

Abstract
The coupling of the human somatostatin sst5 receptor recombinantly expressed in Chinese hamster ovary (CHO-K1) cells to adenylate cyclase was investigated using receptor selective ligands.
Forskolin (10 μM)-stimulated adenosine 3′ : 5′-cyclic monophosphate (cyclic AMP) accumulation was inhibited by somatostatin-14 and a number of receptor-selective agonists with a rank order of agonist potency typical of the sst5 receptor. L-362,855 and BIM-23056 behaved as full agonists. At higher somatostatin-14 concentrations there was sub-maximal inhibition resulting in a bell-shaped concentration-effect relationship. Pertussis toxin (PTx; 100 ng ml−1, 18 h) pre-treatment abolished agonist-mediated inhibition of cyclic AMP accumulation and markedly enhanced stimulation of cyclic AMP at higher agonist concentrations.
The concentration of prostaglandin E2 (PGE2) in the incubation media was raised 14 fold by 1 μM somatostatin-14 but was insufficient to stimulate adenylate cyclase activity via endogenous prostanoid receptors.
Pre-treatment with cholera toxin (ChTx; 20 μg ml−1, 18 h) markedly inhibited sst5 receptor-mediated increases in cyclic AMP formation in intact cells. Somatostatin-14-stimulated cyclic AMP accumulation was also observed in sst5 receptor containing CHO-K1 membranes and was inhibited by the synthetic peptide Gαsacetyl-354-372-amide (100 μM) by 65.9±3.5%, implicating a Gαs protein involvement in this response.
Activation of Gαs proteins by somatostatin-14 could be demonstrated with [35S]-guanosine 5′-[γ-thio]triphosphate ([35S]-GTPγS) binding and subsequent immunoprecipitation of 35S labelled Gαs proteins with anti-Gαs serum.
These data show that the sst5 receptor is very efficiently coupled in a negative manner to adenylate cyclase. However, at higher agonist concentrations the receptor can also mediate activation of adenylate cyclase by a mechanism apparently involving Gαs protein activation.
Keywords: Somatostatin, sst5 receptor, adenylate cyclase, CHO-K1 cells, pertussis toxin, cholera toxin, Gαs proteins, [35S]-GTPγS binding and immunoprecipitation
Introduction
Somatostatin-14 (SRIF; somatotropin release inhibitory factor) is a cyclic tetradecapeptide with a widespread distribution in the CNS and periphery (Mandarino et al., 1981; Schindler et al., 1996). To date, five distinct somatostatin receptor genes have been described (see Reisine & Bell, 1995 for review) which encode receptors sharing predicted topology with other members of the G protein-coupled superfamily. The human and rat sst5 receptors are the only members of this family with a preferential affinity for somatostatin-28, an N-terminally extended form of somatostatin-14 (O'Carroll et al., 1992; Yamada et al., 1993). This receptor, like other recombinant somatostatin receptor types, functionally inhibits adenylate cyclase activity when expressed in cell lines (O'Carroll et al., 1992; 1994; Patel et al., 1994; Raynor et al., 1993; Panetta et al., 1994) but can also activate phosphoinositide hydrolysis and mobilization of intracellular calcium (Akbar et al., 1994; Wilkinson et al., 1996) through interaction with pertussis toxin (PTx)-sensitive G proteins. In contrast, other sst5-mediated effects such as increases in extracellular acidification (Thurlow et al., 1996), stimulation of adenosine 3′ : 5′-cyclic monophosphate (cyclic AMP) formation (Akbar et al., 1994; Williams et al., 1996) and tritium release from [3H]-arachidonic acid pre-loaded cells (unpublished observations) are mediated, at least in part, via PTx-insensitive pathways.
Signalling diversity of somatostatin receptors is also evident in native systems. In addition to inhibition of adenylate cyclase (Jakobs et al., 1983), somatostatin-14 can inhibit voltage-dependent calcium conductances (Lewis et al., 1986) and activate voltage-dependent potassium channels (Yamashita et al., 1987) via PTx-sensitive G proteins. In contrast, somatostatin-induced effects such as inhibition of cell proliferation (Viguerier et al., 1989), and inhibition of Na+/H+ antiport activity (Barber et al., 1989) can occur via PTx-insensitive pathways.
Pleiotropic effects of recombinant G protein coupled receptors have been widely reported. For example recombinant M1, M2, M3 and M5 muscarinic receptors can couple to phosphoinositide turnover and cyclic AMP production (Ashkenazi et al., 1987; Jones et al., 1991; Burford et al., 1995). In addition to coupling to multiple effector systems, the α2-adrenoceptor receptor (Eason et al., 1992), and the muscarinic M4 receptor (Jones et al., 1991) have been reported to have opposing effects on a single effector enzyme, adenylate cyclase. This can occur by direct coupling to both Gi and Gs or indirectly, through coupling of PTx-sensitive and/or PTx-insensitive G proteins to phospholipase A2 (PLA2) and release of bioactive arachidonate metabolites, as demonstrated in CHO cells expressing the α2-adrenoceptor by Fraser and co-workers (1989). The present study describes the ability of the sst5 receptor to mediate both decreases and increases in cyclic AMP accumulation. By using a number of sst5 receptor-selective agonists exhibiting a range of intrinsic activities we have shown that the former, but not the latter response is a highly efficiently coupled receptor-effector mechanism. In addition, we have provided good evidence that the mechanism underlying the increases in cyclic AMP formation involves direct activation of Gs and not other possible indirect mechanisms including the release of arachidonic acid metabolites. A preliminary account of some of these findings has already been published in abstract form (Williams et al., 1996).
Methods
Cell culture
CHO-K1 cells stably expressing the human epitope-tagged sst5 receptor (CHOsst5; Bmax 35.4±3.14 pmol mg protein−1 (n=4) were obtained from Affymax (San Palo, CA, U.S.A.) and cultured in Dulbecco's modified Eagle's medium (DMEM) / Ham's F-12 nutrient (1 : 1) mix supplemented with Glutamax-1, 10% foetal calf serum and G418 (0.5 mg ml−1). Cells were maintained in 225 cm3 flasks at 37°C in a humidified atmosphere (95% air, 5% CO2) and passaged every 4–5 days. The sst5 receptor epitope tag was derived from the HA influenza virus protein (YPYDVPDYA) and was inserted at position-2 from the N-terminus. This mutation did not alter the affinities of ligands in this receptor system (data not shown).
