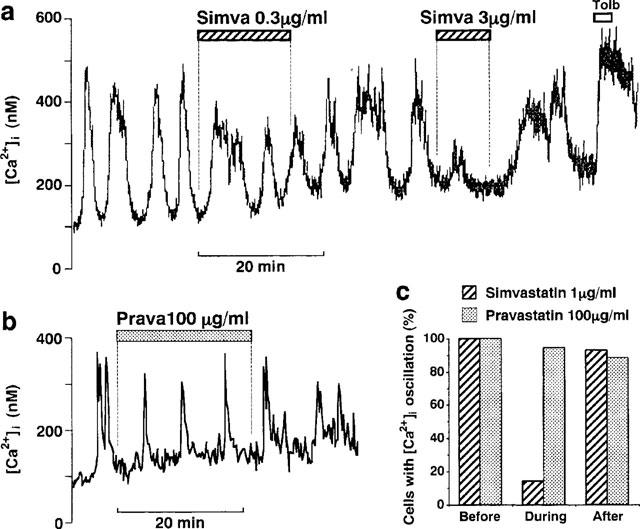
Simvastatin, but not pravastatin, inhibits glucose-induced [Ca2+]i oscillations in single β-cells. (a) Simvastatin (Simva) at 0.3 μg ml−1 partially and at 3 μg ml−1 almost completely inhibited [Ca2+]i oscillations induced by 8.3 mM glucose in a reversible manner. (b) Pravastatin (Prava) at 100 μg ml−1 had no effects on [Ca2+]i oscillations. The results shown are representative of five cells in (a) and 15 in (b). (c) Effects of simvastatin and pravastatin on the incidence of [Ca2+]i oscillations. Among the 14 cells exhibiting [Ca2+]i oscillations in response to 8.3 mM glucose, two cells oscillated during treatment with 1 μg ml−1 simvastatin for about 15 min, and 13 cells oscillated after washing out this drug. In contrast, among the 18 oscillating cells, 17 and 15 cells oscillated during treatment with and after washing out 100 μg ml−1 pravastatin, respectively.
Effects of simvastatin and pravastatin on glucose-induced insulin secretion from islets
Insulin release from rat islets, under static incubation conditions, was stimulated with 8.3 mM glucose. The stimulated insulin release was significantly reduced in the presence of 0.3 μg ml−1 simvastatin (P<0.05) (Figure 5). Simvastatin-acid at 30 μg ml−1, the concentration at which it moderately attenuated the first phase [Ca2+]i response to glucose, mildly attenuated insulin release, though statistically not significant. In contrast pravastatin at 100 μg ml−1 failed to affect insulin release.
Figure 5.
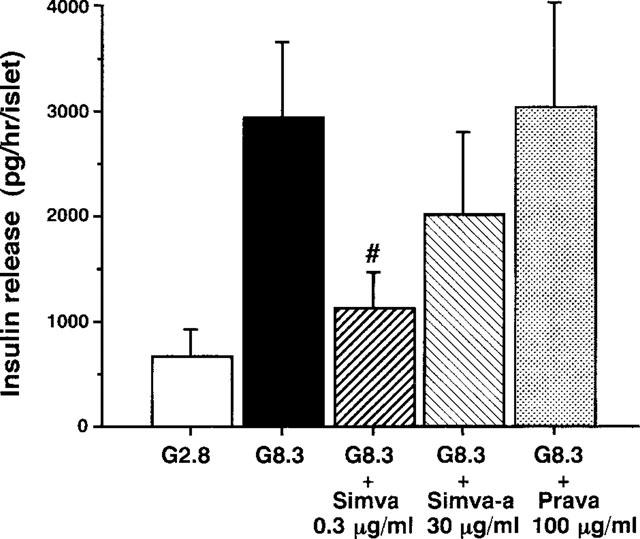
Simvastatin and simvastatin-acid, but not pravastatin, inhibit glucose-induced insulin secretion in islets. Insulin release from islets stimulated with 8.3 mM glucose, under static incubation conditions, was inhibited in the presence of 0.3 μg ml−1 simvastatin (Simva). Glucose-stimulated insulin release was mildly reduced by 30 μg ml−1 simvastatin-acid (Simva-a), but not significantly. Pravastatin (Prava) at 100 μg ml−1 was without effect on glucose-stimulated insulin release. The results are expressed as the mean±s.e.mean of six experiments. #, Significant difference (P<0.05) vs the 8.3 mM glucose (G8.3) group and the pravastatin group.
