

Abstract
The present study addresses the differences in binding profiles and functional properties of the human and rat bradykinin (BK) B2 receptor using various kinin receptor peptide derivatives as well as the non-peptide receptor antagonists WIN 64338 (phosphonium, [[4-[[2-[[bis(cyclohexylamino)methylene]amino]-3-(2-naphtalenyl)1-oxopropyl]amino]-phenyl]-methyl]tributyl, chloride, monohydro-chloride), and FR173657 (E)-3-(6-acetamido-3-pyridyl)-N-[-N-[2,4-dichloro-3-[(2-methyl-8-quinolinyl)oxymethyl]-phenyl]N-methylamino carbonyl methyl] acrylamide.
[3H]-BK bound with a similar affinity to membranes of Chinese hamster ovary cells (CHO-K1) expressing the cloned human (hB2-CHO) or rat (rB2-CHO) B2 receptor, human embryonic intestine cells (INT407) expressing the native B2 receptor, human umbilical vein (HUV) and rat uterus (RU). WIN 64338 and FR173657 bound with a 3.8–6.6 fold and 7.0–16.3 fold higher affinity the rat than the human B2 receptor, respectively. The affinity values of BK derivatives as well as non-peptide antagonists were reduced by 6–23 fold in physiological HBSS compared to low ionic strength TES binding buffer.
BK (0.01–3000 nM) increased inositol triphosphates (IP3) levels in hB2-CHO, rB2-CHO and INT407 cells. The B2 receptor antagonist, Hoe 140 (D-Arg0-[ Hyp3, Thi5, D-Tic7, Oic8]-BK) at 10−7 M, significantly shifted to the right the IP3 response curves to BK giving apparent pKB values of 8.56, 9.79 and 8.84 for hB2-CHO, rB2-CHO and INT407 cells, respectively.
In human isolated umbilical vein, Hoe 140, D-Arg0-[Hyp3, D-Phe7, Leu8]-BK and NPC 567 had a lower potency in functional assays (pKB 8.18, 5.77 and 5.60, respectively) than expected from their affinity in binding studies (pKi 10.52, 8.64 and 8.27, respectively).
FR173657 behaved as a high affinity ligand with pKi values of 8.59 and 9.81 and potent competitive antagonist with pKB values of 7.80 and 8.17 in HUV and RU, respectively. FR173657 bound with a similar affinity the cloned and native bradykinin B2 receptor in human (pKi of 8.66 and 8.59, respectively) and in rat (pKi 9.67 and 9.81, respectively).
In conclusion, we suggest that the binding buffer composition has to be taken into account when screening new compounds and that inter-species differences should be considered when setting up animal models with the aim of developing bradykinin B2 receptor antagonists as therapeutic agents.
Keywords: Bradykinin, kinin B2 receptors, Hoe 140 (icatibant), non-peptide antagonists
Introduction
Activation of B2 receptors by kinins is thought to be involved in a number of diseases associated with inflammation and pain (Bhoola et al., 1992). Therefore, selective B2 receptor antagonists may provide new drugs for the treatment of inflammatory conditions (Stewart, 1995). The bradykinin B2 receptor is a G-protein coupled receptor constitutively expressed in a number of cell types including endothelial cells, smooth muscle cells, fibroblasts, synovioblasts, osteoblasts and astrocytes (Bhoola et al., 1992). Cloning of mouse, rat and human B2 receptor cDNA revealed that the intronless coding sequence for the mouse B2 receptor was 92% identical to the rat B2 receptor and 84% identical to the human B2 receptor (McEachern et al., 1991; Hess et al., 1994). More recently, the rabbit bradykinin B2 receptor amino acid sequence has been shown to be more than 80% identical to the human, rat and mouse B2 receptor sequences (Bachvarov et al., 1995). Despite such a high degree of homology, however the pharmacological profile of B2 receptors differs markedly between species. For instance, large differences in the binding affinity of certain synthetic peptides to mouse and human B2 receptors have been reported (Hess et al., 1994). In addition, a marked dissociation has been observed between the binding affinity of some, but not all, peptide antagonists and their inhibitory effect in functional bioassays (Hall, 1992; Burch et al., 1993). This so-called, binding paradox, remains unexplained.
So far, two non-peptide B2 receptor antagonists have been described; WIN 64338, a compound with a Ki value of approximately 60 nM for the human B2 receptor of IMR-90 cells (Sawutz et al., 1994) and, more recently, FR173657 described as a novel and potent non-peptide B2 receptor antagonist (Aramori et al., 1997; Asano et al., 1997; Griesbacher et al., 1997; Rizzi et al., 1997). We decided to investigate species variations and differences between binding affinity and inhibitory effects in functional tests of peptide and non-peptide compounds. Therefore, we compared the binding properties of the B2 receptor peptide ligands bradykinin (BK), kallidin (KD), Hoe 140 (D-Arg0-[ Hyp3, Thi5, D-Tic7, Oic8]-BK), NPC 567 (D-Arg0 [Hyp3, D-Phe7]-BK) and D-Arg0-[Hyp3, D-Phe7, Leu8]-BK with those of the non-peptide WIN 64338 (phosphonium, [[4-[[2-[[bis(cyclohexylamino)methylene]amino] -3-(2 -naphtalenyl)1-oxopropyl] amino] -phenyl] -methyl]tributyl, chloride, monohydrochloride), and FR173657 (E)-3-(6-acetamido-3-pyridyl) -N-[-N- [2,4-dichloro-3- [(2-methyl-8- quinolinyl) oxymethyl] phenyl]-N-methylaminocarbonylmethyl] acrylamide to rat and human B2 receptors. In addition, the binding paradox issue was addressed by comparison of the binding affinity of both peptide and non-peptide B2 receptor antagonists for native B2 receptors in two different binding buffer conditions. Functional data of antagonists were obtained against BK-induced contractions of isolated human umbilical vein (HUV) and rat uterus (RU).