Cyclic AMP measurements
CHOsst5 cells were harvested with a scraper and suspended in serum-free DMEM, supplemented with bacitracin (0.02 mg ml−1) and 3-isobutyl-1-methylxanthine (IBMX; 0.5 mM). Cyclic AMP determinations in intact cells were performed on 170,000 cells in an assay volume of 300 μl in the presence of forskolin (10 μM) for 10 min at 37°C. Reactions were terminated by the addition of 10 μl 10 M HCl and neutralized with 10 μl 10 M NaOH and 200 μl 1 M Tris-HCl (pH 7.0). Following centrifugation at 8800×g for 20 min, 50 μl of supernatant were added to 100 μl [3H]-cyclic AMP in Tris-HCl 50 mM, NaCl 100 mM, Na2 EDTA 5 mM, pH 7.0 (approximately 1 nM) and binding to the cyclic AMP-binding portion of protein kinase A (100 μl in the above buffer; approximately 2 μg per tube) was measured after 2–4 h at 4°C (Brown et al., 1971). To facilitate manipulation of extracellular calcium levels, cyclic AMP determinations were also performed in HBS (composition mM: NaCl 125, KCl 5.4, NaHCO3 16.2, D-glucose 5.5, (N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid] (HEPES)10, NaH2PO4 1, IBMX 0.5, and either CaCl2 1.3 or ethyleneglycolbis(aminoethylether)tetra-acetic acid 1, 0.02 mg ml−1 bacitracin, (EGTA, pH 7.4). The response profile obtained in this buffer in the presence of 1.3 mM CaCl2 did not differ from that using DMEM media (data not shown). Binding reactions were terminated by rapid vacuum filtration onto pre-wetted (0.5% w/v polyethylenimine) 96 well GF/B filter mats (Top Count™, Packard). Filter mats were dried and bound radioactivity determined after the addition of 50 μl of Microscint-O (Packard) scintillation fluid by counting with a Canberra Packard Topcount Scintillation Counter. Basal levels of cyclic AMP production were 0.008±0.001 nmoles/170,000 cells cyclic AMP, rising to 0.071±0.005 nmoles cyclic AMP/170,000 cells in the presence of 10 μM forskolin representing a 9.7±1.8 fold stimulation (n=4). Following PTx treatment (100 ng ml−1, 18 h), basal cyclic AMP levels were 0.003±0.001 rising to 0.057±0.006 nmoles/170,000 cells representing a 25±6 fold stimulation (n=6). After ChTx treatment or combined ChTx and PTx treatment, basal cyclic AMP levels were 0.134±0.035 and 0.140±0.045 nmoles/170 000 cells, respectively.
Adenylate cyclase activity was also assessed in CHOsst5 membrane preparations. Cells were grown to confluence and harvested with 10 mM HEPES, 0.02% (w v−1) EDTA, 0.9% (w v−1) NaCl, pH 7.4. Cells were homogenized using a polytron homogenizer (speed 5, 4×5 s bursts separated by approximately 30 s) in an ice-cold lysis buffer consisting of 10 mM HEPES, 10 mM EDTA, 10 μg ml−1 leupeptin and 0.2 mg ml−1 bacitracin, pH 7.4. Following centrifugation at 500×g for 2 min to pellet nuclei and unbroken cells, the supernatant was centrifuged at 40,000×g for 20 min. The crude membrane pellet was then resuspended in a cytosol-like assay buffer containing (mM) KCl 120, Na2ATP 2, MgCl2 2.4, KH2PO4 2, sodium succinate 5, HEPES, 20, IBMX 0.5 and GTP 0.05, pH 7.2 with free [Ca2+] buffered to approximately 100 nM with 1 μM EGTA (Burford & Nahorski, 1996). Samples (30 μg protein per tube) were then incubated in a volume of 300 μl with the indicated concentrations of agonist for 20 min at 37°C and processed for cyclic AMP content as described above. Membrane protein was quantified using the copper binichronomic method (Pierce) using BSA as standard.
Measurement of tritium release from [3H]-arachidonic acid pre-loaded cells
CHOsst5 cells were seeded into 16 mm wells (24 well multidishes) and grown to approximately 90% confluency. Cell monolayers were incubated with 1 μCi ml−1 [3H]-arachidonic acid in normal culture media 24 h prior to experimentation. Cells were washed (4×2 ml) in HBS supplemented with 0.1% w v−1 protease-free BSA and cells left to equilibrate for 10 min at 37°C. The wash buffer was removed and replaced with 1 ml of HBS and incubations continued in the presence of the indicated concentrations of somatostatin-14 for 1 h at 37°C. An aliquot (500 μl) of incubation media was then taken and following addition of 2 ml of Ultima Gold XR scintillant (Packard), counted on a Canberra Packard 2500 XR liquid Scintillation Analyser.
Prostaglandin E2 (PGE2) release
PGE2 content of the incubation media after exposure of whole cell monolayers to somatostatin-14 (1 μM) was assessed by enzymeimmunoassay (Amersham). Briefly, cells were plated into 12 well multidishes and grown to approximately 90% confluency. Cell monolayers were then washed (4×2 ml) in HBS and left to equilibrate for 10 min at 37°C. Incubations were performed in a volume of 0.5 ml HBS in either the absence or presence of somatostatin-14 (1 μM) for 10 min and a 50 μl aliquot taken immediately for analysis.
Site-specific synthetic peptides
Synthetic peptides corresponding to amino acids 354–372 of rat Gαs (Jones & Reed, 1987) designated Gαs acetyl-354-372-amide (Rasenick et al., 1994) (sequence: DGRHYCYPHFTCAVDTENI) and residues 345–354 of rat Gαi3 (sequence: NKNLKECGLY), termed Gαi3 345–354, were used as potential competitive inhibitors of sst5 receptor-G protein coupling. Gαs acetyl-354-372-amide has previously been shown to effectively inhibit cyclic AMP formation induced by β-adrenoceptor activation in permeabilized C6 glioma cells and membrane preparations (Rasenick et al., 1994). Peptides were co-incubated with the indicated concentration of agonist for 20 min and sample cyclic AMP content determined as described above.
Somatostatin-14-promoted [35S]-GTPγS binding to Gαs proteins
[35S]-GTPγS binding and immunoprecipitation with antisera to specific Gαs proteins (Wang et al., 1995) was performed as previously described (Burford et al., 1998) with a number of minor modifications. CHOsst5 membranes (75 μg/50 μl) were pre-incubated with the indicated concentrations of somato-statin-14 for 2 min prior to exposure to 2 nM [35S]-GTPγS for 1 min.
ADP-ribosylating toxin treatment
Where indicated, cells were pre-treated with 100 ng ml−1 pertussis toxin (PTx) for 18 h before experimentation. In some experiments, cells were also pre-treated with 20 μg ml−1 cholera toxin either alone or in combination with 100 ng ml−1 PTx for 18 h prior to experimentation.
Data analysis
Cyclic AMP levels following incubation of cells with somatostatin receptor ligands are quoted as the percentage inhibition of the 10 μM forskolin response or as a percentage of the 10 μM forskolin response for stimulation. Maximal inhibition of cyclic AMP accumulation was taken to be the smallest cyclic AMP concentration measured for each ligand on each individual experiment. The concentration of ligand producing this response was taken as the maximum concentration and higher agonist concentrations were excluded from the analysis when calculating a pIC50 value for the inhibition of cyclic AMP accumulation. The pIC50 values for decreasing cyclic AMP accumulation were calculated as the negative log10 of the molar concentration of the agonist producing 50% of the maximal response for that agonist. Similarly, pEC50 values were calculated for the stimulatory effects of the ligands on cyclic AMP accumulation following PTx pre-treatment as the responses reached a maximum effect. Biphasic concentration-effect curves were fitted for presentation purposes only.
