

Abstract
Mycophenolic acid (MPA), is primarily metabolized in the liver to 7-O-MPA-β-glucuronide (MPAG). Using RP-h.p.l.c. we observed three further MPA metabolites, M-1, M-2, M-3, in plasma of transplant recipients on MMF therapy. To obtain information on the structure and source of these metabolites: (A) h.p.l.c. fractions containing either metabolite or MPA were collected and analysed by tandem mass spectrometry; (B) the metabolism of MPA was studied in human liver microsomes in the presence of UDP-glucuronic acid, UDP-glucose or NADPH; (C) hydrolysis of metabolites was investigated using β-glucosidase, β-glucuronidase or NaOH; (D) cross-reactivity of each metabolite was tested in an immunoassay for MPA (EMIT).
Mass spectrometry of M-1, M-2, MPA and MPAG in the negative ion mode revealed molecular ions of m/z 481, m/z 495, m/z 319 and m/z 495 respectively.
Incubation of microsomes with MPA and UDP-glucose produced M-1, with MPA and UDP-glucuronic acid MPAG and M-2 were formed, while with MPA and NADPH, M-3 was observed.
β-Glucosidase hydrolysed M-1 completely. β-Glucuronidase treatment led to a complete disappearance of MPAG whereas the amount of M-2 was reduced by approximately 30%. Only M-2 was labile to alkaline treatment.
M-2 and MPA but not M-1 and MPAG cross-reacted in the EMIT assay.
These results suggest that: (i) M-1 is the 7-OH glucose conjugate of MPA; (ii) M-2 is the acyl glucuronide conjugate of MPA; (iii) M-3 is derived from the hepatic CYP450 system.
Keywords: Mycophenolic acid, metabolites, acyl glucuronide, human microsomes, mass spectrometry, high performance liquid chromatography
Introduction
Mycophenolic acid (MPA), the active metabolite of mycophenolate mofetil (MMF) was developed in the 1960s as a potential antibiotic, antineoplastic, and antipsoriatic drug. Because of its immunosuppressive properties, the drug has now been introduced into immunosuppressant protocols after solid organ transplantation and in autoimmune diseases (Hood & Zarembski, 1997; Simmons et al., 1997; Sievers et al., 1997; Shaw et al., 1995; Nowack et al., 1997; Enk & Knop 1997; Briggs et al., 1998; Zimmer-Molsberger et al., 1997). Its immunosuppressant action residues in the uncompetitive, selective and reversible inhibition of inosine monophosphate dehydrogenase (IMPDH), thus preventing the de novo synthesis of guanosine nucleotides in lymphocytes leading to a loss of proliferation and cell function (Wu, 1994). Even though MMF seems to be well tolerated toxic side effects associated with this drug include diarrhoea, nausea, and leucopenia (Bullingham et al., 1998). Following oral or intravenous administration, the prodrug MMF is rapidly converted to MPA by plasma and tissue esterases such that no measurable concentrations of MMF have been observed in plasma of patients or experimental animals (Shaw et al., 1995). MPA is primarily metabolized by glucuronidation to form a phenolic glucuronide (MPAG), which is the major urinary excretion product of the drug (Bullingham et al., 1998; Shaw & Nowak, 1995). MPAG is thought to undergo deglucuronidation and reabsorption as MPA during the enterohepatic circulation in humans (Shaw et al., 1995; Bullingham et al., 1998; Shaw & Nowak, 1995).
We now describe the isolation and characterization of further MPA metabolites that have been observed in the plasma of kidney, liver and heart transplant recipients on immunosuppressive therapy with MMF.
Methods
Chemicals
Nicotinamide adenine dinucleotide phosphate hydrate (NADPH), ethylenediaminetetraacetic acid (EDTA), UDP-glucose, UDP-glucuronic acid, glycine, cysteine, 1-aminobenzotriazole, troleandomycin, Brij 58, and β-glucosidase (EC 3.2.1.21, recombinant cloned into E. Coli) were from Sigma Chemical Co. (St. Louis, MO, U.S.A.), Sucrose, HC1O4, NaCl, K2HPO4, KH2PO4, H3PO4, Na2WO4.2H2O, MgCl2, and NaOH were purchased from Merck (Darmstadt, Germany). 2[4-(2-hydroxyethyl)-1-piperazinol]-ethane sulphonic acid (HEPES) was from Aldrich, (Steinheim, Germany). Acetonitrile and methanol were obtained from J.T. Baker B.B. (Deventer, Netherlands). All reagents were analytical grade. β-Glucuronidase (EC 3.2.1.3 from E. coli K12,) was from Boehringer Mannheim (Mannheim, Germany). MPA, MPAG, and the internal standard carboxybutoxy ether of MPA (MPAC) were gifts from Hofmann-LaRoche (Basel, Switzerland).
H.p.l.c. analysis
MPA, MPAG and the putative metabolites were purified from plasma or microsomal incubations through a modification of an h.p.l.c procedure (Shipkova et al., 1998) recently developed in our laboratory for simultaneous quantification of MPA and MPAG. Briefly, 200 μl plasma or microsomal preparation and 100 μl acetonitrile containing the internal standard MPAC 0.036 mM (15 mg l−1) were mixed by vortexing in a 1.5 ml polypropylene tube for 5 s. This was followed by the sequential addition of 20 μl of 1.5 M perchloric acid and 20 μl of 0.76 M sodium tungstate dihydrate. The tube was mixed by vortexing for 15 s after each addition. The sample was then centrifuged for 5 min at 10,000 × g and 80 μl of the supernatant were removed for chromatography.