Effects of simvastatin and pravastatin on L-type Ca2+ channels in single β-cells
It has been shown that the glucose-induced increase in [Ca2+]i in β-cells is due mainly, if not solely, to the Ca2+ influx through the L-type Ca2+ channels in the β-cell plasma membrane (Henquin, 1994; Ashcroft & Rorsman, 1989). Therefore, we investigated the effect of HMG-CoA reductase inhibitors on the Ca2+ currents of single pancreatic β-cells with the whole-cell voltage-clamp configuration. In the control external solution containing 2.8 mM glucose, a depolarizing pulse from a holding potential of −70 to 0 mV evoked a long-lasting inward current with an amplitude of about −70 pA (Figure 6Aa,B and C), the properties of which fit well with those of the current passing through the L-type Ca2+ channels in β-cells (Ashcroft & Rorsman, 1989). Upon exposure to 3 μg ml−1 simvastatin, the current decreased rapidly and markedly (Figure 6Ab and B), and upon washing out simvastatin the current was partially restored (Figure 6Ac and B). Current-voltage relationships before and during exposure of β-cells to 3 μg ml−1 simvastatin are shown in Figure 6C. Simvastatin reduced the Ca2+ current amplitude without shifting the current-voltage relationship along the voltage axis. In contrast, pravastatin at 100 μg ml−1 was without effect on the Ca2+ current (data not shown).
Figure 6.
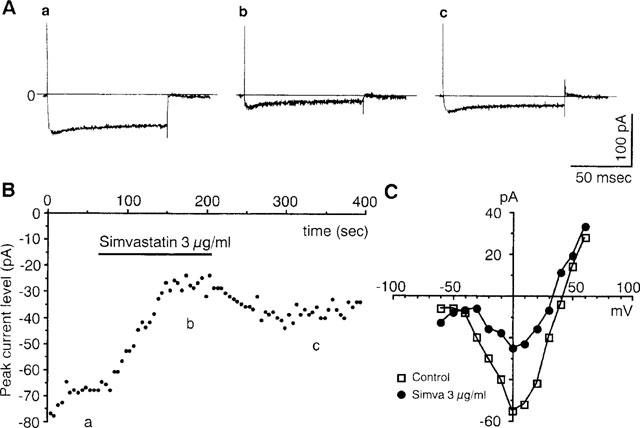
Simvastatin inhibits Ca2+ currents in single β-cells. (A) Whole-cell Ca2+ currents in a single β-cell depolarized to 0 mV from the holding potential of −70 mV before (a), during (b) and after exposure to 3 μg ml−1 simvastatin (c) under superfusion conditions. (B) Recording of the temporal change of the whole-cell Ca2+ currents. a, b and c, specify the time points at which the current traces in Aa, Ab and Ac were taken. (C) The current-voltage relationship of the peak Ca2+ current amplitude in the control and in the presence of 3 μg ml−1 simvastatin. The results shown are representative of five cells in (A) and (B) and three in (C).
Inhibition of arginine- and KCl-induced [Ca2+]i increases in single cells by simvastatin
The Ca2+ influx through the L-type Ca2+ channels is thought to be the principal mechanism not only for the glucose- but also for the L-arginine- and KCl-induced release of insulin (Ashcroft & Rorsman, 1989). Arginine, a cationic amino acid, at relatively high concentrations (1–20 mM), directly depolarizes the β-cell plasma membrane when it is transported across it, resulting in a voltage-dependent activation of L-type Ca2+ channels and an increase in [Ca2+]i (Henquin & Meissner, 1981; Blachier et al., 1989; Yada, 1994; Smith et al., 1997). KCl, via a direct depolarization, also induces an activation of L-type Ca2+ channels and a sustained increase in [Ca2+]i (Yaekura et al., 1996). The result that simvastatin inhibited the L-type Ca2+ channels urged us to examine whether this drug could inhibit the increases in [Ca2+]i induced by arginine and KCl. Sequential administration of 10 mM arginine in brief pulses evoked a repetitive increase in [Ca2+]i, and in the presence of simvastatin the [Ca2+]i response to arginine was markedly inhibited in a reversible manner (Figure 7a). A sustained increase in [Ca2+]i was induced by 25 mM KCl and it was markedly reduced by simvastatin (Figure 7b). The inhibition started at about 20 s after administration of the drug.
Figure 7.