Materials
[2,8,-3H]-adenosine 3′:5′-cyclic monophosphate (25–40 Ci mmol−1); [5,6,8,9,11,12,14,153H]-arachidonic acid, guanosine 5′[γ-35S] thiotriphosphate (1000 Ci mmol−1), bacitracin and PGE2 enzymeimmunoassay kit were purchased from Amersham International, (Little Chalford, U.K.). PTx, ChTx, IBMX, GTP (dilithium salt), cyclic AMP and PKA (cyclic AMP-binding regulatory subunit) were all obtained from Sigma Chemical (Poole, U.K.). Gαs acetyl-354–372-amide peptide was custom synthesised by Genosys Biotechnologies (Cambridge, U.K.) and the Gαi3 C-terminal (345–354) synthetic peptide obtained from Calbiochem (Nottingham, U.K.). Gαs antiserum was purchased from Santa Cruz (Calne, U.K.). Unless indicated, all cell culture media and reagents were from Gibco/Life Technologies (Paisley, U.K.) And all plasticware obtained from Costar (High Wycombe, U.K.). All other chemicals were of analytical grade from established commercial sources. Somatostatin-14 and somatostatin-28 were obtained from Peninsula Laboratories, Europe (St Helens, U.K.); BIM-23056 (D-Phe-Phe-Tyr-D-Trp-Lys-Val-Phe-D-Nal-NH2), BIM-23027 (c[N-Me-Ala-Tyr-D-Trp-Lys-Abu-Phe]) and L-362,855 (c[Aha-Phe-Trp-D-Trp-Lys-Thr-Phe]), in which Na1 is β-(2-naphthyl)alanine, Abu is aminobutyric acid and Aha is 7-amino-heptanoic acid, were custom synthesized by Peptide and Protein Research Consultants (Exeter, U.K.). All of the peptides, with the exception of BIM-23056, were prepared in distilled water at 1 mM and stored at −20°C. BIM-23056 was initially dissolved in 10% dimethylsulphoxide (DMSO).
Results
Characteristics of sst5 receptor-mediated cyclic AMP synthesis
Coupling of the sst5 receptor to adenylate cyclase was investigated in whole cells. Somatostatin-14 concentration-dependently (pIC50: 9.93±0.19, n=4, Figure 1A) decreased forskolin (10 μM) cyclic AMP production by 75±3% at 10 nM. However, at higher agonist concentrations, there was a concentration-dependent reduction in the degree of inhibition, and at 10 μM somatostatin, the highest agonist concentration tested, an increase of 515±22% of basal forskolin-stimulated cyclic AMP accumulation was observed (Figure 1B). Consistent with the reported preferential affinity of somatostatin-28 over somatostatin-14 at the human sst5 receptor (Panetta et al., 1994), somatostatin-28 inhibited cyclic AMP accumulation with approximately 6 fold higher potency than somatostatin-14 (pIC50: 10.7±0.22, maximum inhibition 81±4%, n=4, Figure 1A) and displayed a similar profile for the stimulation of cyclic AMP accumulation at higher agonist concentrations (Figure 1B). L-362,855 reduced forskolin (10 μM)-stimulated cyclic AMP production with similar intrinsic activity and potency as somatostatin-14 (pIC50: 10.0±0.1, maximum, inhibition 84±6%, n=4), but its ability to reverse the inhibition of cyclic AMP accumulation at higher agonist concentrations was diminished (Figure 1B). The cyclic hexapeptide BIM 23027, a reported sst2 receptor selective agonist (Raynor et al., 1993), behaved as a full agonist for inhibition of cyclic AMP (pIC50: 8.35±0.15, maximum inhibition 76±3%, n=4, Figure 1A) but was less effective than somatostatin-14 in stimulating cyclic AMP accumulation (Figure 1B). The linear peptide BIM-23056, previously been shown to act as an antagonist at the human recombinant sst5 receptor (Wilkinson et al., 1996; Williams et al., 1997), also inhibited forskolin-stimulated cyclic AMP accumulation with similar intrinsic activity, but with reduced potency to somatostatin-14 and somatostatin-28 (pIC50: 9.65±0.28, maximum inhibition 76±2%, n=4, Figure 1A). High concentrations of BIM-23056 did not stimulate cyclic AMP formation (Figure 1B).
Figure 1.

(A) Concentration-dependency of somatostatin-28, somatostatin-14, L-362,855, BIM-23027 and BIM-23056-mediated inhibition of forskolin (10 μM)-stimulated cyclic AMP formation in CHOsst5 cells. (B) Depicts both the inhibitory and stimulatory response phases of the above agonists on an expanded ordinate scale from the same data set. For subsequent measurement of cyclic AMP, CHOsst5 cells were suspended in DMEM supplemented with 0.5 mM IBMX, 0.02 mg ml−1 bacitracin and 10 μM forskolin and the indicated concentration of agonist for 10 min. Cyclic AMP levels were measured as described in the Methods. Data are expressed as a percentage of the 10 μM forskolin response and are the means ±s.e.mean of 4–5 experiments performed in triplicate.
Effects of PTx
The treatment of CHOsst5 cells for 18 h with 100 ng ml−1 PTx abolished the inhibition of cyclic AMP accumulation elicited by somatostatin-14 and other ligands (Figure 2A). However, pre-treatment of cell monolayers with PTx was associated with a marked enhancement of somatostatin-induced cyclic AMP formation. Somatostatin-14 and somatostatin-28 increased forskolin (10 μM)-stimulated cyclic AMP accumulation by 927±15% (pIC50: 7.14±0.1, n=4) and 928±65% (pIC50: 7.86±0.04, n=4), respectively (Figure 2A). Although L-362,855 exhibited a reduced maximal response in comparison to either somatostatin-14 and somatostatin-28 (694±73%, n=3), it was of similar potency (pIC50: 7.53±0.04, Figure 2A). The effect of BIM-23027 was weaker and was manifest as a smaller increase at 10 μM (418±4%, Figure 2A). In contrast, BIM-23056 was inactive for stimulation of cyclic AMP (Figure 2A) in contrast to it behaving as a full agonist for inhibition of cyclic AMP formation (Figure 1A). In another CHOsst5 clone expressing lower levels of receptor (1.8 pmol mg protein−1), somatostatin-28 and somatostatin-14 stimulated cyclic AMP accumulation in whole cells by 254±5 and 238±8% respectively following PTx pre-treatment (n=5, data not shown).
Figure 2.

(A) Concentration-dependency of somatostatin-28, somatostatin-14, L-362,855, BIM-23027 and BIM-23056-mediated stimulation of forskolin (10 μM)-stimulated cyclic AMP formation in PTx (100 ng ml−1, 18 h) pre-treated CHOsst5 cells. (B) Somatostatin-14-stimulated cyclic AMP accumulation following PTx pre-treatment (100 ng ml−1, 18 h) in the presence of 100 nM, 300 nM, 1 μM and 3 μM BIM-23056. CHOsst5 cells were pre-incubated with the indicated concentrations of BIM-23056 for 15 min and then co-incubated with somatostatin-14 in the presence of 10 μM forskolin for 10 min. Data are expressed as a percentage of the 10 μM somatostatin-14 response and are the means±s.e.mean of 5–6 separate experiments performed in triplicate.
Antagonist effects of BIM-23056
The ability of BIM-23056 to antagonize somatostatin-14-induced cyclic AMP accumulation in PTx-treated CHOsst5 cells was investigated (Figure 2B). The concentration-effect curves to somatostatin-14 were shifted to the right with increasing concentrations of BIM-23056 (0.1, 0.3, 1 and 3 μM), yielding agonist concentration ratios of 8.2±2.9 (n=5), 26.5±10.0 (n=6), 65.5±16.0 (n=6) and 429.1±267 (n=4), respectively. Analysis of these data using the Gaddum-Schild equation yielded mean pKB estimates of 7.86, 7.93, 7.81 and 8.15, respectively, which were not significantly different from one another. Thus, an average mean pKB value for BIM-23056 of 7.94±0.07 was calculated.