The h.p.l.c. system consisted of a chromatographic pump (M450), an automatic injector (GINA 50), a diode array detector (UVD 340S), a computer interface system controller linked to a PC (Gynkotek, Germering, Germany) and a 250 mm × 4.6 mm Hypersil C-18 reversed-phase column (MZ Analysentechnik, Mainz, Germany). The column was maintained at 42°C. The mobile phase consisted of solution A (250 ml acetonitrile, 750 ml phosphate buffer - pH 3.0, 20 mM final concentration) and solution B (450 ml acetonitrile and 550 ml phosphate buffer - pH 6.5, 40 mM final concentration) that formed the following gradient: 7%B (0–2 min); 7–35%B (2–17 min); 35%B (17–18 min); 35–100%B (18–18.5 min); 100%B (18.5–20 min); 100–7%B (20–20.5 min). The flow rate was 1.2 ml min−1 and the compounds were detected at 215 nm and identified by their retention times and u.v. spectra. The u.v. spectra were recorded using the diode array detector.
Preparation of human liver microsomes
Human liver samples were obtained from the Department of Transplant Surgery as excess material removed during transplantation of a partial liver from a 39-year-old male donor (subarachnoidal haematoma) to a paediatric recipient. Tissue was homogenized in HEPES 5 mM, sucrose-buffer 250 mM pH 7.4 containing EDTA 0.5 mM using a Polytron PT3000 homogenizer (Kinematica, Kriens, Switzerland). Microsomal vesicles were isolated by removal of the nuclear and mitochondrial fractions at 10,000 × g for 20 min, 4°C using a L8-55 ultracentrifuge and a 50T13 rotor (Beckman, Irvine, CA, U.S.A.). The microsomal fraction was sedimented at 105,000 × g for 60 min, 4°C. The pellet was washed once in HEPES 50 mM, KCl 150 mM pH 7.4 and collected again at 105,000 × g for 30 min. The resulting microsomal pellet was suspended in HEPES/KCl, pH 7.4 by careful sonication in ice and stored in portions (10 mg protein ml−1) at −80°C until use.
Incubation of microsomes with MPA, co-substrates, and cytochrome P-450 inhibitors.
Microsomal preparations (0.25 ml, 1 mg protein ml−1) were incubated in the presence of 0.1 mM MPA for 30 min at 37°C in HEPES/KCl/EDTA/MgCl2 buffer in mM: (HEPES 50, KCl 150, EDTA 1, MgCl2 5) adjusted to either pH 7.0 or 7.4. In separate experiments either NADPH, UDP-glucuronic acid, or UDP-glucose (final concentrations: 4 mM) were added to each of these buffers. Before incubation with UDP-glucuronic acid or UDP-glucose the microsomal preparations were always preincubated in the presence of the activator Brij 58 (0.1 mg mg protein−1) (Raoof et al., 1996; Radominska et al., 1993) for 10 min on ice. Control incubations contained either MPA and microsomes without any other additions or MPA plus co-substrates but no microsomes. In addition, incubations with MPA and NADPH were performed in the presence of the general CYP450 inhibitor 1-aminobenzotriazole (2 mM), and the CYP3A4 inhibitor troleandomycin (2 mM).
Isolation of putative MPA metabolites
Fractions (n=5) containing either the putative MPA metabolites, MPA or MPAG obtained through h.p.l.c. of a single plasma sample preparation from a kidney transplant recipient were separately pooled, and concentrated by evaporation (60 min, 37°C) using a vacuum centrifuge. To remove phosphate the concentrated h.p.l.c. fractions (0.5 ml) were passed through a 100 mg RP18 Adsorbex column (Merck, Darmstradt, Germany) which had been activated by washing with 2×2 ml methanol followed by flushing with 4 ml H2O. After applying the concentrated fraction, the column was flushed with 2×4 ml H2O and the metabolite was then eluted with 2×0.5 ml acetonitrile/H2O (80/20 v/v). After evaporation of acetonitrile the eluate was subjected to analysis with the EMIT immunoassay for MPA (Haley et al., 1995) to test for cross-reactivity and the rest was evaporated to dryness for mass spectrometry (MS) analysis. For MS analysis the sample was reconstituted using methanol/water (1/1 v/v). An aliquot was rerun on the h.p.l.c. to verify the purity of the putative MPA metabolite.
Electrospray ionization mass spectrometry (ESI-MS)
ESI-MS was performed on a PE Sciex API 365 triple quadropole mass spectrometer (PE Sciex, Concord, Ontario, Canada) using an electrospray ion source (PE Sciex, Concord, Ontario, Canada) both in the positive and in the negative ionization mode. Samples were introduced into the ion source via an infusion syringe pump (Harvard Apparatus, Inc., South Natick, MA, U.S.A.) at a flow rate of 6 μl min−1. MS-experiments (Q1-scans) were carried out at a spray voltage of +5000 or −4500 V, an orifice voltage from ±30 to ±50 V and a ring electrode voltage from ±150 to ±250 V depending on the ionization mode. Further MS/MS-experiments (product ion scans) were carried out at a collision offset voltage of ±20 to ±60 V using nitrogen as collision gas at a pressure of 3.95×10−3 mTorr in the collision cell.