Methods
Cloning and expression of the human and rat B2 receptor in CHO cells
The coding region of the human receptor was isolated by Polymerase Chain Reaction (PCR) using Goldstar polymerase (Eurogentec, Serain, Belgium) and the human genomic DNA from HepG2 cells as a template as well as the 5′-primer GCGCGAATTCTTTCAGCGCCGAC ATG CTC AAT GTC and the 3′-primer GCGCTCTAGATGTCCCTCAATCCTTACACAAATTCACAGC. The primers were designed so that the PCR fragment begins 14 bp upstream of the initiator methionine and ends 13 bp downstream of the stop codon. The PCR product was subcloned into the EcoRI and XbaI sites of the vector pBlueScript SK− (Stratagene, Ozyme, Montigny le Bretoneux, France). The DNA sequence analysis of the subcloned PCR product confirmed that it was identical to that published by Hess et al. (1992). The recombinant plasmid was digested with EcoRI and XbaI and the insert was subcloned into the eukaryotic expression vector pcDNA3 (Invitrogen, Leek, Netherlands).
The cDNA of the rat B2 receptor subcloned in pRC/CMV was kindly provided by Dr J. Navarro (University of Texas Medical Branch, Galveston, Texas).
Chinese Hamster ovary cells (CHO-K1, ATCC CRL 61) were maintained in HAM F12 containing 10% foetal calf serum, 100 mg 1−1 streptomycin and 105 units 1−1 penicillin. Cells were transfected with the two different cDNA containing vectors (10 μg per plate of 150 mm in diameter) using the calcium phosphate precipitation method (Chen & Okayama, 1987). Transfected cells were allowed to recover 3 days and were then subjected to selection pressure with 500 μg ml−1 geneticin (Gibco, Cergy-Pontoise, France). Resistant cells were propagated and individual cell clones were isolated by limiting dilution plating. Cell clones were screened for receptor expression and then propagated.
Binding assays
CHO cells were grown in Ham F12 containing 10% foetal calf serum, 4.5 g 1−1 glucose, 100 mg 1−1 streptomycin and 105 units 1−1 penicillin. INT407 cells were cultured in 4.5 g 1−1 glucose DMEM containing 10% foetal calf serum, 100 mg 1−1 streptomycin and 105 units 1−1 penicillin. CHO and INT407 cells were scrapped in TES binding buffer solution of the following composition: TES (N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid; pH 6.8) (20 mM), 1,10-phenantroline (1 mM), 140 μg ml−1 bacitracine and 0.1% bovine serum albumin. Cells were homogenized for 10 s using a Polytron homogenizer (Kinematica GmBh, Luzern, Switzerland) (setting 6). Membranes were pelleted at 40,000×g for 15 min and resuspended in ice cold TES binding buffer.
Human umbilical cords were collected after spontaneous delivery and immediately placed at 4°C in a Krebs solution of the following composition (in mM): NaCl 119, KCl 4.7, KH2PO4 1.18, MgSO4 1.17, NaHCO3 25, CaCl2 2.5, ethylenediaminetetracetic acid (EDTA) 0.026, glucose 5.5 for no longer than 12 h. The umbilical vein was dissected out and cleared of surrounding connective and fat tissues whilst maintained in Krebs solution. Rings (3–4 mm in length) were prepared and the endothelium was rubbed off by gently moving a catheter (0.7 mm in outside diameter, Biotrol-Merck, Paris, France) back and forth several times. The collected veins were stored at −70°C for less than 30 days. Female Sprague-Dawley rats weighing 250–300 g (Iffa Credo, L'Arbresles, France) were pretreated with diethylstilboestrol at 0.1 mg kg−1 subcutaneously. Eighteen hours later, rats were sacrificed by a blow on the neck and the uterus was dissected out. Human or rat tissues were homogenized in ice-cold TES binding buffer with a Polytron homogenizer (setting 10) for 30 s. The homogenate was then centrifuged at 500×g for 20 min at 4°C. The supernatant was collected and the membranes were pelleted by centrifugation at 40,000×g for 20 min at 4°C. Membranes were resuspended in ice-cold TES binding buffer. Protein concentration was determined according to the method of Bradford (1976) using a Bio-Rad protein assay kit.
In a set of experiments, cells and membranes were processed as described above except that TES binding buffer was replaced by a Hank's balanced saline solution (HBSS) containing (HEPES 25 mM), 1,10-phenanthroline (1 mM), 140 μg ml−1 bacitracin, and 0.1% bovine serum albumin (BSA), at pH 7.4.
Saturation isotherms were obtained with [3H]-BK (0.1–5 nM) in a total volume of 0.5 ml for 90 min at room temperature. Non specific binding was evaluated by adding BK at 10 μM. Reactions were terminated by filtration using a Brandel Tissue Harvester onto GF/B filters that had been previously soaked for 2 h in 0.1% (w v−1) polyethyleneimine. Filters were washed with ice-cold 50 mM Tris, at pH 7.4. Dry filters were then counted in a Beckman liquid scintillation counter.