Stimulation of tritium release from [3H]-arachidonic acid pre-loaded cells
Tritium release was stimulated by somatostatin-14 with a pEC50: of 7.04±0.03, (1656±183% over basal at 10 μM somatostatin-14, n=8, Figure 3A). In PTx pre-treated cultures, somatostatin-14-mediated tritium release was reduced by 58% (728±49% over basal; pEC50: 6.78±0.05, n=5). Exposure of cell monolayers to ChTx (20 μg ml−1, 18 h) did not modify agonist-mediated responses (1840±329% over basal at 10 μM somatostatin-14; pEC50: 6.83±0.03, n=3) compared to control (Figure 3A). Similarly, basal or somatostatin-14-stimulated tritium release was unaffected by co-incubation with forskolin (10 μM) or 8-bromo-cyclic AMP (500 μM; data not shown).
Figure 3.
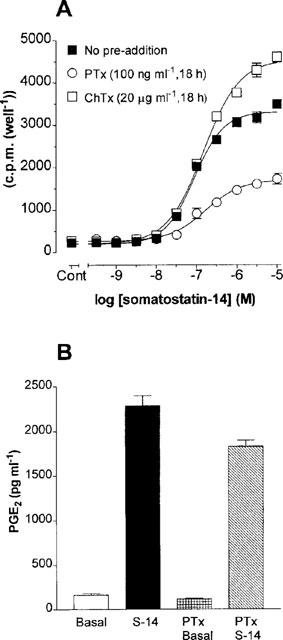
(A) Somatostatin-14-induced tritium release from [3H]-arachidonic acid pre-loaded cells. Cell monolayers were labelled with 1 μCi ml−1 [3H]-arachidonic acid in normal culture media for 18 h and then incubated in HBS supplemented with 0.2% (w v−1) BSA for 1 h in the presence of the indicated concentrations of somatostatin-14. Data represent means±s.e.means of 3–6 experiments performed in triplicate. (B) PGE2 release from CHOsst5 cells. Whole cell monolayers were incubated in the presence of somatostatin-14 (1 μM) in HBS for 10 min and samples analysed by enzymeimmunoassay. Data are the means ±s.e.mean of three experiments performed in triplicate.
Under calcium-free conditions (omission of CaCl2 and addition of 1 mM EGTA to buffer), somatostatin-14 (3 μM)-stimulated tritium release was abolished, although basal release was unchanged. Pre-treatment of PTx-treated cells with indomethacin (10 μM, 40 min) failed to modify somatostatin-14 (3 μM) stimulated cyclic AMP accumulation (97.4±0.3% of control, n=3). A possible role for calcium underlying the residual cyclic AMP stimulation seen after PTx and ChTx treatment was tested directly by use of calcium chelating agents. The magnitude of the 10 μM somatostatin-14 effect was unaffected in the presence of 1 mM EGTA (control: 161±6.4%, +EGTA: 164±18% over basal, n=4) although the concentration-dependency of the response was shifted rightward (pEC50s: control 7.48±0.50, +EGTA 6.39±0.57). Pre-incubation of PTx and ChTx-treated cells for 60 min with the intracellular Ca2+ chelator, BAPTA/AM (100 μM) failed to alter responses to 10 μM somatostatin-14 compared to vehicle-treated control cells (control: 176±13% over basal, pEC50: 7.55±0.30, n=4; BAPTA/AM: 160±7% over basal, pEC50: 7.54±0.20, n=4).
Somatostatin-14 induced PGE2 release from CHOsst5 cells
Exposure of CHOsst5 cell monolayers to 1 μM somatostatin-14 for 10 min increased the PGE2 content of the incubation media by 1409±98% compared to non-stimulated cells (Figure 3B, n=3). After PTx pre-treatment, resting PGE2 levels were slightly reduced although somatostatin-14-induced PGE2 release was unaffected when assessed on a percentage stimulation basis (1605±152% over basal, n=3).
Cholera toxin sensitivity
The mechanism(s) underlying the somatostatin-14-induced increases in forskolin-stimulated cyclic AMP accumulation in PTx-treated cells were investigated with cholera toxin. Cholera toxin (ChTx) ADP-ribosylates and constitutively activates Gαs resulting in a functional occlusion of receptor-Gs protein mediated events. In the present study, an 18 h incubation of CHOsst5 monolayers with a high concentration of ChTx (20 μg ml−1) in the presence of PTx produced a marked elevation of basal cyclic AMP levels (see Methods). Increasing the ChTx concentration to 100 μg ml−1 did not further increase basal cyclic AMP accumulation confirming the adequacy of the toxin concentration in use in the present study (data not shown). In CHOsst5 cells treated with ChTx alone (20 μg ml−1) the stimulatory limb of the response to somatostatin-14 (see Figure 1B) was reduced but inhibition was maintained (Figure 4). In ChTx and PTx-treated cells a modest percentage increase in cyclic AMP formation (168±7.5% over basal at 10 μM somatostatin-14) was the equivalent data in evidence although this was much reduced compared to non-ChTx-treated cells (Figure 1B).
Figure 4.
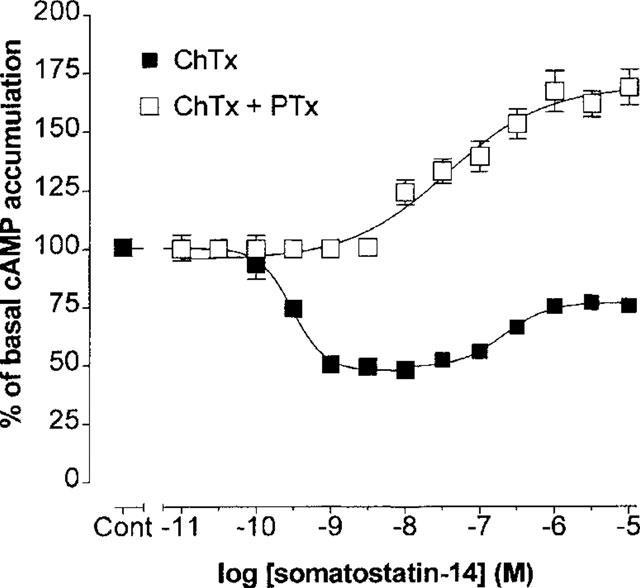
Effect of ChTx pre-treatment (20 μg ml−1, 18 h) on somatostatin-14-induced cyclic AMP formation in the absence or in combination with PTx pre-treatment (100 ng ml−1, 18 h) in CHOsst5 cells. Data are expressed as means±s.e.mean of four separate experiments performed in triplicate.
Somatostatin-14 and PGE2-stimulated cyclic AMP accumulation in CHOsst5 membranes
To examine whether a major component of sst5-mediated cyclic AMP accumulation in whole cells reflects direct stimulation of Gs and adenylate cyclase or an indirect effect attributable to phosphoinositide hydrolysis/Ca2+-PKC signalling, somatostatin-14 stimulation of cyclic AMP accumulation was examined in membrane preparations. Following inactivation of Gi/Go-type G proteins by pre-exposure of cells to PTx (100 ng ml−1, 18 h), membranes were prepared and experiments conducted in the presence of cytosolic levels of free Ca2+ (approximately 100 nM) and 50 μM GTP, a concentration previously found to be optimal for promoting α2-adrenoceptor stimulation of adenylate cyclase activity in PTx-pre-treated CHO membrane preparations (Eason et al., 1994). Under these conditions, 10 μM somatostatin-14 concentration-dependently increased adenylate cyclase activity by 227±23% (pEC50: 6.75±0.12, n=3, Figure 5A). In addition, the ability of PGE2 to stimulate cyclic AMP formation via endogenous EP (presumably EP2-like) prostanoid receptors in CHO-K1 cells was also demonstrated. PGE2 produced concentration-dependent increases in adenylate cyclase activity, the threshold for this effect was 100 nM and at 10 μM PGE2 the increase was 281±11% over basal, n=4 (Figure 5A). In intact cells, 100 μM PGE2 increased 10 μM forskolin-stimulated cyclic AMP accumulation by 375±91% (pEC50: 6.69±0.12, n=3) (data not shown).