Incubation of plasma with β-glucuronidase or β-glucosidase
A plasma pool obtained from four kidney transplant recipients under therapy with MMF containing putative MPA metabolites was divided into four 0.1 ml portions. The first portion was incubated for 24 h at 37°C with β-glucosidase (15 u ml−1) after adjusting the pH to 5.0 using 0.61 M H3PO4. The second portion was incubated for 75 min at 37°C in the presence of β-glucuronidase (33 u ml−1) after adjusting the pH to 6.5 using 0.61 M H3PO4. The third and the fourth portions served as controls and were incubated under identical conditions but without the added enzymes for 24 h or 75 min respectively. Incubations were stopped by using the h.p.l.c. pre-treatment procedure and analysed by h.p.l.c.
Alkaline hydrolysis of metabolites
Plasma was pre-treated as described for the h.p.l.c. analysis. To 100 μl of the supernatant 10 μl of 2 M NaOH was added and the sample was then left at room temperature for 2 h. A control sample of supernatant was stored at room temperature for 2 h without addition of NaOH.
Protein measurement
Protein was determined using the BCA protein assay reagent kit supplied by Pierce (Rockford, IL, U.S.A.).
Ethics committee
The use of excess human liver tissue for scientific investigations was approved by the local Ethics Committee.
Results
Using the modified h.p.l.c. procedure we were able to identify (Figure 1) three putative MPA metabolites (M-1, M-2, M-3), in addition to MPAG, in plasma samples from transplant recipients on MMF therapy. The metabolites were identified on the basis of the similarity of their u.v. spectra to those of MPA and MPAG (Figure 2). A typical chromatogram is shown in Figure 1 after pre-treatment of a plasma sample obtained from a kidney transplant recipient under MMF therapy (1.2 g m−2). The MPAG and MPA concentrations in the original plasma sample were 0.33 mM (163 mg l−1) and 0.073 mM (23.4 mg l−1) respectively as determined using the h.p.l.c. procedure recently developed in our laboratory (Shipkova et al., 1998). The metabolites eluted after 4.4 min (M-1), 6.2 min (M-2) and 11.2 min (M-3). As can be seen in Figure 2, the u.v. spectrum of M-1 was very similar to that of MPAG with three absorption maxima at around 294, H251 and 214 nm. The u.v. spectrum of M-2 was virtually identical to that of MPA with characteristic absorption maxima at around 306, 250 and 216 nm. In the case of M-3 the u.v. spectrum was similar to that of MPA.
Figure 1.
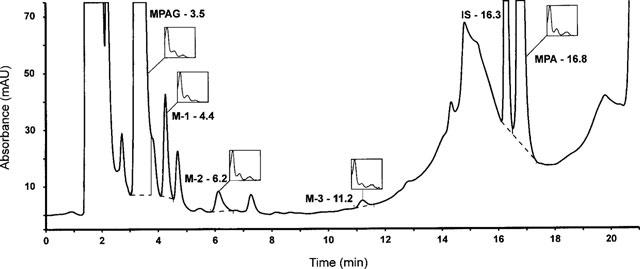
Representative chromatogram of a plasma sample obtained from a kidney transplant recipient 75 min after administration of 0.6 g m−2 MMF. The sample was pre-treated as described in Methods and applied to a 250 mm×4.6 mm Hypersil C-18 column maintained at 42°C. The column was eluted at a flow rate of 1.2 ml min−1 with a mobile phase consisting of solution A (250 ml acetonitrile, 750 ml phosphate buffer–pH 3.0, 20 mM final concentration) and solution B (450 ml acetonitrile and 550 ml phosphate buffer–pH 6.5, 40 mM final concentration) that formed the following gradient: 7%B (0–2 min); 7–35%B (2–17 min); 35%B (17–18 min); 35–100%B (18–18.5 min); 100%B (18.5–20 min); 100–7%B (20–20.5 min). Compounds eluting from the column were monitored continuously with a diode array detector. This chromatogram illustrates the u.v. absorption at 215 nm. The 7-O glucuronide metabolite (MPAG) eluted after 3.5 min and mycophenolic acid (MPA) after 16.8 min. The metabolites M-1, M-2 and M-3 which eluted after 4.4, 6.2, and 11.2 min respectively were identified by comparison of their u.v. spectra (shown in the insets) to those of MPAG and MPA. The internal standard carboxybutoxy ether of MPA (IS) eluted after 16.3 min.
Figure 2.