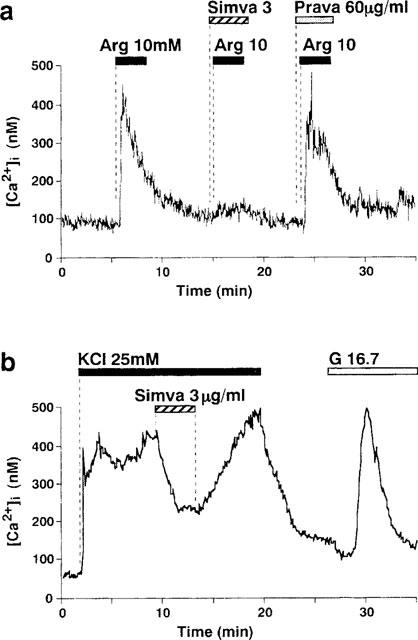
Simvastatin (Simva) inhibits [Ca2+]i increases in response to L-arginine (Arg) and KCl in single β-cells. (a) An increase in [Ca2+]i induced by 10 mM L-arginine was inhibited in the presence of 3 μg ml−1 simvastatin, but not 60 μg ml−1 pravastatin (Prava). (b) A sustained increase in [Ca2+]i induced by 25 mM KCl was almost instantaneously inhibited by the addition of 3 μg ml−1 simvastatin, and it was restored upon washing out the drug. The glucose concentration was 8.3 mM in (a) and 2.8 mM in (b). G16.7: change to 16.7 mM glucose. The results shown are representative of five cells in (a) and 14 in (b).
Discussion
The novel findings of our study were: (1) Among the HMG-CoA reductase inhibitors, simvastatin was about two orders of magnitude more potent than simvastatin-acid in inhibiting glucose-induced [Ca2+]i signalling in rat islet β-cells, whereas pravastatin was without effect; (2) Simvastatin inhibited glucose-induced insulin secretion from islets more potently than simvastatin-acid, whereas pravastatin was ineffective; (3) Simvastatin, but not pravastatin, inhibited L-type Ca2+ channels in β-cells; and (4) Simvastatin also inhibited the [Ca2+]i increases induced by L-arginine and KCl in β-cells.
Only the HMG-CoA reductase inhibitors that attenuated the glucose-induced first phase [Ca2+]i increases and [Ca2+]i oscillations, i.e., simvastatin and simvastatin-acid, attenuated the glucose-induced insulin secretion, whereas pravastastin affected neither of these parameters. The rank order of potency for the inhibition of the [Ca2+]i increase (simvastatin>simvastatin-acid≫pravastatin) was identical to that for the inhibition of insulin secretion. Simvastatin at 0.3 μg ml−1 partially inhibited both the [Ca2+]i increase and insulin secretion, and the extent of inhibition was somewhat larger for insulin secretion, which may fit with the longer period of simvastatin treatment for insulin secretion (30 min) than for [Ca2+]i (10–15 min). The increase in [Ca2+]i is the principal signal that triggers insulin secretion (Wollheim & Sharp, 1981; Gilon et al., 1993; Ammala et al., 1993; Yada et al., 1994; Henquin, 1994). These observations taken together suggest that the inhibition of the [Ca2+]i increase accounts for the inhibition of insulin secretion.
The whole-cell patch-clamp study clearly demonstrated that simvastatin inhibits L-type Ca2+ channels in β-cells. This notion was further strengthened by the finding that simvastatin inhibited the [Ca2+]i increases induced by arginine and KCl, the insulin secretagogues that activate L-type Ca2+ channels and, consequently, increase [Ca2+]i (Henquin & Meissner, 1981; Blachier et al., 1989; Yada, 1994; Smith et al., 1997; Yaekura et al., 1996). The temporal profiles of the inhibition of Ca2+ currents by simvastatin and its recovery upon washout of the drug were similar to those of the inhibition of [Ca2+]i increases (Figure 6B vs Figure 7b). Simvastatin inhibited both L-type Ca2+ channels and [Ca2+]i increases while pravastatin had no effect on either; therefore, the two activities changed in parallel. The Ca2+ influx through L-type Ca2+ channels is thought to be the major route of glucose-induced [Ca2+]i increases in β-cells (Henquin, 1994; Ashcroft & Rorsman, 1989). Accordingly, our results indicate that the inhibition of L-type Ca2+ channels is the major, if not the only, cause for the inhibition of [Ca2+]i increases. Alternatively, it is possible that the inhibitory action of HMG-CoA reductase inhibitors is caused by the reduced level of mevalonate, that results from the inhibition of HMG-CoA reductase which catalyzes the formation of mevalonate from HMG-CoA (Alberts, 1988). However, in the presence of 100 μM mevalonic acid lactone for both preincubation (30 min) and experimental periods, simvastatin inhibited the first phase [Ca2+]i response to glucose in a manner similar to the control experiments without mevalonate (data not shown). The result that inhibition of Ca2+ currents and [Ca2+]i increases started as soon as 10–20 s after administration of simvastatin suggests that inhibition of L-type Ca2+ channels and [Ca2+]i increases is not mediated by the metabolic change that reduces mevalonate levels, but is due to a more direct interaction of the drug with the plasma membrane L-type Ca2+ channels.