Figure 5.

Somatostatin-14 and PGE2-induced cyclic AMP formation (A) and inhibition of somatostatin-14 (1 μM)-stimulated adenylate cyclase activity by the site-specific synthetic peptide Gαs-acetyl-354–372-amide (B) in PTx (100 ng ml−1, 18 h) pre-treated CHOsst5 membranes. Experiments were performed without forskolin and basal cyclic AMP production was 138±16.8 nmoles mg protein−1 (n=10). Assay conditions were as described in the Methods. Values are means±s.e. mean of 3–4 experiments performed in triplicate.
Effect of site-specific synthetic peptides on somatostatin-14 stimulation of adenylate cyclase in CHOsst5 membranes
In membranes prepared from PTx-pre-treated CHOsst5 cells, a synthetic peptide derived from the C-terminal region of rat Gαs, Gαs-acetyl-354–372-amide concentration-dependently decreased somatostatin-14 (1 μM) stimulation of cyclic AMP formation. At the highest peptide concentration tested (100 μM), Gαs-acetyl-354–372-amide inhibited somatostatin-14-stimulated adenylate cyclase activity by 65.9±3.5%, n=3 (Figure 5B). To confirm the specificity of action of Gαs-acetyl-354–372-amide, responses to somatostatin-14 (1 μM) were also examined in the presence of the peptide Gαi3 345–354. Under these conditions, responses to 1 μM somatostatin-14 were unaffected by 100 μM Gαi3 345–354 (103.4±16% of control, n=3).
[35S]-GTPγS binding to G proteins and immunoprecipitation of [35S]-GTPγS bound G proteins with antiserum to Gαs proteins
Activation of sst5 receptors induced concentration-dependent increases in [35S]-GTPγS binding to Gαs in non-PTx pre-treated CHOsst5 membranes (Figure 6). Somatostatin-14 (10 μM) evoked a 327±44% increase in labelling with a pEC50 of 7.75±0.2 (n=3).
Figure 6.

Concentration-dependency of somatostatin-14-stimulated [35S]-GTPγS binding to Gαs proteins in CHOsst5 membranes. Membranes were incubated with the indicated concentrations of somatostatin-14 for 2 min and then in the presence of 2 nM [35S]-GTPγS for a further minute at 30°C. Immunoprecipitated 35S-labelled Gαs proteins were then counted. Basal [35S]-GTPγS binding to Gαs was 198±27 c.p.m. Data are the means±s.e.mean of three experiments performed in triplicate.
Discussion
The ability of the human sst5 receptor to couple to multiple transduction processes is well established (Akbar et al., 1994; Wilkinson et al., 1997; Thurlow et al., 1996). The present study has investigated the inhibitory and stimulatory effects of sst5 receptor activation on adenylate cyclase activity in CHOsst5 cells and in addition, the operational characteristics of a number of agonists have been assessed on both transduction events. Moreover, the use of ligands that possess a wide spectrum of intrinsic activity in other experimental paradigms such as [35S]-GTPγS binding (Williams et al., 1997) and stimulation of phosphoinositide hydrolysis (Wilkinson et al., 1997) have provided insight into the efficiency of different somatostatin5 receptor transduction pathways.
In confirmation of the preferential affinity of somatostatin-28 over somatostatin-14 at the human sst5 receptor, somatostatin-28 inhibited cyclic AMP formation with higher potency than somatostatin-14. Moreover, adenylate cyclase inhibition profiles were highly correlated with relative affinity estimates for [125I]-Tyr11-somatostatin-14 binding at the sst5 receptor (Williams et al., 1997). The agonist rank order for inhibition of cyclic AMP formation was somatostatin-28>L-362,855>somatostatin-14>BIM-23056>BIM-23027. However, in the present study, BIM-23056, previously shown to act as a competitive antagonist of somatostatin-14-induced activation of phosphoinositide hydrolysis (Wilkinson et al., 1997) behaved as a full agonist (76% inhibition at 10 nM) for inhibition of cyclic AMP formation. Similarly, L-362,855 also behaved as a full agonist for adenylate cyclase inhibition, findings that accord with [35S]-GTPγS studies under low sodium conditions (Williams et al., 1997). At higher agonist concentrations, the reversal of inhibition to that of stimulation of adenylate cyclase followed a similar rank order profile but under these conditions, L-362,855 behaved as a partial agonist and BIM-23027 and BIM-23056 were devoid of agonist activity. In the case of BIM-23056, the lack of intrinsic activity for stimulatory but not the inhibitory pathway may have its basis in conformational changes that are conferred by this ligand to the receptor in the presence of a given G protein population.
After inactivation of G proteins by exposure of cells to PTx, the inhibitory effect of somatostatin receptor ligands was abolished, implicating Gi/Go type G proteins in these responses. The stimulatory effects of all agonists (except BIM-23056) were enhanced suggesting that functional antagonism occurs between the inhibitory and the stimulatory response phases. Agonist rank order of potencies for stimulation of adenylate cyclase activity were identical to that for inhibition except that BIM-23056 was inactive (somatostatin-28>L-362,855>somatostatin-14>BIM-23027). This profile is similar to that obtained for [35S]-GTPγS binding (Williams et al., 1997) and measurement of extracellular acidification (Thurlow et al., 1996). Thus, ligand intrinsic activity appears to reflect the receptor-effector coupling efficiency in a given system. BIM-23056 exhibited high affinity binding at the sst5 receptor but appeared inactive in stimulating cyclic AMP accumulation in PTx-treated cells; rather it behaved as an antagonist and shifted the concentration-effect curve to somatostatin-14 rightward. Gaddum-Schild analysis was consistent with competitive antagonism. The calculated pKB value for BIM-23056 of 7.9 was similar to that obtained for blockade of somatostatin-14-induced increases in intracellular Ca2+ mobilization and [35S]-GTPγS binding (Wilkinson et al., 1996; Williams et al., 1997).
Significantly, a recent study has identified types VI and VII to be the major adenylate cyclase mRNAs present in CHO cells (Varga et al., 1998). It is therefore unlikely that the calcium-dependent types I and III or the diacylglycerol-activated type II isozymes contribute appreciably to the observations made in the present study (Iyengar, 1993). Moreover, the reported PTx-sensitivity of calcium mobilization in CHOsst5 cells (Wilkinson et al., 1997) is incompatible with the observed PTx-insensitive stimulation of cyclic AMP accumulation in the present study. For the same reason, a role for Gαi/Gαo derived βγ subunits that can activate adenylate cyclase types II, IV and VII in the presence of activated Gs (Tang & Gilman, 1991; Federman et al., 1992; Iyengar, 1993) can also be discounted.