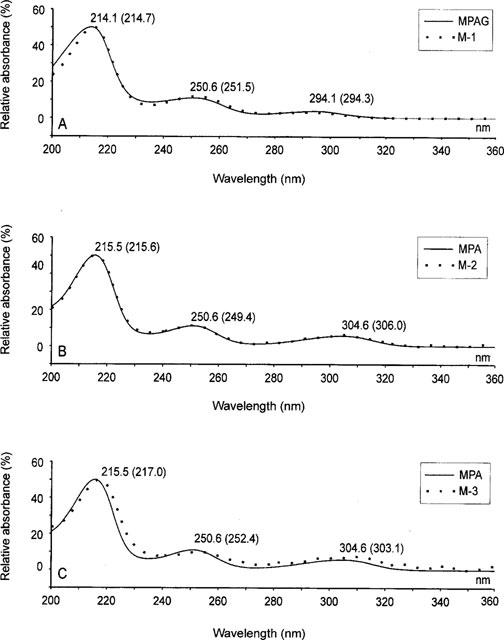
Comparison of the u.v. spectra of M-1 (A), M-2 (B) and M-3 (C) with those of either MPA (B and C); or MPAG (A). The spectra were determined with a diode array detector and the highest relative absorption maximum for each compound was arbitrarily set at 50%. Spectra were then normalized according to the highest relative maximum. The wavelengths of each absorption maximum are illustrated in the Figure. Values in parentheses correspond to the metabolites M-1, M-2 or M-3 and values not in parentheses either MPA or MPAG.
To further characterize the putative MPA metabolites, fractions containing either M-1 or M-2 as well as MPAG and MPA were separately collected from the h.p.l.c. column. Further investigations to characterize M-3 were not performed since the very low concentrations in human plasma samples and in microsomal incubations precluded its isolation. After removal of the phosphate through elution from a RP18 Adsorbex column and of acetonitrile by vacuum centrifugation, the fractions were diluted with distilled water to give an absorbance of 1.27 at 215 nm and were tested for cross-reactivity in an immunoassay for MPA (EMIT, Dade-Behring, Liederbach, Germany). Neither M-1 nor MPAG displayed any cross-reactivity. In contrast M-2 gave a response equivalent to a MPA concentration of 0.044 mM (14 mg l−1). MPA at the same absorbance revealed a concentration of 0.028 mM (8.8 mg l−1). Rechromatography of M-2 over the h.p.l.c column showed the expected retention time for this metabolite and confirmed that it had not been hydrolyzed to MPA during the purification procedure.
Mass spectrometric analysis of M-1 and M-2
MPA, MPAG and the metabolites M-1 and M-2 were analysed by tandem mass spectrometry (Figure 3). In the negative ion mode MPA and MPAG revealed (M-H)−-ions of m/z 319 and m/z 495 respectively. Fragmentation of the precursor ion of MPA produced characteristic fragment ions of m/z 287 and m/z 275. These can be explained by the loss of either CH3OH or CO2 from the precursor ion. In the case of MPAG, fragmentation of the precursor ion m/z 495 led to the formation of one major product ion with m/z 319, presumably due to loss of the dehydroglucuronic acid moiety. Further fragmentation produced the product ions m/z 287 and m/z 275 both typical for MPA. The mass spectrum of metabolite M-1 showed a characteristic ion at m/z 481. The fragmentation pattern of this ion revealed product ions with m/z 319, m/z 287 and m/z 275, that are characteristic for MPA. These results suggested that the metabolite M-1 could be a glucoside conjugate of MPA which would have a molecular weight of 482 Da. The spectrum of M-2 revealed a precursor ion peak of m/z 495, identical to that seen for MPAG. As with MPAG, fragmentation of this precursor ion led to the fragment ions with m/z 319, m/z 287 and m/z 275. Since MPA has two potential sites for glucuronidation, one at the phenolic 7-hydroxyl group and one at the aliphatic carboxyl group of the acyl side chain (Figure 4), we inferred that M-2 could be the acyl glucuronide conjugate of MPA.
Figure 3.
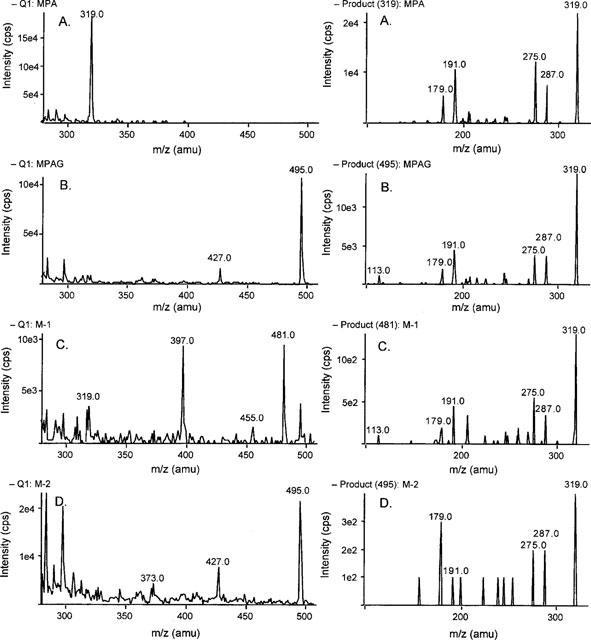
Mass spectra of MPA (A) and its metabolites MPAG (B), M-1 (C), and M-2 (D) in the negative ionization mode: Q1 scans (left) and product ions scans (right). The mass spectra were measured with a PE Sciex API 365 triple quadrople mass spectrometer using electrospray ion source. Samples were introduced into the ion source via an infusion syringe pump at a flow rate of 6 μl min−1. MS-experiments (Q1-scans) were carried out at a spray voltage of +5000 or −4500 V, an orifice voltage from ±30 to ±50 V and a ring electrode voltage from ±150 to ±250 V depending on the ionization mode. Further MS/MS-experiments (product ion scans) were carried out at a collision offset voltage of ±20 to ±60 V using nitrogen as collision gas at a pressure of 3.95×10−3 mTorr in the collision cell. All mass spectra scans are displayed after normalization to the highest detectable mass peak in each spectrum.