Competition experiments were carried out by incubating membranes with 11 concentrations of competitor ligands and concentrations of [3H]-BK equal to KD in a final volume of 0.5 ml for 90 min at room temperature.
Measurement of inositol phosphates (IPs)
CHO and INT407 cells grown in 12-well plates were labelled for 18 h with 1 μCi ml−1 [3H] myo-inositol in serum free medium 199 (Gibco, Cergy-Pontoise, France). Cells were washed with phosphate buffer solution and then incubated in 500 μl of IPs assay buffer of the following composition (in mM): NaCl 116, KCl 4.7, MgSO4 1.2, CaCl2 2.5, KH2PO4 1.2, NaHCO3 5, glucose 11, HEPES 20, captopril 0.01, LiCl 10 and 140 μg ml−1 bacitracin for 15 min at 37°C. Cells were incubated with 10−7 M of the antagonist, Hoe 140, 20 min before addition of the increasing concentrations of BK from 10−11 to 3×10−6 M and the incubation was continued for an additional 15 min period. The reaction medium was then removed and the reactions were stopped by adding 0.5 ml of an ice-cold solution of 5% perchloric acid containing 50 μg ml−1 phytic acid. After 15 min on ice, the mixture was neutralized with a 2 M K2CO3 solution. Different IPs components were then separated by anion exchange chromatography according to the method as described by Berridge et al. (1982).
Isolated organs experiments
Human umbilical vein were dissected out and rings were prepared as described above. Vein rings were set up in 8-ml jacketed organ baths containing Krebs solution and maintained at 37°C and bubbled with 95% O2 plus 5% CO2. Rings were left unstretched for 2 h and were then stretched in a stepwise fashion by 250 mg tension increments up to 1 g. After a 1 h resting period, Krebs solution of the organ bath was replaced by a high potassium containing Krebs solution (KPSS) in which NaCl was replaced by KCl in order to assess the contractile capacity of the tissue. After washing twice with normal Krebs and return to the baseline, the following compounds were added into the organ bath: mepyramine (1 μM), atropine (1 μM), indomethacin (3 μM), NG-nitro-L-arginine (L-NOARG, 30 μM), captopril (10 μM), DL-thiorphan (1 μM), DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid (MERGETPA, 5 μM) and nifedipine (0.1 μM). Mepyramine and atropine were used to block histaminergic and muscarinic receptors. Indomethacin and L-NOARG inhibited prostanoids formation and nitric oxide-synthase pathways, respectively. MERGETPA, captopril and thiorphan were used to prevent the degradation of BK by carboxypeptidases, angiotensin converting enzyme and neutral endopeptidase (EC 3.4.24.11), respectively. Nifedipine blocked the occurrence of spontaneous contractions without affecting the tonic response to BK (see Results section). Thirty minutes later the concentration-response curve to BK or KD was obtained. In another series of experiments, responses to cumulative BK were obtained in the presence or the absence of Hoe 140, NPC 567, D-Arg0-[Hyp3, D-Phe7, Leu8]-BK, WIN 64338, FR173657 or des-Arg9-[Leu8]-BK added 15 min before BK. At the end of the experiments, after washing and return to the baseline level the maximal contraction of each vein segment was obtained by adding the thromboxane A2 mimetic, U46619 (1 μM). We found that even after repeated washings, WIN 64338 at 100 μM significantly reduced the contractile response to U46619. Therefore, the contraction response curve in the presence of WIN 64338 was expressed as per cent of the initial KPSS-induced contraction.
Uterus from female Sprague-Dawley rats pretreated with diethylstilboestrol was dissected out and immediately placed in a Jalon's solution of the following composition (in mM): NaCl 154, KCl 5.6, NaHCO3 1.7, MgCl2 1.4, glucose 5.5 and CaCl2 0.3. Four segments, 10 mm in length, were prepared and suspended in jacketed organ baths containing 8 ml of a Jalon's solution maintained at 30°C and bubbled with 95% O2 and 5% CO2. Uterus segments were gradually stretched to a resting tension of 1 g. After a 90 min resting period the bath solution was changed for a Jalon's solution containing captopril (1 μM), atropine (1 μM), indomethacin (3 μM), mepyramine (1 μM) and DL-thiorphan (1 μM).
The antagonists or their respective vehicles were added at various concentrations 15 min before cumulative addition of BK excepted for Hoe 140 which was added 1 h before the addition of the agonist. A single concentration-response curve to BK was obtained for each uterus segment. At the end of the experiment, after washing and return to the baseline angiotensin II (3 μM) was added in order to obtain the maximal contractile response of each segment.
Analysis of data
Binding competition data and concentration-response curves for IPs hydrolysis and BK or KD-induced contractions were analysed using GraphPADInPlot (GraphPAD Software, San Diego, CA, U.S.A.). The maximal binding of [3H]-BK at equilibrium (Bmax) and the equilibrium dissociation constant (KD) were derived from saturation curves fitted with one site ligand binding model.
Values of inhibitory binding constants (Ki) were obtained from the Cheng-Prusoff equation (Cheng & Prusoff, 1973):
where L and KD are the concentration and equilibrium dissociation constant of the radioligand, respectively and IC50 is the concentration of competing ligand reducing specific binding by 50%.
In functional assays, EC50 was the concentration of agonist needed to reach 50% of the maximal response and was calculated using least-square analysis (Tallarida & Murray, 1981). pKB value (−log KB) was obtained according to the equation:
where [A] is the concentration of the antagonist and concentration ratio is the EC50 in the presence of the antagonist divided by the EC50 in the absence of antagonist. The potency of the agonists is expressed as a pD2 value representing −log (EC50).