Although ChTx treated cells contained high basal levels of cyclic AMP, somatostatin-14-induced cyclic AMP formation was inhibited implicating a role for Gs proteins. However, there was a remaining stimulation for which there is no obvious explanation. However, we can exclude phosphoinositidase C activation as neither BAPTA or EGTA had any significant effects on somatostatin-14-mediated cyclic AMP formation. This ChTx-resistant effect could be due to either incomplete ADP-ribosylation of Gs substrate or transduction via other PTx-insensitive and ChTx-insensitive G proteins such as G13, a species known to be present in CHO-K1 cells (Strathmann & Simon, 1991; Berg et al., 1998).
Evidence suggesting that sst5-mediated increases in adenylate cyclase activity are predominantly due to direct coupling to Gs-type G proteins was obtained in membrane preparations. Under these conditions, responses to somatostatin-14 were maintained in the absence of forskolin and in the presence of nanomolar levels of Ca2+. Somatostatin-14 evoked an increase in cyclic AMP formation, suggesting that a direct coupling to Gαs and not elevated Ca2+ levels underlie responses in whole cells. Since ChTx-treated cells had high basal cyclic AMP levels which might have masked a stimulatory effect of somatostatin-14, further evidence of functional sst5 receptor-Gαs coupling in CHOsst5 membranes was provided using the Gαs-acetyl-354–372-amide site-specific synthetic peptide (Jones & Reed, 1987). The significance of the Gαs C-terminal domain in receptor-G protein interactions is widely appreciated (Masters et al., 1988) and in previous studies this peptide has been shown to block isoproterenol-stimulated adenylate cyclase activity by 50% in C6 glioma cell membranes (Rasenick et al., 1994). In the present study, 100 μM Gαs-acetyl-354–372-amide inhibited 1 μM somatostatin-14-induced cyclic AMP formation by 65%. The inhibitory effect of Gαs-acetyl-354–372-amide could not be attributed to an antagonistic effect at the sst5 receptor. In [125I]-Tyr11-somatostatin-14 binding studies, Gαs-acetyl-354–372-amide did not inhibit binding and at 100 μM a small increase in specific binding was observed (data not shown). More significantly, evidence of sst5-Gαs coupling in membranes was obtained using [35S]-GTPγS binding and immunoprecipitation of 35S-labelled Gαs proteins by specific antisera. Using this strategy, concentration-dependent increases in labelling (>3 fold) could be demonstrated with a pEC50 value (7.7) which is close to that for somatostatin-14-mediated increases in cyclic AMP seen in pertussis toxin-treated whole cells (7.1).
We examined the possibility that the somatostatin-14 mediated increase in cyclic AMP involving Gαs may be partially mediated via release of arachidonic acid and its metabolites. The present study provides evidence that sst5 receptor activation stimulates tritium release from [3H]-arachidonic acid preloaded cells with apparent pEC50 value of 7.04, similar to that of stimulation of cyclic AMP accumulation (7.1). It is notable that in many studies of this type, it is assumed that [3H]-arachidonic acid is liberated intact after preloading (Bito et al., 1994; Sakanaka et al., 1994). In this regard, it has been shown that PGE2 is the major arachidonic acid metabolite in CHO cells expressing the rat D2 receptor (DiMarzo & Piomelli, 1992). In the present study, we demonstrated a 14–16 fold increase in PGE2 levels following exposure to 10 μM somatostatin-14 for 10 min. This would represent a final media PGE2 concentration of about 4 nM, a level below the threshold to stimulate adenylate cyclase activity in both intact cells and membranes. It could be argued that within the microenvironment of the plasmalemmal membrane, localized concentrations might be higher particularly over longer incubation periods than that used in the adenylate cyclase assay. However, an indirect mechanism involving release arachidonic acid metabolites cannot account for the somatostatin-14-stimulated cyclic AMP formation because of the lack of effect of ChTx on tritium release. Furthermore, the amount of somatostatin-14-induced PGE2 release was insufficient to stimulate cyclic AMP and somatostatin-14-induced increases in cyclic AMP were not modified by indomethacin.
It is unclear whether there is any physiological significance of the positive coupling of the sst5 receptor to adenylate cyclase and consequent increases in cyclic AMP formation. In HT-29cl.19A colonocytes (Warhurst et al., 1995) inhibition by somatostatin-14 of PGE2-stimulated cyclic AMP formation was shown to be monophasic although there is a report of biphasic effects of somatostatin-14 but not SMS-201-995 on cyclic AMP formation in the rat hippocampus and substantia nigra (Markstein et al., 1989). In these brain regions, high concentrations (0.3 μM), somatostatin-14 caused a 20 and 10% increase in cyclic AMP formation, respectively, although it is unclear whether this reflects a direct or indirect action of somatostatin. Amongst recombinant somatostatin receptor types, the marked stimulation of cyclic AMP formation appears to be restricted to human sst2 and sst5 receptor types in CHO-K1 cells expressing similar levels of receptor (unpublished observations). The fact that similar observations have not been made in rat (O'Carroll et al., 1992) or murine (Baumeister et al., 1998) sst5 receptors may be due to the low sequence identities of these receptors with the human orthologue (80.5 and 81.7% respectively). However, the human sst5 receptor described by Panetta et al. (1994) was expressed in COS-7 cells at a much lower density (162±30 fmol mg protein−1) and no stimulation of cyclic AMP was observed, although effects after PTx-treatment were not tested. Similarly, in the study of O'Carroll and co-workers (1994), a low receptor expression of 196 fmol mg protein−1 was determined in CHO-K1 cells with no reversal to stimulation of cyclic AMP formation. In the light of these observations, it is likely that this phenomenon may indeed be receptor expression-dependent, although it is notable that the observations made by Akbar et al. (1994) were seen at expression levels that can be considered to be physiological (∼400 fmol mg protein−1). It bears emphasis that this phenomenon does not occur in COS-7 cells expressing sst3 or sst4 (Akbar et al., 1994) and that endogenous sst receptor expression in some cell lines e.g. AR4 2J can be as high as 2.48 pmol mg protein−1 (Taylor et al., 1994).
In conclusion, the human somatostatin sst5 receptor expressed in CHO-K1 cells potently mediated inhibition of forskolin-stimulated adenylate cyclase activity such that even low efficacy agonists exhibited maximal intrinsic activity. At much higher agonist concentrations the receptor mediated stimulation of cyclic AMP accumulation, indicating a lower receptor-effector coupling efficiency for the latter transduction pathway. Evidence has been presented that suggests that this stimulation of cyclic AMP accumulation is mediated predominantly by a direct interaction with Gαs-type G proteins, since we have precluded a number of plausible indirect mechanisms including the release of prostaglandin E2 which occurs at concentrations too low to be involved.