Figure 4.
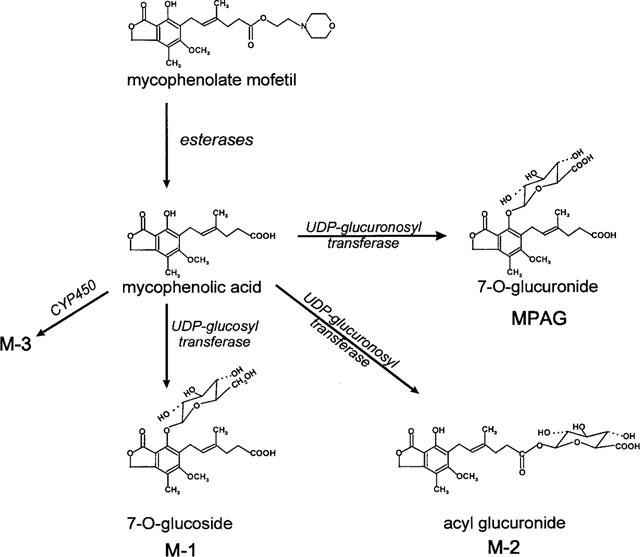
Metabolic pathways of mycophenolic acid in humans. MMF is hydrolyzed to MPA the immunosuppressive metabolite of MMF by plasma and tissue esterases. MPA is primarily converted by glucuronidation to the major metabolite product, the phenolic MPA glucuronide (MPAG) as well as to its newly detected acyl glucuronide (M-2). In addition a phenolic 7-O-glucoside (M-1) and a CYP450 oxidation product (M-3) were observed.
To confirm the structures of these metabolites two sets of experiments were carried out. First we tried to generate the metabolites by incubation of MPA with human liver microsomes and secondly we investigated the cleavage of these putative MPA conjugates by enzyme treatment as well as by alkaline hydrolysis.
Incubations with microsomes
Incubation of MPA with human liver microsomes in the presence of UDP-glucose led to the formation of a metabolite showing the same u.v. spectrum and an identical retention time as M-1. When UDP-glucuronic acid was used instead of UDP-glucose two metabolites were formed, MPAG and a metabolite with a retention time and u.v. spectrum identical to that of M-2 from plasma of transplant recipients on MMF. Control incubations in the absence of microsomes or in the presence of microsomes but without UDP-glucose, or UDP glucuronic acid did not generate any metabolites. The formation of MPAG, M-1 and M-2 was facilitated at pH 7.0.
Incubation of MPA with human liver microsomes at pH 7.4 in the presence of NADPH led to formation of a metabolite with a retention time and a u.v. spectrum identical with M-3. The formation of M-3 was prevented to >90% (n=3) by the addition of the general CYP450 inhibitor 1-aminobenzotriazole whereas the specific CYP450 3A4 inhibitor troleandomycin was less efficient (<40%, n=3). No metabolites were formed in control incubations. Because M-3 was only present in trace amounts it was not further investigated.
Hydrolysis experiments
In order to further investigate the structures of the metabolites M-1 and M-2, plasma samples containing these metabolites were subjected to enzymatic of alkaline hydrolysis. In one set of experiments plasma samples were incubated in the presence or absence (control) of either β-glucosidase or β-glucuronidase. In a second set of experiments the metabolite containing supernatant from the h.p.l.c. pre-treatment procedure was incubated in the presence and absence (control) of 0.18 M NaOH. After incubation, the amounts of the metabolites in each of the samples were estimated by h.p.l.c. The results are expressed as the percentage of metabolite that was hydrolyzed relative to the control. As shown in Table 1, β-glucosidase degraded M-1 completely. Treatment with β-glucuronidase led to a complete disappearance of MPAG in the plasma sample, whereas the amount of M-2 was reduced by approximately 30% (Table 1). Only M-2 proved to be highly alkali-labile.
Table 1.
Hydrolysis of MPAG, M-1 and M-2

Discussion
MPA, the active metabolite of MMF, is increasingly used for immunosuppressive therapy after solid organ transplantation and is also proposed for treatment of autoimmune diseases. Its mode of action is thought to reside in the uncompetitive inhibition of inosine monophosphate dehydrogenase (IMPDH), thereby depleting intracellular guanine nucleotide pools and thus preventing proliferation of functionally active B and T lymphocytes. The major metabolite of MPA in human plasma is MPAG, a glucuronic acid conjugate at position seven of the phenolic ring of the MPA molecule (Figure 4). MPAG has no inhibitory activity towards IMPDH (Nowak & Shaw, 1997) and shows virtually no cross reactivity (<0.2%) with an immunoassay which uses a monoclonal antibody against MPA (Shaw et al., 1995). This suggests that the epitope recognized by the anti MPA antibody includes the free hydroxyl group at position seven of the phenol ring. Thus any adduct moiety at this position is likely to prevent binding to the antibody. This phenolic hydroxyl group was also found to be essential for the potent inhibition of IMPDH, the immunosuppression target of MPA (Nelson et al., 1996).