Simvastatin at 0.1–3 μg ml−1, administered under superfusion conditions, concentration-dependently attenuated the glucose-induced first phase [Ca2+]i increase and [Ca2+]i oscillation, in a fully or partially reversible manner. The less lipophilic simvastatin-acid in this concentration range was without effect, but at the higher doses of 30 and 100 μg ml−1, it attenuated the first phase [Ca2+]i increases. Thus, simvastatin-acid was about two orders of magnitude less potent than simvastatin. The hydrophilic pravastatin at concentrations up to 100 μM affected neither the first phase [Ca2+]i increase nor the [Ca2+]i oscillation induced by glucose. The overwhelming potency of simvastatin over pravastatin has also previously been reported for worsening of segment shortening in the reperfused myocardium in dogs (Ichihara et al., 1993) and for inhibition of proliferation of human smooth muscle cells (Negre-Arninou et al., 1997). In our study and these reports, the inhibitory potencies of the HMG-CoA reductase inhibitors appeared to parallel their lipophilicities. It was shown that the partition coefficient between water and n-octanol, an indicator of lipophilicity, is 25118 for simvastatin, 75.8 for simvastatin-acid, and 0.34 for pravastatin (Serajuddin et al., 1991), yielding a relative lipophilic ratio of about 70000 : 2000 : 1. This ratio fits well with the relative potency of the HMG-CoA reductase inhibitor for the inhibition of [Ca2+]i increases and insulin secretion observed in the present study. Therefore, it is likely that the highly lipophilic simvastatin has a strong affinity for the cell membrane and, consequently, may also have an easy access to the intracellular space, and that these properties are related to the ability of simvastatin to effectively inhibit Ca2+ channels in the β-cell plasma membrane and, thereby, inhibit [Ca2+]i and insulin responses to glucose. In contrast, hydrophilic pravastatin has a limited rate of access to the plasma membrane and the intracellular space, which likely accounts for the lack of immediate effects of this drug on Ca2+ channels, [Ca2+]i increases and insulin secretion. In support of this idea, in β-cells which had been cultured for 1 day with a high concentration of pravastatin (100 μg ml−1), the amplitude of the first phase [Ca2+]i response to 8.3 mM glucose was mildly attenuated in comparison to that in the paired β-cells cultured under control conditions (T. Yada, unpublished data).
It has been reported that in healthy volunteers, after oral administration of simvastatin, the serum concentration of the sum of simvastatin and its active metabolites reaches a level around 0.01 μg ml−1 (Koga et al., 1995). In our results, the concentration at which simvastatin exhibited an immediate and significant inhibition of glucose-induced [Ca2+]i increase was 0.1 μg ml−1 and, therefore, one order of magnitude higher. However, since HMG-CoA reductase inhibitors are often used for a long period of several years or more (Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995; Byington et al., 1995; Sacks et al., 1996), the inhibitory effect might be evoked by simvastatin at lower concentrations when used chronically. Hypercholesterolaemia often occurs in diabetic patients. Since functional alterations of β-cells are known to be associated with type 2 diabetes (O'Meara & Polonsky, 1994; Porte & Kahn, 1995), the possibility cannot be excluded that β-cells in some type 2 diabetic patients are more susceptible to the inhibition by lipophilic HMG-CoA reductase inhibitors. In type 2 diabetic patients with a reduced insulin secretory ability (Perley & Kipnis, 1967; Kosaka et al., 1977; Pfeifer et al., 1981), a further attenuation of the β-cell activity by simvastatin, even to a mild extent could have a pathophysiologically significant influence on glucose metabolism. It is also possible that the serum simvastatin reaches higher levels than 0.01 μg ml−1 in the patients with reduced rates of drug metabolism and/or extrusion due to more or less impaired liver and/or kidney functions, which is the case in some patients with type 2 diabetes. A recent report (Neuvonen et al., 1998) showed that when simvastatin was combined with the CYP3A4 inhibitor itraconazole, the serum concentration of simvastatin and simvastatin-acid increased one order of magnitude higher and exceeded 0.1 μg ml−1, the level at which the inhibition of β-cells were observed in the present study. It has also been reported that serum concentrations of fluvastatin, a HMG-CoA reductase inhibitor which has a similar lipophilicity to simvastatin-acid, reach a level as high as 0.25 μg ml−1 in healthy subjects (Appel et al., 1995).