Amongst the antagonists tested, WIN 64338 and FR173657 appeared insurmontable by depressing significantly the maximum response in HUV and RU, respectively. In order to evaluate the potency of these antagonists, we have calculated a pKB value and its s.e.mean by applying the following equation:
in which slope is that of the double-reciprocal plot of equieffective concentrations of agonist (A) in the absence (1/A) and in the presence (1/A′) of the antagonist (B) and [B] represents the antagonist concentration (Kenakin, 1993).
Schild analysis was used to calculate pKB values when Schild plot slopes did not differ from unity and when maximum responses to BK were not significantly affected whatever the concentration of antagonist.
Statistical analysis were performed using Statview (Abacus Concept, Palo Alto, CA, U.S.A.). A one-way analysis of variance followed by a Student's t-test was used to establish significant differences between Ki values. A P value less than 0.05 was considered as statistically significant.
Drugs
[3H]-Bradykinin (90–120 Ci mmol−1) was from New England Nuclear (Les Ullis, France). FR173657 and WIN 64338 were synthesized by Dr P. Dodey (Laboratoires Fournier S.A., Daix, France). Hoe 140 was obtained from Pr J. Martinez (CNRS URA 1852, Montpellier, France). D-Arg0 [Hyp3, D-Phe7, Leu8]-BK and D-Arg0 [Hyp3, D-Phe7]-BK (NPC 567) were from Bachem (Bubendorf, Switzerland). MERGETPA (DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid) was obtained from Calbiochem (La Jolla, CA, U.S.A.). All molecular biology and cell culture reagents were purchased from Life Technologies (Cergy-Pontoise, France). Other chemicals were from Sigma Chemical Co. (St. Louis, MO, U.S.A.).
Results
Saturation binding experiments
CHO cells stably transfected with an expression vector containing the cloned human or rat B2 receptor sequence bound [3H]-BK with a high affinity at a single, saturable binding site (Table 1). [3H]-BK did not bind to mock-transfected CHO cells (data not shown). Scatchard analysis gave KD values of 644±80 pM and 459±90 pM for the human and rat B2 receptor, respectively. The calculated average densities of binding sites were 2283±325 fmol mg−1 protein and 2265±526 fmol mg−1 protein for the human and rat B2 receptor, respectively. Binding properties of the human umbilical vein and rat uterus B2 receptor were evaluated by measuring the saturation of [3H]-BK. [3H]-BK labelled a single class of high-affinity binding sites in both membrane preparations. Scatchard plot, as well as computer curve fitting of saturation data yielded KD values of 495±150 and 597±160 pM and Bmax values of 90±20 and 264±86 fmol mg−1 of protein for human umbilical vein and rat uterus membrane preparations, respectively (Table 1).
Table 1.
Saturation analysis of [3H]-bradykinin binding to different human or rat bradykinin B2 receptor preparations

Saturation experiments with [3H]-BK were carried out on membrane preparations from INT407 cells in order to investigate the influence of the binding buffer on both the affinity and the binding capacity of the native B2 receptor. The binding of [3H]-BK was saturable giving a Bmax value of 648±146 and 122±43 fmol mg−1 protein in TES and HBSS, respectively (P<0.05). Scatchard analysis of the saturation isotherms revealed a single class of high-affinity binding sites with different KD values (P<0.05) of 422±79 and 3390±1230 pM in TES and HBSS, respectively.
Measurements of inositol phosphates
BK caused concentration-dependent increases in IP3 levels in INT407 cells and CHO cells expressing the human or rat B2 receptor (Figure 1). These responses were mediated by B2 receptors since the B2 receptor antagonist Hoe 140 produced rightward shifts of the concentration-response curves to BK (Figure 1) with calculated apparent pKB values of 8.84±0.15, 8.56±0.16 and of 9.79±0.55 for INT407 cells, hB2-CHO and rB2-CHO, respectively.
Figure 1.
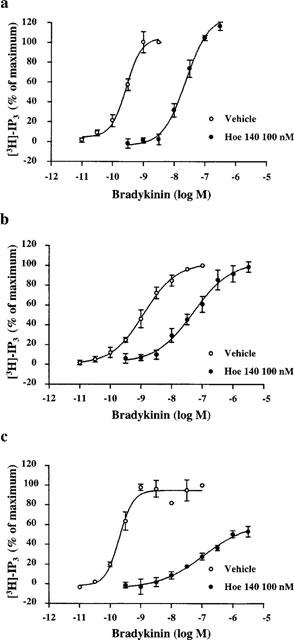
Effects of D-Arg, [Hyp3, Thi5, D-Tic7, Oic8]-BK (Hoe 140) on bradykinin-evoked increased in inositol triphosphate (IP3) in CHO cells stably expressing the human (a) or rat (c) B2 receptor and INT407 cells (b). The 100% value indicates the control response to 100 nM (for the cloned rat B2 receptor or INT407 cells) or 30 nM (for the cloned human B2 receptor BK). Values represent means±s.e.mean of three independent experiments. Vehicle is the IPs buffer solution alone.