We now describe the identification and structural characterization of two further metabolites of MPA (Figure 4) that have been regularly observed (Shipkova et al., 1999 in press; Schütz et al., 1998) in the plasma of kidney, liver and heart transplant recipients on immunosuppressive therapy with MMF. The metabolites M-1 and M-2 were identified as the 7-0- glucoside conjugate and the acyl glucuronide conjugate of MPA respectively. Clarification of the structures of these metabolites was based on LC/MS/MS analysis, comparison of their u.v. spectra to those of MPA and MPAG, in vitro formation from MPA and the corresponding co-substrates using human liver microsomes, enzymatic cleavage experiments and cross-reactivity with an immunoassay specific for MPA but not MPAG. Mass spectrometry analysis of M-2 in the negative mode revealed a precursor ion with m/z 495, identical to that of MPAG and a similar fragmentation pattern to that seen with MPAG. In contrast to MPAG, M-2 cross-reacted with the EMIT immunoassay for MPA, had a different retention time on the h.p.l.c., and also had inhibitory activity towards IMPDH (Shipkova et al., 1999 in press; Schütz et al., 1998). Furthermore, the u.v. spectrum of M-2 more closely resembled that of MPA rather than that of MPAG. MPA has two potential sites of glucuronidation, the phenolic 7-hydroxyl group and the aliphatic carboxyl group of the acyl side chain (Figure 4). Our results are, therefore, consistent with M-2 being the acyl glucuronide conjugate of MPA. Mojarrhabi & Mackenzie (1997) observed three glucuronidation products of MPA when MPA was incubated in vitro with COS-7 cells transfected with UDP glucuronyltransferase from human colon. They proposed one of these metabolites to be the acyl glucuronide of MPA but did not confirm its structure. In addition, Bullingham et al. (1998) recently suggested an acyl glucuronide of MPA to be present in urine of patients under MMF therapy. Acyl glucuronides have been reported for a number of carboxylic acids. At physiological pH they are relatively unstable and undergo intramolecular acyl group migration to form isomers that are stable to hydrolysis by β-glucuronidase. Such acyl group migration has been shown for the carboxylic acid glucuronides of zomepirac, valproate, and bilirubin (Newton et al., 1992; Panfil et al., 1992; Dickinson et al., 1984). The fact that M-2 was relatively stable to β-glucuronidase hydrolysis would suggest that it is primarily present in plasma in the form of the non-C-1 esters of glucuronic acid. However, M-2 was labile to alkaline hydrolysis as would be expected for such esters (Newton et al., 1992; Dickinson et al., 1984).
Evidence that M-1 is a glucose conjugate of MPA is as follows: MS analysis revealed a precursor ion of m/z 481 that would be expected for a glucose conjugate of molecular weight 482 Da; formation of M-1 in vitro required the presence of UDP-glucose; M-1 was cleaved to MPA by incubation with β-glucosidase (Table 1). Conjugation of the glucose moiety to the phenolic ring at the 7 OH position is suggested by its lack of cross-reactivity in the EMIT immunoassay, and the fact that its u.v. spectrum more closely resembles that of MPAG and not that of MPA. In mammals, the conjugation of drugs to glucose generally represents a minor metabolic pathway if glucuronidation is possible (Tang, 1990). Conjugation can occur through both N-, S- and O-linkages. According to Kirkman et al. (1998), approximately a dozen xenobiotics are known to form conjugates with glucose in mammalian systems or models for mammalian systems. N-linked glucoside metabolites of phenobarbitone, amobarbital, sulphonamides (Tang, 1990) and 5-aminosalicyclic acid (Tjørnelund et al., 1989) have been identified in man. To our knowledge the 7-O- glucoside of MPA is the first drug O-glucoside metabolite to be found in man. This conjugate has also been observed in dogs (Hoffman-La Roche, investigators brochure) that had been given MMF.
In addition to M-1 and M-2 a third putative MPA metabolite M-3 has been seen in the plasma of patients on MMF. However, M-3 which is only present in trace amounts in human plasma could be produced in vitro by incubation of MPA with human liver microsomes in the presence of NADPH, suggesting MPA can also be partially metabolized by the microsomal CYP450 system. This is further supported by the finding that formation of M-3 is inhibited by 1-aminobenzotriazole, a general CYP450 inhibitor. Data derived from incubations in the presence of troleandomycin suggest that CYP3A4 is less important for the metabolism of MPA than for the metabolism of other immunosuppressants such as cyclosporin A. The pharmacological and toxicological significance of the MPA metabolites characterized in the present investigation remains to be clarified. However, glucuronides formed from carboxylate containing xenobiotics are more chemically reactive than most phase II conjugates. They have been shown to form adducts by covalent binding to tissue and plasma proteins (Spahn-Langguth & Benet, 1992). This may be of importance and relevant with respect to the toxicity observed in patients treated with MMF. Furthermore, both the acyl glucuronide of MPA as well as the glucose conjugate of MPA may be of pharmacological significance if they have inhibitory activity towards IMPDH. Preliminary experiments suggest that M-2 but not M-1 can inhibit IMPDH (Schütz et al., 1998). The cross reactivity of M-2 in the EMIT assay for MPA determination in plasma will have an impact on the monitoring of MPA when immunochemical methods such as the EMIT are used.