The effect of HMG-CoA reductase inhibitors on glucose metabolism and blood glucose level is an issue of great clinical importance. Impairment in insulin secretion and/or insulin action (insulin sensitivity) are considered to be the two major causes for the impaired glucose tolerance and type 2 diabetes. Regarding the effects of HMG-CoA reductase inhibitors on insulin action, it has been shown that lovastatin, a lipophilic HMG-CoA reductase inhibitor, disrupts the early events of insulin signalling, which include tyrosine phosphorylation of the insulin receptor β subunit and MAP kinase, association of the p85 subunit of PI-3 kinase with insulin receptor substrate-1, and activation of MAP kinase (Xu et al., 1996; McGuire et al., 1994). It has been reported that administration of simvastatin (Ohrvall et al., 1995; Nielsen et al., 1993) or pravastatin (Sheu et al., 1994) in patients with hypercholesterolaemia does not improve or somewhat worsens insulin resistance despite their marked LDL-cholesterol-lowering effect. Regarding the effects on blood glucose, it has been shown that plasma glucose concentrations from 08.00–16.00 h in response to breakfast and lunch were moderately but significantly higher after pravastatin treatment (Sheu et al., 1994). A study in patients with type 2 diabetes and hyperlipidaemia has demonstrated that simvastatin treatment significantly elevated plasma glucose concentrations both at fasting and during an intravenous glucose tolerance test (IVGTT), which was accompanied by an increment of fasting insulin concentrations but not of insulin responses to IVGTT (Ohrvall et al., 1995). This result may imply that the plasma insulin concentration increases to compensate for the impairment of insulin sensitivity but not to a sufficient extent to maintain blood glucose at a low level, thereby resulting in hyperglycaemia (Ohrvall et al., 1995). Although the number of reports are limited, these in vitro and in vivo studies indicate reduction of insulin sensitivity by treatment with HMG-CoA reductase inhibitors. In addition, the present study revealed a novel action of simvastatin to directly inhibit the β-cell insulin secretory response to glucose. Thus, it appears that lipophilic HMG-CoA reductase inhibitors are able to impair secretion and may also affect insulin sensitivity and, consequently, could accelerate the pathogenesis and progression of type 2 diabetes.
In conclusion, our results reveal that simvastatin, the lipophilic HMG-CoA reductase inhibitor, has an ability to inhibit glucose-induced insulin secretion by inhibiting L-type Ca2+ channel and [Ca2+]i signalling in pancreatic β-cells. Caution should be taken with this finding when HMG-CoA reductase inhibitors are used clinically, especially in patients with type 2 diabetes. Since L-type Ca2+ channels and [Ca2+]i signalling regulate a variety of β-cell functions including insulin biosynthesis, possible effects of HMG-CoA reductase inhibitors on the β-cell functions other than secretion also need to be investigated.
Acknowledgments
This work was supported by grants from the Ministry of Education, Science, Sports and Culture of Japan (to T.Y. and M.K.).
Abbreviations
- [Ca2+]i
cytosolic calcium concentration, FBS, foetal bovine serum
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- IVGTT
intravenous glucose tolerance test
- KRB
Krebs-Ringer bicarbonate buffer
- TEA
tetraethylammonium
References
- ALBERTS A.W. Discovery, biochemistry and biology of lovastatin. Am. J. Cardiol. 1988;62:10J–15J. doi: 10.1016/0002-9149(88)90002-1. [DOI] [PubMed] [Google Scholar]