Competition binding studies on human bradykinin B2 receptor
Competition binding curves were obtained with different peptide and non-peptide kinin derivatives in cell and tissue membranes. Bradykinin B2 receptor ligands fully compete with [3H]-BK to its binding site in various membrane preparations. The corresponding data are given in Table 2. The affinity of BK was 15 fold higher for hB2 receptor expressed in CHO cells than for the native receptor from human umbilical vein, whereas kallidin exhibited similar affinity for both preparations. Hoe 140, NPC 567 and D-Arg0-[Hyp3, D-Phe7, Leu8]-BK had a similar affinity (expressed as Ki value) for the human and rat recombinant or native B2 receptor. WIN 64338 had a higher affinity for the recombinant than for the native human B2 receptor (P<0.05) whilst FR173657 bound on both receptors with a similar affinity (Table 2). Des-Arg9-BK and des-Arg9-[Leu8]-BK which have been described as specific ligands of B1 receptor had weak affinities for the human cloned or native B2 receptor (Table 2).
Table 2.
Comparison of binding profiles of the human umbilical vein and cloned human and of the rat uterus and recombinant rat bradykinin B2 receptor

Competition binding studies in TES and HBSS buffers
To study the influence of ionic strength and pH of the binding buffer, binding experiments were conducted either in TES or HBSS buffer. The results of Table 3 show that there was approximately a 20 fold reduction in the affinity of bradykinin, kallidin and Hoe 140 when assayed in the physiological HBSS buffer whilst the affinity of NPC 567 and D-Arg0-[Hyp3, D-Phe7, Leu8]-BK for in HBSS was reduced by 8 and 6 fold. The affinity of the non-peptide antagonists FR173657 and WIN 64338 was similar in TES and HBSS buffers.
Table 3.
Inhibition of [3H]-bradykinin binding to membranes from INT407 cells in TES (N-tris[hydroxymethyl]-methyl-2-aminoethonesulphonic acid) or HBSS (Hank's balanced saline solution) binding buffer

Competition binding studies on rat bradykinin B2 receptor
The pharmacological characterization of the rat bradykinin B2 was made on membranes of rB2-CHO or uterus by using different peptide kinin derivatives and the two non-peptides WIN 64338 and FR173657. The corresponding data are given in Table 2. The affinities of kinin peptides ligands: BK, kallidin, Hoe 140, NPC 567 and D-Arg0-[Hyp3, D-Phe7, Leu8]-BK were similar for the native compared to the recombinant rat B2 receptor. When assayed on membrane preparations from rat uterus, WIN 64338 had a lower affinity compared to the cloned receptor expressed in CHO cells. The order of potency of the antagonists in displacing [3 H]-BK binding was: Hoe 140>D-Arg0-[Hyp3, D-Phe7, Leu8]-BK = FR173657> NPC 567>WIN 64338 and Hoe 140 = FR173657>D-Arg0-[Hyp3, D-Phe7, Leu8]-BK = NPC 567>WIN 64338 for the recombinant and the native B2 receptor, respectively. Both kinin B1 receptor ligands, des-Arg9-BK and des-Arg9-[Leu8]-BK displayed low affinity values (>100 μM).
Isolated organ experiments
BK induced large all-or-none rhythmical contractions in arterial rings which precluded the building of a concentration-response curve to BK (data not shown). Similarly to what has been previously shown in the human coronary artery (Cocks et al., 1993), nifedipine at 0.1 μM was efficient to abolish phasic BK-induced contractions of the human umbilical vein. Under these conditions, BK produced concentration-dependent tonic contractions.
We found pD2 values of 8.13±0.14 and 8.53±0.19 for BK and KD, respectively. The maximal contraction to BK represented 81.0±5.7% of the response to U46619 (n=6). Hoe 140, NPC 567, D-Arg0-[Hyp3, D-Phe7, Leu8]-BK, WIN 64338 and FR173657 antagonized the contractile response to BK in HUV whilst the bradykinin B1 receptor antagonist, des-Arg9-[Leu8]-BK, was inactive (Table 4). Hoe 140, NPC 567 and D-Arg0-[Hyp3, D-Phe7, Leu8]-BK behaved as competitive antagonists since these compounds did not significantly affect the maximum response to BK and the slope of the Schild plot was not significantly different from the unity. The corresponding values of pKB are given in Table 4. WIN 64338 produced a rightward shift of the concentration-response curve to BK and significantly depressed the maximum (Figure 2a). A pKB value of 6.06±0.12 was subsequently calculated according to Kenakin (1993). In contrast, FR173657 behaved as a competitive antagonist with a pKB of 7.8±0.3 (Figure 2b).
Table 4.
Values of pD2 and pKB for peptide and non-peptide bradykinin B2 receptor agonists or antagonists obtained against BK-induced contractions in human umblical vein (HUV) and rat uterus (RU)

Figure 2.
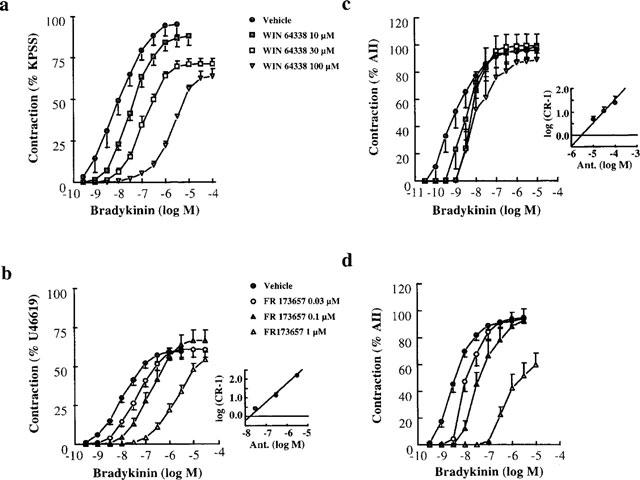
Effect of WIN 64338 and FR173657 on the concentration-response curve to bradykinin in the human umbilical vein (a and b) and in the rat uterus (c and d). Schild plot for FR173657 in the human umbilical vein and Schild plot for WIN 64338 for the rat uterus are inserted. Values represent means±s.e.mean of 5–6 experiments for the human umbilical vein and means±s.e.mean of 6–8 experiments for the rat uterus. Vehicle is a 1 : 1000 (v/v) dimethylsulfoxide solution in assay buffer. KPSS, High Potassium containing Krebs Solution; AII, Angiotensin II; U46619, Thromboxane A2 mimetic.