Abbreviations
- BAPTA-AM
[1,2-bis(o-amino-5-fluorophenoxy)ethane-N,N,N′N′-tetraacetic acid tetra(acetoxymethyl)ester]
- ChTx
cholera toxin
- cyclic AMP
adenosine 3′ : 5′-cyclic monophosphate
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethyleneglycolbis(aminoethyl-ether)-tetra-acetic acid
- GTP
guanosine 5′-triphosphate
- [35S]-GTPγS
guanosine 5′[γ-35S]thiotriphosphate
- HEPES
(N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulphonic acid]
- IBMX
3-isobutyl-1-methylxanthine
- PTx
pertussis toxin
References
- AKBAR M., OKAJIMA F., TOMURA H., MAJID M.A., YAMADA Y., SEINO S., KONDO Y. Phospholipase C activation and Ca2+ mobilization by cloned human somatostatin receptor subtypes 1–5, in transfected COS-7 cells. FEBS Lett. 1994;348:192–196. doi: 10.1016/0014-5793(94)00603-2. [DOI] [PubMed] [Google Scholar]
- ASHKENAZI A., WINSLOW J.W., PERALTA E.G., PETERSON G.L., SCHIMERLIK M.I., CAPON D.J., RAMACHANDRAN J. An M2 muscarinic receptor subtype coupled to both adenylyl cyclase and phosphoinositide turnover. Science. 1987;238:672–675. doi: 10.1126/science.2823384. [DOI] [PubMed] [Google Scholar]
- BARBER D.L., MCGUIRE M.E., GANZ M.B. β-adrenergic and somatostatin receptors regulate Na-H exchange independent of cAMP. J. Biol. Chem. 1989;264:21038–21042. [PubMed] [Google Scholar]
- BAUMEISTER H., KREUZER O.J., ROOSTERMAN D., SCHAFER J., MEYERHOF W. Cloning, expression, pharmacology and tissue distribution of mouse somatostatin receptor subtype 5. J. Neuroendocrinol. 1998;10:283–290. doi: 10.1046/j.1365-2826.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- BERG K.A., MAAYANI S., GOLDFARB J., SCARAMELLINI C., LEFF P., CLARKE W.P. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: Evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- BITO H., MORI M., SAKANAKA C., TAKANO T., HONDA Z., GOTOH Y., NISHIDA E., SHIMIZU T. Functional coupling of SSTR4, a major hippocampal somatostatin receptor, to adenylate cyclase inhibition, arachidonate release, and activation of the mitogen-activated protein kinase cascade. J. Biol. Chem. 1994;269:12722–12730. [PubMed] [Google Scholar]
- BROWN B.L., ALBANO J.D.M., ELKINS R.P., SGHERZI A.M., TAMPION W. A simple and sensitive saturation assay method for the measurement of adenosine 3′,5′-cyclic monophosphate. Biochem. J. 1971;121:561–562. doi: 10.1042/bj1210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURFORD N.T., NAHORSKI S.R. Muscarinic M1 receptor-stimulated adenylate cyclase activity in Chinese hamster ovary cell is mediated by Gsα and is not a consequence of phosphoinositide C activation. Biochem. J. 1996;315:883–888. doi: 10.1042/bj3150883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURFORD N.T., TOBIN A.B., NAHORSKI S.R. Differential coupling of m1, m2 and m3 muscarinic receptor subtypes to inositol-1,4,5-trisphosphate and adenosine 3′,5′-cyclic monophosphate accumulation in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 1995;274:134–142. [PubMed] [Google Scholar]
- BURFORD N.T., TOLBERT L.M., SADEE W. Specific G protein activation and μ-opioid receptor internalization caused by morphine, DAMGO and endomorphin I. Eur. J. Pharmacol. 1998;342:123–126. doi: 10.1016/s0014-2999(97)01556-2. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., PIOMELLI D. Participation of prostaglandin E2 dopamine D2 receptor-dependent potentiation of arachidonic acid release. J. Neurochem. 1992;59:379–382. doi: 10.1111/j.1471-4159.1992.tb08915.x. [DOI] [PubMed] [Google Scholar]
- EASON M.G., JACINTO M.T., LIGGETT S.B. Contribution of ligand structure to activation of α2-adrenergic receptor subtype coupling to Gs. Mol. Pharmacol. 1994;45:696–702. [PubMed] [Google Scholar]
- EASON M.G., KUROSE H., HOLT B.D., RAYMOND J.R., LIGGETT S.B. Simultaneous coupling of α2-adrenergic receptors to two G-proteins with opposing effects. J. Biol. Chem. 1992;267:15795–15801. [PubMed] [Google Scholar]
- FEDERMAN A.D., CONKLIN B.R., SCHRADER K.A., REED R.R., BOURNE H.R. Hormonal stimulation of adenylyl cyclase through Gi-protein beta gamma subunits. Nature. 1992;356:159–161. doi: 10.1038/356159a0. [DOI] [PubMed] [Google Scholar]
- FRASER C.M., ARAKAWA S., MCCOMBIE W.R., VENTER J.C. Cloning, sequence analysis and permanent expression of a human α2-adrenergic receptor in Chinese hamster ovary cells. J. Biol. Chem. 1989;264:11754–11761. [PubMed] [Google Scholar]
- IYENGAR R. Molecular and functional diversity of mammalian Gs-stimulated adenylyl cyclases. FASEB. J. 1993;7:768–775. doi: 10.1096/fasebj.7.9.8330684. [DOI] [PubMed] [Google Scholar]
- JAKOBS K.H., AKTORIES K., SCHULTZ G. A nucleotide regulatory site for somatostatin inhibition of adenylate cyclase in S49 lymphoma cells. Nature. 1983;303:177–178. doi: 10.1038/303177a0. [DOI] [PubMed] [Google Scholar]
- JONES D.T., REED R.R. Molecular cloning of five GTP-binding protein cDNA species from rat olfactory neuroepithilium. J. Biol. Chem. 1987;262:14241–14249. [PubMed] [Google Scholar]
- JONES S.V.P., HEILMAN C.J., BRANN M.R. Functional responses of cloned muscarinic receptors expressed in CHO-K1 cells. Mol. Pharmacol. 1991;40:242–247. [PubMed] [Google Scholar]
- LEWIS D.F., WEIGHT F., LUNI A. A guanine nucleotide-binding protein mediating the inhibition of voltage-dependent calcium current by somatostatin in a pituitary cell line. Proc. Natl. Acad. Sci U.S.A. 1986;83:9035–9039. doi: 10.1073/pnas.83.23.9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANDARINO L., STENNER D., BLANCHARD W., NISSEN S., GERICH J., LING N., BRAZEAU P., BOHLEN P., ESCH F., GUILLEMIN R. Selective effects of somatostatin-14,-25 and -28 on in vitro insulin and glucagon secretion. Nature. 1981;291:76–77. doi: 10.1038/291076a0. [DOI] [PubMed] [Google Scholar]
- MARKSTEIN R., STOCKLI K.A., REUBI J.-C. Differential effects of somatostatin on adenylate cyclase as functional correlates for different brain somatostatin receptor subpopulations. Neurosci. Lett. 1989;104:13–18. doi: 10.1016/0304-3940(89)90321-2. [DOI] [PubMed] [Google Scholar]
- MASTERS S.B., SULLIVAN K.A., MILLER R.T., BEIDERMAN B., LOPEZ N.G., RAMACHANDRAN J., BOURNE H.R. Carboxyl terminal domain of Gsα specifies coupling of receptors to stimulation of adenylyl cyclase. Science. 1988;241:448–451. doi: 10.1126/science.2899356. [DOI] [PubMed] [Google Scholar]