- AMMALA C., ELIASSON L., BOKVIST K., LARSSON O., ASHCROFT F.M., RORSMAN P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J. Physiol. 1993;472:665–688. doi: 10.1113/jphysiol.1993.sp019966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APPEL S., RUFENACHT T., KALAFSKY G., TETZLOFF W., KALLAY Z., HITZENBERGER G., KUTZ K. Lack of interaction between fluvastatin and oral hypoglycemic agents in healthy subjects and in patients with non-insulin-dependent diabetes mellitus. Am. J. Cardiol. 1995;76:29A–32A. doi: 10.1016/s0002-9149(05)80012-8. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., RORSMAN P. Electrophysiology of the pancreatic β-cell. Prog. Biophys. Mol. Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- BERGSTEN P. Slow and fast oscillations of cytoplasmic Ca2+ in pancreatic islets correspond to pulsatile insulin release. Am. J. Physiol. 1995;268:E282–E287. doi: 10.1152/ajpendo.1995.268.2.E282. [DOI] [PubMed] [Google Scholar]
- BLACHIER F., MOURTADA A., SENER A., MALAISSE W.J. Stimulus-secretion coupling of arginine-induced insulin release. Uptake of metabolized and nonmetabolized cationic amino acids by pancreatic islets. Endocrinology. 1989;124:134–141. doi: 10.1210/endo-124-1-134. [DOI] [PubMed] [Google Scholar]
- BYINGTON R.P., JUKEMA J.W., SALONEN J.T., PITT B., BRUSCNKE A.V., HOEN H., FURBERG C.D., MANCINI G.B. Reduction in cardiovascular events during pravastatin therapy. Pooled analysis of clinical events of the Pravastatin Atherosclerosis Intervention Program. Circulation. 1995;92:2419–2425. doi: 10.1161/01.cir.92.9.2419. [DOI] [PubMed] [Google Scholar]
- DOLMETSCH R.E., XU K., LEWIS R.S. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- FUJITANI S., OKAZAKI K., YADA T. The ability of a new hypoglemic agent, A-4166, compard to sulphonylureas, to increase cytosolic Ca2+ in pancreatic β-cells under metabolic inhibition. Br. J. Pharmacol. 1997;120:1191–1198. doi: 10.1038/sj.bjp.0701017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILON P., SHEPHERD R.M., HENQUIN J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidenced in single pancreatic islets. J. Biol. Chem. 1993;268:22265–22268. [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HELLMAN B., GYLFE E., GRAPENGIESSER E., LUND P.E., BERTS A. Cytoplasmic Ca2+oscillations in pancreatic β-cells. Biochim. Biophys. Acta. 1992;1113:295–305. doi: 10.1016/0304-4157(92)90003-s. [DOI] [PubMed] [Google Scholar]
- HENQUIN J.C.Cell biology of insulin secretion Joslin's Diabetes Mellitus 199456–80.Kahn, C.R. & Weir, G.C., eds. Philadelphia: Lea & Febigar, pp
- HENQUIN J.C., MEISSNER H.P. Effects of amino acids on membrane potential and 86Rb+ fluxes in pancreatic β-cells. Am. J. Physiol. 1981;240:E245–E252. doi: 10.1152/ajpendo.1981.240.3.E245. [DOI] [PubMed] [Google Scholar]
- HOEG J.M. The HMG CoA reductase inhibitors. Curr. Opin. Lipidol. 1990;1:29–33. doi: 10.1097/00041433-199402000-00010. [DOI] [PubMed] [Google Scholar]
- HURMINGHAKE D.B. HMG CoA reductase inhibitors. Curr. Opin. Lipidol. 1992;3:22–28. doi: 10.1097/00041433-199402000-00010. [DOI] [PubMed] [Google Scholar]
- ICHIHARA K., SATOH K., ABIKO Y. Influences of pravastatin and simvastatin, HMG-CoA reductase inhibitors, on myocardial stunning in dogs. J. Cardiovasc. Pharmacol. 1993;22:852–856. doi: 10.1097/00005344-199312000-00012. [DOI] [PubMed] [Google Scholar]
- KENNEDY E.D., RIZZUTO R., THELER J.M., PRALONG W.F., BASTIANUTTO C., POZZAN T., WOLLHEIM C.B. Glucose-stimulated insulin secretion correlates with changes in mitochondrial and cytosolic Ca2+ in aequorin-expressin INS-1 cells. J. Clin. Invest. 1996;98:2524–2538. doi: 10.1172/JCI119071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOGA T., KAWABATA K., ARAI K., MATSUSHIMA N., KOIKE H., KOMAI T., TEI M., NAKAMURA H. Comparative pharmacokinetics and pharmacodynamics of pravastatin and simvastatin. Bull. Mol. Biol. Med. 1995;20:103–105. [Google Scholar]