As shown in Figure 2c and d, RU contracted in response to BK (pD2, 8.48±0.14, n=13). Hoe 140, NPC 567 and D-Arg0-[Hyp3, D-Phe7, Leu8]-BK were competitive antagonists giving pKB values of 8.94±0.38, 6.24±0.28 and 6.44±0.30, respectively. WIN 64338 appeared less potent to inhibit the BK-induced response in RU than HUV (Figure 2c; Table 4). FR173657 was a slightly more potent competitive antagonist of BK-induced responses in RU than in HUV (Figure 2d; Table 4).
Discussion
The present study shows that binding affinity of kinin B2 receptor ligands may differ between receptors from transfected cells and isolated tissues, depends on the binding buffer and may also vary according to the species. In addition, the functional inhibitory potency of the antagonists against a B2 receptor-mediated response appears species-dependent and may not match the respective binding affinity of the compounds.
A saturable single site of [3H]-BK binding was found in membranes of CHO cells transfected with the human recombinant B2 receptor, human umbilical vein and INT407 cells with an affinity KD varying from 0.42–0.64 nM. These data are consistent with previous studies on human native B2 receptor from different cultured human cell lines including foetal lung fibroblasts (Goldstein & Wall, 1984; Phagoo et al., 1996), human epidermoid carcinoma (Liebmann et al., 1996), synovial cells (Bathon et al., 1992) and human recombinant receptor expressed in CHO cells (Hess et al., 1994; Eggerickx et al., 1992).
Whilst KD, Hoe 140, D-Arg0-[Hyp3, D-Phe7, Leu8]-BK, NPC 567 and FR173657 displayed similar affinity for the hB2-CHO and human umbilical vein receptor, BK and WIN 64338 bound respectively with a 15 and 7.5 fold lower affinity to umbilical vein than to hB2-CHO cell membranes. A similar trend was obtained when comparing the rB2-CHO and the rat uterus. The lower affinity of BK in HUV membrane preparations could not be attributed to an abnormally high number of receptors in CHO cells because BK had a similar affinity on hB2-CHO and INT407 cell membranes which markedly differ in receptor expression levels. Such a discrepancy in the binding affinity of BK was previously reported in competition studies using [3H]-BK in human WI38 fibroblasts giving a Ki value of 0.15 nM (Phagoo et al., 1996) and human epidermoid carcinoma cells giving a Ki value of 1.1 nM (Liebmann et al., 1996). In these cells, the expression of the kinin B2 receptor was low and a similar TES-type binding buffer was used. On another hand, degradation of the radioligand by proteases could not occur in our experiments since we used bacitracin as well as 1-10-phenanthroline which fully inhibits the degradation of [3H]-BK as described by Falcone et al. (1993) in guinea-pig gall bladder preparations. Recently, a phenomenon of negative cooperativity was reported to occur with BK (Pizard et al., 1998). It may provide a possible explanation for the present findings although further adequate experiments would be required to investigate this possibility. The binding affinity of WIN 64338 was also consistently reduced in HUV compared to h-B2 CHO or INT407 cell membranes. These results are in accordance with those from Gessi et al. (1997) and from Sawutz et al. (1994) who reported Ki values of 1450 and 64 nM for WIN 64338 in HUV membranes and IMR-90 cells, respectively. Therefore, it appears that, for unknown reasons, WIN 64338 displays a peculiar affinity towards cloned bradykinin B2 receptors.
FR173657 was recently described as a potent non-peptide B2 receptor antagonist (Aramori et al., 1997; Griesbacher et al., 1997; Rizzi et al., 1997). In the present study, FR173657 had a high affinity for the cloned human B2 receptor giving a pKi value of 8.66±0.08. These results are in agreement with a previously reported binding IC50 of 1.7 nM for FR173657 in membranes of IMR-90 cells which constitutively express the B2 receptor (Asano et al., 1997). Interestingly, this compound bound equally well to cloned and native bradykinin B2 receptors in human and rat preparations.
The buffer used in most of the binding studies is a low ionic strength TES buffer which is non physiological. As postulated by Félétou et al. (1995), the absence of physiological concentrations of monovalent and divalent cations, which are essential for functional studies, may explain some discrepancies observed between binding and functional data. In order to assess the validity of this hypothesis, the affinity of kinin receptor ligands towards B2 receptors from INT407 cell membranes was determined in TES and physiological HBSS buffers. Consistent with the results from Ransom et al. (1992), the affinity of peptide agonists and antagonists was reduced by 6 to 23 fold when assayed under physiological conditions whilst the affinity of the two non-peptide antagonists, FR173657 and WIN 64338 was unchanged. These data might explain, at least in part, the so-called binding paradox observed with some compounds (Hall, 1992). For example, the inhibitory potency of Hoe 140 as determined in inositol phosphate experiments (pKB, 8.93) or isolated organ tests (pKB, 8.2) matches better its affinity in HBSS binding buffer (pKi, 9.01) than in a low ionic strength TES buffer (pKi, 10.4). Nevertheless, the cause of divergence between affinity data and functional effects of some other B2 receptor antagonists remains unexplained. Although binding assays on membranes and functional tests in isolated organs can certainly not be directly compared, it remains that compounds such as D-Arg0-[Hyp3, D-Phe7, Leu8]-BK and NPC 567 having pKi values of 7.52 and 6.91 for the human B2 receptor in HBSS solution should have higher pKB values than 5.77 and 5.60 in HUV, respectively.