- O'CARROLL A.-M., LOLAIT S.J., KONIG M., MAHAN L.C. Molecular cloning and expression of a pituitary somatostatin receptor with preferential affinity for somatostatin-28. Mol. Pharmacol. 1992;42:939–946. [PubMed] [Google Scholar]
- O'CARROLL A.-M., RAYNOR K., LOLAIT S.J., REISINE T. Characterization of cloned human somatostatin receptor SSTR5. Mol. Pharmacol. 1994;46:291–298. [PubMed] [Google Scholar]
- PANETTA R., GREENWOOD M.T., WARSZYNSKA A., DEMCHYSHYN L., DAY R., NIZNIK H.B., SRIKANT C.B., PATEL Y.C. Molecular cloning, functional characterization, and chromosomal localisation of a human somatostatin receptor (somatostatin receptor type 5) with preferential affinity for somatostatin-28. Mol. Pharmacol. 1994;45:417–427. [PubMed] [Google Scholar]
- PATEL Y.C., GREENWOOD M.T., WARSZYNSKA A., PANETTA R., SRIKANT C.B. All five cloned human somatostatin receptors (hSSTR1-5) are functionally coupled to adenylyl cyclase. Biochem. Biophys. Res. Comm. 1994;198:605–612. doi: 10.1006/bbrc.1994.1088. [DOI] [PubMed] [Google Scholar]
- RASENICK M.M., WATANABE M., LAZEREVIC M.B., HATTA S., HAMM H.E. Synthetic peptides as probes for G protein function. J. Biol. Chem. 1994;269:21519–21525. [PubMed] [Google Scholar]
- RAYNOR K., MURPHY W.A., COY D.H., TAYLOR J.E., MOREAU J., YASUDA K., BELL G.I., REISINE T. Cloned somatostatin receptors: identification of subtype-selective peptide and demonstration of high affinity binding of linear peptides. Mol. Pharmacol. 1993;43:838–844. [PubMed] [Google Scholar]
- REISINE T., BELL G.I. Molecular properties of somatostatin receptors. Neuroscience. 1995;67:777–790. doi: 10.1016/0306-4522(95)00072-q. [DOI] [PubMed] [Google Scholar]
- SAKANAKA C., FERBY I., WAGA I., BITO H., SHIMIZU T. On the mechanism of cytosolic phospholipase A2 activation in CHO cells carrying somatostatin receptor: Wortmannin-sensitive pathway to activate mitogen-activated protein kinase. Biochem. Biophys. Res. Comm. 1994;205:18–23. doi: 10.1006/bbrc.1994.2623. [DOI] [PubMed] [Google Scholar]
- SCHINDLER M., HUMPHREY P.P.A., EMSON P.C. Somatostatin receptors in the central nervous system. Prog. Neurobiol. 1996;50:9–47. doi: 10.1016/0301-0082(96)00030-5. [DOI] [PubMed] [Google Scholar]
- STRATHMANN M.P., SIMON M.I. Gα12 and Gα13 subunits define a fourth class of G protein α subunits. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5582–5586. doi: 10.1073/pnas.88.13.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG W.J., GILMAN A.G. Type-specific regulation of adenylyl cyclase by G protein βγ subunits. Science. 1991;254:1500–1503. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- TAYLOR J.E., THEVENIAU M.A., BASHIRZADEH R., REISINE T., EDEN P.A. Detection of somatostatin receptor subtype 2 (SSTR2) in established tumors and tumor cell lines: Evidence for SSTR2 heterogeneity. Peptides. 1994;15:1229–1236. doi: 10.1016/0196-9781(94)90146-5. [DOI] [PubMed] [Google Scholar]
- THURLOW R.T., SELLERS L., COOTE J.E., FENIUK W., HUMPHREY P.P.A. Human recombinant sst5 receptors expressed in CHO-K1 cells mediate increases in extracellular acidification by pertussis toxin-sensitive and -insensitive pathways. Br. J. Pharmacol. 1996;117:9P. [Google Scholar]
- VARGA E.V., STROPOVA D., RUBENIK M., WANG M., LANDSMAN R.S., ROESKE W.R., YAMAMURA H.I. Identification of adenylyl cyclase isoenzymes in CHO and B82 cells. Eur. J. Pharmacol. 1998;348:R1–R2. doi: 10.1016/s0014-2999(98)00258-1. [DOI] [PubMed] [Google Scholar]
- VIGUERIER N., TAHIN-JOUTI N., AYRIL A.M., CAMBILLAN C., SCEMAMA J.L., BASTIE M.J., KRUHTESEN S., ESTERIE J.P., PAYDAYROL L., SUSINI C., VAYSSE N. Direct inhibitory effects of a somatostatin analogue, SMS 201–995, on AR4-2J cell proliferation via a pertussis toxin-sensitive guanosine triphosphate-binding protein-independent mechanism. Endocrinology. 1989;124:1017–1025. doi: 10.1210/endo-124-2-1017. [DOI] [PubMed] [Google Scholar]
- WANG H.-Y., UNDUE A.S., FRIEDMAN E. Evidence for the coupling of Gq protein into Di-like dopamine sites in rat striatum: Possible role in dopamine-mediated inositol phosphate formation. Mol. Pharmacol. 1995;48:988–994. [PubMed] [Google Scholar]
- WARHURST G., BARBEZAT G.O., HIGGS N.B., REYL-DESMAR F., LEWIN M.J.M., COY D.H., ROSS I., GRIGOR M.R. Expression of somatostatin receptor genes and their role in inhibiting Cl secretion in HT-29cl.19A colonocytes. Am. J. Physiol. 1995;269:G729–G736. doi: 10.1152/ajpgi.1995.269.5.G729. [DOI] [PubMed] [Google Scholar]
- WILKINSON G.F., FENIUK W., HUMPHREY P.P.A. Characterization of human recombinant somatostatin sst5 receptors mediating activation of phosphoinositide metabolism. Br. J. Pharmacol. 1997;121:91–96. doi: 10.1038/sj.bjp.0701116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILKINSON G.F., THURLOW R.J., SELLERS L.A., COOTE J.E., FENIUK W., HUMPHREY P.P.A. Potent antagonism by BIM-23056 at the human recombinant somatostatin sst5 receptor. Br. J. Pharmacol. 1996;118:445–447. doi: 10.1111/j.1476-5381.1996.tb15423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS A.J., MICHEL A.D., FENIUK W., HUMPHREY P.P.A. The human recombinant sst5 receptor couples to pertussis toxin-sensitive and -insensitive G-proteins. Br. J. Pharmacol. 1996;119:11P. [Google Scholar]
- WILLIAMS A.J., MICHEL A.D., FENIUK W., HUMPHREY P.P.A. Somatostatin5 receptor-mediated binding: [35S]guanosine-5′-0-(3-thio)triphosphate binding: Agonist potencies and the influence of sodium chloride on intrinsic activity. Mol. Pharmacol. 1997;51:1060–1069. doi: 10.1124/mol.51.6.1060. [DOI] [PubMed] [Google Scholar]
- YAMADA Y., KAGIMOTO S., KUBOTA A., YASUDA K., MASUDA K., SOMEGA Y., IHARA Y., LI Q., IMURA H., SEINO S., SEINO Y. Cloning, functional expression and pharmacological characterization of a fourth (hSSTR4) and a fifth (hSSTR5) human somatostatin receptor subtype. Biochem. Biophys. Res. Comm. 1993;195:844–852. doi: 10.1006/bbrc.1993.2122. [DOI] [PubMed] [Google Scholar]
- YAMASHITA N., SHIBUYA N., OGATA E. Hyperpolarisation of the membrane potential caused by somatostatin in dissociated human pituitary adenoma cells that secrete growth hormone. Proc. Natl. Acad. Sci. U.S.A. 1987;83:6198–6202. doi: 10.1073/pnas.83.16.6198. [DOI] [PMC free article] [PubMed] [Google Scholar]