- KOSAKA K., HAGURA R., KUZUYA T. Insulin responses in equivocal and definite diabetes, with special reference to subjects who had mild glucose intolerance but later developed definite diabetes. Diabetes. 1997;26:944–952. doi: 10.2337/diab.26.10.944. [DOI] [PubMed] [Google Scholar]
- LI W., LLOPIS J., WHITNEY M., ZLOKARNIK G., TSIEN R.Y. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 1998;392:936–941. doi: 10.1038/31965. [DOI] [PubMed] [Google Scholar]
- MATTHEWS D.R., NAYLOR B.A., JONES R.G., WARD G.M., TURNER R.C. Pulsatile insulin has greater hypoglycemic effect than continuous delivery. Diabetes. 1983;32:617–621. doi: 10.2337/diab.32.7.617. [DOI] [PubMed] [Google Scholar]
- MCGUIRE T.F., XU X.-Q., COREY S.J., ROMERO G.G., SEBTI S.M. Lovastatin disrupts early events in insulin signaling: a potential mechanism of lovastatin's anti-mitogenic activity. Biochem. Biophys. Res. Commun. 1994;204:399–406. doi: 10.1006/bbrc.1994.2472. [DOI] [PubMed] [Google Scholar]
- NAKAHARA K., KURIYAMA M., YOSHIDOME H., NAGATA K., NAGADO T., NAKAGAWA M., ARIMURA K., HIGUCHI I., OSAME M. Experimental simvastatin-induced myopathy in rabbits. J. Neurol. Sci. 1992;113:114–117. doi: 10.1016/0022-510x(92)90273-n. [DOI] [PubMed] [Google Scholar]
- NAKAHARA K., YADA T., KURIYAMA M., OSAME M. Cytosolic Ca2+ increase and cell damage in L6 rat myoblasts by HMG-CoA reductase inhibitors. Biochem. Biophys. Res. Commun. 1994;202:1579–1585. doi: 10.1006/bbrc.1994.2112. [DOI] [PubMed] [Google Scholar]
- NAKAZAKI M., ISHIHARA H., KAKEI M., INUKAI K., ASANO T., MIYAZAKI J.-I., TANAKA H., KIKUCHI M., YADA T., OKA Y. Repetitive mitochondrial Ca2+ signals synchronize with cytosolic Ca2+ oscillations in the pancreatic beta-cell line, MIN6. Diabetologia. 1998;41:279–286. doi: 10.1007/s001250050904. [DOI] [PubMed] [Google Scholar]
- NEGRE-ARNINOU P., VAN-VLIET A.K., VAN-ERIC M., VAN-THIEL G.C., VAN-LEEUWEN R.E., COHEN L.H. Inhibition of proliferation of human smooth muscle cells by various HMG-CoA reductase inhibitors; comparison with other human cell types. Biochim. Biophys. Acta. 1997;1345:259–268. doi: 10.1016/s0005-2760(96)00184-1. [DOI] [PubMed] [Google Scholar]
- NEUVONEN P.J., KANTOLA T., KIVISTO K.T. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin. Pharmacol. Ther. 1998;63:332–341. doi: 10.1016/S0009-9236(98)90165-5. [DOI] [PubMed] [Google Scholar]
- NIELSEN S., SCHMITZ O., MOLLER N., PORKSEN N., KLAUSEN I.C., ALBERTI K.G., MOGENSEN C.E. Renal function and insulin sensitivity during simvastatin treatment in type 2 (non-insulin-dependent) diabetic patients with microalbuminuria. Diabetologia. 1993;36:1079–1086. doi: 10.1007/BF02374502. [DOI] [PubMed] [Google Scholar]
- OHRVALL M.L., LITHELL H., JOHANSSON J., VESSBY B. A comparison between the effects of gemfibrozil and simvastatin on insulin sensitivity in patients with non-insulin-dependent diabetes mellitus and hyperlipoproteinemia. Metabolism. 1995;44:212–217. doi: 10.1016/0026-0495(95)90267-8. [DOI] [PubMed] [Google Scholar]
- O'MEARA N.M., POLONSKY K.S.Insulin secretion in vivo Joslin's Diabetes Mellitus 199481–96.Kahn, C.R. & Weir, G.C., eds. Philadelphia: Lea & Febigar, pp
- O'RAHILLY S., TURNER R.C., MATTHEWS D.R. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N. Engl. J. Med. 1988;318:1225–1230. doi: 10.1056/NEJM198805123181902. [DOI] [PubMed] [Google Scholar]
- PERLEY M.J., KIPNIS D.M. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J. Clin. Invest. 1967;46:1954–1962. doi: 10.1172/JCI105685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PFEIFER M.A., HALTER J.B., PORTE D., JR Insulin secretion in diabetes mellitus. Am. J. Med. 1981;70:579–588. doi: 10.1016/0002-9343(81)90579-9. [DOI] [PubMed] [Google Scholar]
- PORTE D., JR, KAHN S.E. The key role of islet dysfunction in type II diabetes mellitus. Clin. Invest. Med. 1995;18:247–254. [PubMed] [Google Scholar]