We found pronounced differences for some antagonists in both the affinity and the inhibitory potency between human and rat bradykinin B2 receptors. In accordance with previous data (Eggerickx et al., 1992; Hess et al., 1994), Hoe 140 bound with a similar affinity to the human and rat B2 receptor and inhibited BK-mediated response in both species with the same potency. D-Arg0-[Hyp3, D-Phe7, Leu8]-BK and NPC 567 which were 11.3 and 2.6 times more potent in displacing [3H]-BK from rat than from human B2 receptors, respectively, were also better antagonists in rat than in human isolated tissues. The affinity of WIN 64338 to human B2 receptor was 16 times higher than for the rat B2 receptor whilst, FR173657 behaved the opposite way with a ten times higher affinity for the rat than the human B2 receptor. These results suggest that WIN 64338 and FR173657 which have unrelated chemical structures do not bind the same site of the B2 receptor. Again, the functional results obtained with WIN 64338 and FR173657 were consistent with the binding data. It must be pointed out that WIN 64338 significantly depressed the maximal responses to BK in HUV, thus indicating an apparent insurmountable antagonism. However, the calculated pKB of 6.06±0.12 of WIN 64338 is in agreement with the results of Marceau et al. (1994) who reported a pKB value of 6.0 for WIN 64338. When tested at the highest concentration (100 μM), the inhibition of the contraction of the human umbilical vein produced by WIN 64338 was apparently not fully B2-dependent since WIN 64338 significantly reduced the contractile response to U46619. As expected, both B1 receptor ligands, des-Arg9-BK and des-Arg9-[Leu8]-BK, had a weak affinity for the human B2 receptor and did not bind to the rat B2 receptor up to concentrations of 100 μM. Taken together, these data confirmed that species differences in amino-acid composition of the B2 receptor may account for the differential pharmacology observed with some antagonists.
Taken together, these results illustrate the species differences in the pharmacological profile of the B2 receptor. We have also shown that the binding buffer may affect the affinity of bradykinin peptide derivatives towards the human bradykinin B2 receptor. In addition, we have provided evidence that despite a high binding affinity towards the receptor some compounds display a lower potency in functional tests than expected. Thus, we conclude that both a binding assay on the human B2 receptor and a functional test on human isolated tissue should be performed when looking for B2 receptor antagonists with the aim of treating human diseases. In this respect, FR173657 appears as a promising new drug with a high potency for the human B2 receptor.
Acknowledgments
We wish to thank Dr T. Cocks for helpful comments and corrections regarding this manuscript.
Abbreviations
- BK
bradykinin
- TES
N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid
- PBS
phosphate buffered solution
- HBSS
Hank's balanced saline solution
- CHO-K1
Chinese hamster ovary cell
References
- ARAMORI I., ZENKOH J., MORIKAWA N., O'DONNELL N., ASANO M., NAKAMURA K., IWAMI M., KOJO H., NOTSU Y. Novel subtype-selective non-peptide bradykinin antagonists FR167344 and FR173657. Mol. Pharmacol. 1997;51:171–176. doi: 10.1124/mol.51.2.171. [DOI] [PubMed] [Google Scholar]
- ASANO M., INAMURA N., HATORI C., SAWAI H., FUJIWARA T., KATAYAMA A., KAYAKIRI H., SATOH S., ABE Y., INOUE T., SAWADA Y., NAKAHARA K., OKU T., OKUHARA M. The identification of an orally active, nonpeptide bradykinin B2 receptor antagonist, FR173657. Br. J. Pharmacol. 1997;120:617–624. doi: 10.1038/sj.bjp.0700955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BACHVAROV D.R., SAINT-JACQUES E., LARRIVÉE J.F., LEVESQUE L., RIOUX F., DRAPEAU G., MARCEAU F. Cloning and pharmacological characterization of the rabbit bradykinin B2 receptor. J. Pharmacol. Exp. Ther. 1995;275:1623–1630. [PubMed] [Google Scholar]
- BATHON J.M., MANNING D.C., GOLDMAN D.W., TOWNS M.C., PROUD D. Characterization of kinin receptors on human synovial cells and upregulation of receptor number by interleukin-1. J. Pharmacol. Exp. Ther. 1992;260:384–392. [PubMed] [Google Scholar]
- BERRIDGE M.J., DAWSON R.M.C., DOWNES C.P., HESLOP J.P., IRVINE R.F. Lithium amplifies agonist-dependent phosphatidyl inositol responses in brain and salivary gland. Biochem. J. 1982;206:587–595. doi: 10.1042/bj2060587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHOOLA K.D., FIGUEROA C.D., WORTHY K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992;44:1–79. [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;721:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BURCH R.M., KYLE D.J., STORMANN T.M.Structural insight into bradykinin receptor via conformationally constrained peptides Molecular biology and pharmacology of bradykinin receptors 1993Austin: RG Landes Company; 93–104.Eds. Burch, R.M., Kyle, D.J. & Stormann, T.M. pp [Google Scholar]