We have identified two new metabolites M-1 and M-2 of the immunosuppressant mycophenolic acid (MPA) commonly present in plasma samples of transplant recipients under therapy with mycophenolate mofetil. As shown by (i) mass spectrometry, (ii) incubation of microsomes with MPA plus co-substrates, and (iii) hydrolysis experiments, M-1 turned out to be the 7-O-glucoside of MPA and M-2 a carboxyl linked glucuronide conjugate of MPA. To our knowledge the 7-O-glucoside of MPA is the first drug O-glucoside metabolite observed in man. The carboxyl-linked glucuronide conjugate may have an impact on the immunosuppressive therapy using MMF since it inhibits IMPDH activity. In addition, acyl glucuronides are known to be highly reactive thus possibly causing adverse effects.
Acknowledgments
The skillful technical assistance of Christina Wiese and Tanja Schneider is gratefully acknowledged. We thank Dr H Luthe for fruitful discussions. Maria Shipkova was supported by a grant from the Volkswagenstiftung.
Abbreviations
- CYP450
cytochrome P-450
- EDTA
ethylenediaminetetraacetic acid
- EMIT
enzyme multiplied immunoassay technique
- ESI
electrospray ionisation
- IMPDH
inosine monophosphate dehydrogenase
- MMF
mycophenolate mofetil
- MPA
mycophenolic acid
- MPAC
mycophenolic acid carboxybutoxy ether
- MPAG
mycophenolic acid 7-O-phenolic glucuronide
- MS
mass spectrometry
- NADPH
nicotinamide adenine dinucleotide phosphate hydrate
- RP
reversed phase
- UDP
uridine diphosphate
References
- BRIGGS W.A., CHOI M.J., SCHEEL P.J., JR Successful mycophenolate mofetil treatment of glomerular disease. Am. J. Kidney Dis. 1998;31:213–217. doi: 10.1053/ajkd.1998.v31.pm9469489. [DOI] [PubMed] [Google Scholar]
- BULLINGHAM R.E.S., NICHOLLS A.J., KAMM B.R. Clinical pharmacokinetics of mycophenolate mofetil. Clin. Pharmacokinet. 1998;34:429–455. doi: 10.2165/00003088-199834060-00002. [DOI] [PubMed] [Google Scholar]
- DICKINSON R.G., HOOPER W.D., EADIE M.J. pH-dependent rearrangement of the biosynthetic ester glucuronide of valproic acid to β-glucuronidase-resistant forms. Drug Metab. Dispos. 1984;12:247–252. [PubMed] [Google Scholar]
- ENK A.H., KNOP J. Treatment of pemphigus vulgaris with mycophenolate mofetil [letter] Lancet. 1997;350:494. doi: 10.1016/S0140-6736(05)63084-X. [DOI] [PubMed] [Google Scholar]
- HALEY C.J., JAKLITSCH A., MCGOWAN B., WIEDER-BUNGER T., KAMPE T., ALEXANDER S.Feasibility of an Emit assay for mycophenolic acid in plasma Ther. Drug Monit. 199517431et al [Google Scholar]
- HOOD K.A., ZAREMBSKI D.G. Mycophenolate mofetil: a unique immunosuppressive agent. Am. J. Health Syst. Pharm. 1997;54:285–294. doi: 10.1093/ajhp/54.3.285. [DOI] [PubMed] [Google Scholar]
- KIRKMAN S.K., ZHANG M.Y., HORWATT P.M., SCATINA J. Isolation and identification of bromfenac glucoside from rat bile. Drug Metab. Dispos. 1998;26:720–723. [PubMed] [Google Scholar]
- MOJARRABI B., MACKENZIE P.I. The human UDP glucuronosyltransferase, UGT1A10, glucuronidates mycophenolic acid. Biochem. Biophys. Res. Comm. 1997;238:775–778. doi: 10.1006/bbrc.1997.7388. [DOI] [PubMed] [Google Scholar]
- NELSON P.H., CARR S.F., DEVENS B.H., EUGUI E.M., FRANCO F., GONZALEZ C., HAWLEY R.C., LOUGHHEAD D.G., MILAN D.J., PAPP E., PATTERSON J.W., RHOUHAFJA S., SJOGREN E.B., SMITH D.B., STEPHENSON R.A., TALAMAS F.X., WALTOS A.M., WEIKERT R.J., WU J.C. Structure-activity relationships for inhibition of inosine monophosphate dehydrogenase by nuclear variants of mycophenolic acid. J. Med. Chem. 1996;39:4181–4196. doi: 10.1021/jm9603633. [DOI] [PubMed] [Google Scholar]
- NEWTON J.F., STRAUB K.M., DEWEY R.H., PERCHONOCK C.D., MCCARTHY M.E., GLEASON J.G., LYNN R.K. Metabolism of the leukotriene receptor antagonist 5-(2-(8-phenyloctyl)phenyl)-4,6-dithianonanedioic acid (SK&F 102922) in the guinea pig. Rearrangement of the acyl glucuronide. Drug Metab. Dispos. 1992;20:479–484. [PubMed] [Google Scholar]