- SACKS F.M., PFEIFER M.A., MOYE L.A., ROULEAU J.L., RUTHERFORD J.D., COLE T.G., BROWN L., WARNICA J.W., ARNOLD J.M., WUN C.C., DAVIS B.R., BRAUNWALD E. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- SCANDINAVIAN SIMVASTATIN SURVIVAL STUDY GROUP Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- SERAJUDDIN A.T., RANADIVE S.A., & MAHONEY E.M. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. J. Pharm. Sci. 1991;80:830–834. doi: 10.1002/jps.2600800905. [DOI] [PubMed] [Google Scholar]
- SHEPHERD J., COBBE S.M., FORD I., ISLES C.G., LORIMER A.R., MACFARLANE P.W., MCKILLOP J.H., PACKARD C.J. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N. Engl. J. Med. 1995;333:1301–1307. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- SHEU W.H., SHIEH S.M., SHEN D.D., FUH M.M., JENG C.Y., CHEN Y.D., REAVEN G.M. Effect of pravastatin treatment on glucose, insulin, and lipoprotein metabolism in patients with hypercholesterolemia. Am. Heart J. 1994;127:331–336. doi: 10.1016/0002-8703(94)90121-x. [DOI] [PubMed] [Google Scholar]
- SMITH P.A., SAKURA H., COLES B., GUMMERSON N., PROKS P., ASHCROFT F.M. Electrogenic arginine transport mediates stimulus-secretion coupling in mouse pancreatic β-cells. J. Physiol. 1997;499:625–635. doi: 10.1113/jphysiol.1997.sp021955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOBERT J.A. Efficacy and long-term adverse effect pattern of lovastatin. Am. J. Cardiol. 1988;62:28J–34J. doi: 10.1016/0002-9149(88)90004-5. [DOI] [PubMed] [Google Scholar]
- TOBERT J.A. HMG-CoA reductase inhibitors, gemfibrozil, and myopathy. Am. J. Cardiol. 1995;75:862. doi: 10.1016/s0002-9149(99)80435-4. [DOI] [PubMed] [Google Scholar]
- WOLLHEIM C.B., SHARP G.W.G. Regulation of insulin release by calcium. Physical. Rev. 1981;61:914–973. doi: 10.1152/physrev.1981.61.4.914. [DOI] [PubMed] [Google Scholar]
- XU X.Q., MCGUIRE T.F., BLASKOVICH M.A., SEBTI S.M., ROMERO G. Lovastatin inhibits the stimulation of mitogen-activated protein kinase by insulin in HIRcB fibroblasts. Arch. Biochem. Biophys. 1996;326:233–237. doi: 10.1006/abbi.1996.0070. [DOI] [PubMed] [Google Scholar]
- YADA T.Action mechanisms of amino acids in pancreatic B-cells Insulin Secretion and Pancreatic B-cell Research 1994129–135.Flatt, P.R. & Lenzen, S., eds. London: Smith-Gordon and Company Limited, pp
- YADA T., HAMAKAWA N., YAEKURA K. Two distinct modes of Ca2+ signalling by ACh in rat pancreatic β-cells: concentration, glucose dependence and Ca2+ origin. J. Physiol. 1995;488:13–24. doi: 10.1113/jphysiol.1995.sp020942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YADA T., KAKEI M., TANAKA H. Single pancreatic β-cells from normal rats exhibit an initial decrease and subsequent increase in cytosolic free Ca2+ in response to glucose. Cell Calcium. 1992;13:69–76. doi: 10.1016/0143-4160(92)90031-m. [DOI] [PubMed] [Google Scholar]
- YADA T., SAKURADA M., IHIDA K., NAKATA M., MURATA F., ARIMURA A., KIKUCHI M. Pituitary adenylate cyclase activating polypeptide is an extraordinarily potent intra-pancreatic regulator of insulin secretion from islet β-cells. J. Biol. Chem. 1994;269:1290–1293. [PubMed] [Google Scholar]
- YADA T., SAKURADA M., ISHIHARA H., NAKATA M., SHIODA S., YAEKURA K., HAMAKAWA N., YANAGIDA K., KIKUCHI M., OKA Y. Pituitary adenylate cyclase-activating polypeptide (PACAP) is an islet substance serving as an intra-islet amplifier of glucose-induced insulin secretion in rats. J. Physiol. 1997;505:319–328. doi: 10.1111/j.1469-7793.1997.319bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAEKURA K., KAKEI M., YADA T. cAMP-signaling pathway acts in selective synergism with glucose or tolbutamide to increase cytosolic Ca2+ in rat pancreatic β-cells. Diabetes. 1996;45:295–301. doi: 10.2337/diab.45.3.295. [DOI] [PubMed] [Google Scholar]