- NOWAK I., SHAW L.M. Effect of mycophenolic acid glucuronide on inosine monophosphate dehydrogenase activity. Ther. Drug Monit. 1997;19:358–360. doi: 10.1097/00007691-199706000-00018. [DOI] [PubMed] [Google Scholar]
- NOWACK R., BIRCK R., VAN DER WOUDE F.J. Mycophenolate mofetil for systemic vasculitis and IgA nephropathy [letter] Lancet. 1997;349:774. doi: 10.1016/S0140-6736(05)60198-5. [DOI] [PubMed] [Google Scholar]
- PANFIL I., LEHMAN P.A., ZIMNIAK P., ERNST B., FRANZ T., LESTER R., RADOMINSKA A. Biosynthesis and chemical synthesis of carboxyl-linked glucuronide of lithocholic acid. Biochem. Biophys. Acta. 1992;1126:221–228. doi: 10.1016/0005-2760(92)90294-6. [DOI] [PubMed] [Google Scholar]
- RADOMINSKA A., LITTLE J., PYREK J.S., DRAKE R.R., IGARI Y., FOURNEL-GIGLEUX S., MAGDALOU J., BURCHELL B., ELBEIN A.D., SIEST G., LESTER R. A novel UDP-Glc-specific glucosyltransferase catalyzing the biosynthesis of 6-O-glucosides of bile acids in human liver microsomes. J. Biol. Chem. 1993;268:15127–15135. [PubMed] [Google Scholar]
- RAOOF A.A., VAN OBBERGH L.J., DE GOYET J., VERBEECK R.K. Extrahepatic glucuronidation of propofol in man: possible contribution of gut wall and kidney. Eur. J. Clin. Pharmacol. 1996;50:91–96. doi: 10.1007/s002280050074. [DOI] [PubMed] [Google Scholar]
- SCHÜTZ E., SHIPKOVA M., ARMSTRONG V.W., NIEDMANN P.D., WEBER L., TÖNSHOFF B., PETHIG K., WAHLERS T., BRAUN F., RINGE B., OELLERICH M. Therapeutic drug monitoring of mycophenolic acid: comparison of h.p.l.c. and immunoassay reveals new MPA metabolites. Transplant. Proc. 1998;30:1185–1187. doi: 10.1016/s0041-1345(98)00201-2. [DOI] [PubMed] [Google Scholar]
- SHAW L.M., NOWAK I. Mycophenolic acid: measurement and relationship to pharmacologic effects. Ther. Drug Monit. 1995;17:685–689. doi: 10.1097/00007691-199512000-00024. [DOI] [PubMed] [Google Scholar]
- SHAW L.M., SOLLINGER H.W., HALLORAN P., MORRIS R.E., YATSCOFF R.W., RANSOM J., TSINA I., KEOWN P., HOLT D.W., LIEBERMAN R., JAKLITSCH A., POTTER J. Mycophenolate mofetil: a report of the consensus panel. Ther. Drug Monit. 1995;17:690–699. doi: 10.1097/00007691-199512000-00025. [DOI] [PubMed] [Google Scholar]
- SHIPKOVA M., NIEDMANN P.D., ARMSTRONG V.W., SCHÜTZ E., WIELAND E., OELLERICH M. Simultaneous determination of mycophenolic acid and its glucuronide in human plasma using a simple high-performance liquid chromatographic procedure. Clin. Chem. 1998;44:1481–1488. [PubMed] [Google Scholar]
- SHIPKOVA M., SCHÜTZ E., ARMSTRONG V.W., NIEDMANN P.D., WIELAND E., OELLERICH M.Overestimation of mycophenolic acid (MPA) by EMIT correlates with MPA metabolite Transplant. Proc. 1999. in press [DOI] [PubMed]
- SIEVERS T.M., ROSSI S.J., GHOBRIAL R.M., ARRIOLA E., NISHIMURA P., KAWANO M., HOLT C.D. Mycophenolate mofetil. Pharmacotherapy. 1997;17:1178–1197. [PubMed] [Google Scholar]
- SIMMONS W.D., RAYHILL S.C., SOLLINGER H.W. Preliminary risk-benefit assessment of mycophenolate mofetil in transplant rejection. Drug Saf. 1997;17:75–92. doi: 10.2165/00002018-199717020-00001. [DOI] [PubMed] [Google Scholar]
- SPAHN-LANGGUTH H., BENET L.Z. Acyl glucuronides revisited: is the glucuronidation process a toxification as well as a detoxification mechanism. Drug Metab. Rev. 1992;24:5–48. doi: 10.3109/03602539208996289. [DOI] [PubMed] [Google Scholar]
- TANG B.K. Drug glucosidation. Pharmacol. Ther. 1990;46:53–56. doi: 10.1016/0163-7258(90)90034-y. [DOI] [PubMed] [Google Scholar]
- TJORNELUND J., HANSEN S.H., CORNETT C. New metabolites of the drug 5-aminosalicyclic acid. I: N-beta-D-glucopyranosyl-5-aminosalicylic acid. Xenobiotica. 1989;19:891–899. doi: 10.3109/00498258909043149. [DOI] [PubMed] [Google Scholar]
- WU J.C. Mycophenolate mofetil: molecular mechanism of action. Perspect Drug Discov. Design. 1994;2:185–204. [Google Scholar]
- ZIMMER-MOLSBERGER B., KNAUF W., THIEL E. Mycophenolate mofetil for severe autoimmune haemolytic anemia [letter] Lancet. 1997;350:1003–1004. doi: 10.1016/S0140-6736(05)64068-8. [DOI] [PubMed] [Google Scholar]