- CHEN C., OKAYAMA H. High efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- COCKS T.M., KEMP B.K., PRUNEAU D., ANGUS J.A. Comparison of contractile responses to 5-hydroxytryptamine and sumatriptan in human isolated coronary artery: synergy with the thromboxane A2-receptor agonist, U46619. Br. J. Pharmacol. 1993;110:360–368. doi: 10.1111/j.1476-5381.1993.tb13818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EGGERICKX D., RASPE E., BERTRAND D., VASSART G., PARMENTIER M. Molecular cloning, functional expression and pharmacological characterization of a human bradykinin B2 receptor gene. Biochem. Biophys. Res. Commun. 1992;187:1306–1313. doi: 10.1016/0006-291x(92)90445-q. [DOI] [PubMed] [Google Scholar]
- FALCONE R.C., HUBBS S.J., VANDERLOO J.D., PROSSER J.C., LITTLE J., GOMES B., AHARONY D., KRELL R.D. Characterization of bradykinin receptors in guinea pig gall bladder. J. Pharmacol. Exp. Ther. 1993;266:1291–1299. [PubMed] [Google Scholar]
- FÉLÉTOU M., ROBINEAU P., LONCHAMPT M., BONNARDEL E., THURIEAU C., FAUCHÉRE J.-L., WIDDOWSON P., MAHIEU J.-P., SERKIZ B., VOLLAND J.-P., MARTIN C., NALINE E., ADVENIER C., PROST J.-F., CANET E. S 16118 (p-Guanidobenzoyl-[Hyp3, Thi5, D-Tic7, Oic8]Bradykinin) is a potent and long-acting bradykinin B2 receptor antagonist, in vitro and in vivo. J. Pharmacol. Exp. Ther. 1995;273:1071–1077. [PubMed] [Google Scholar]
- GESSI S., RIZZI A., CALO G., AGNELLO G., JORIZZO G., MOLLICA G., BOREA P.A., REGOLI D. Human vascular kinin receptors of the B2 type characterized by radioligand binding. Br. J. Pharmacol. 1997;122:1450–1454. doi: 10.1038/sj.bjp.0701536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDSTEIN D.J., WALL M. Activation of protein formation and cell division by bradykinin and des-Arg9-bradykinin. J. Biol. Chem. 1984;259:9623–9268. [PubMed] [Google Scholar]
- GRIESBACHER T., SAMETZ W., LEGAT F.J., DIETHART S., HAMMER S., JUAN H. Effects of the non-peptide B2 antagonist FR173657 on kinin-induced smooth muscle contraction and relaxation, vasoconstriction and prostaglandin release in vitro and in vivo. Br. J. Pharmacol. 1997;121:469–476. doi: 10.1038/sj.bjp.0701159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL J.M. Bradykinin receptors: pharmacological properties and biological roles. Pharmac. Ther. 1992;56:131–190. doi: 10.1016/0163-7258(92)90016-s. [DOI] [PubMed] [Google Scholar]
- HESS J.F., BORKOWSKI J.A., MACNEIL T., STONESIFER G.Y., FRAHER J., STRADER C.D., AND , RANSOM R.W. Differential pharmacology of cloned human and mouse B2 bradykinin receptors. Mol. Pharmacol. 1994;45:1–8. [PubMed] [Google Scholar]
- HESS J.F., BORKOWSKI J.A., YOUNG G.S., STRADER C.D., RANSOM R.W. Cloning and pharmacological characterization of a human bradykinin BK2 receptor gene. Biochem. Biophys. Res. Commun. 1992;184:260–268. doi: 10.1016/0006-291x(92)91187-u. [DOI] [PubMed] [Google Scholar]
- KENAKIN T.Allotopic, noncompetitive, and irreversible antagonism Pharmacologic Analysis of Drug-Receptor Interaction 1993New York: Raven Press; 323–343.Ed. Kenakin, T. pp [Google Scholar]
- LIEBMANN C., GRANESS A., LUDWIG B., ADOMEIT A., BOEHMER A., BOEHMER F.D., NÜRNBERG B., WETZKER R. Dual bradykinin B2 receptor signalling in A431 human epidermoid carcinoma cells: activation of protein kinase C is counteracted by a Gs-mediated stimulation of the cyclic AMP pathway. Biochem. J. 1996;313:109–118. doi: 10.1042/bj3130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCEAU F., LEVESQUE L., DRAPEAU G., RIOUX F., SALVINO J.M., WOLFE H.R., SEOANE P.R., SAWUTZ D.G. Effects of peptide and non-peptide antagonists of bradykinin B2 receptors on the venoconstrictor action of bradykinin. J. Pharmacol. Exp. Ther. 1994;269:1136–1143. [PubMed] [Google Scholar]
- MCEACHERN A.E., SHELTON E.R., BHAKTA S., OBERNOLTE R., BACH C., ZUPPAN P., FUJISAKI J., ALDRICH R.W., JARNAGIN K. Expression cloning of a rat B2 bradykinin receptor. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7724–7728. doi: 10.1073/pnas.88.17.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHAGOO S.B., YAQOOB M., BROWN M.C.S., BURGESS G.M. Selective labelling of bradykinin receptor subtypes in WI38 human lung fibroblasts. Br. J. Pharmacol. 1996;119:863–868. doi: 10.1111/j.1476-5381.1996.tb15752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIZARD A., MARCHETTI J., ALLEGRINI J., ALHENC-GELAS F., RAJERISON R.M. Negative cooperativity in the human bradykinin B2 receptor. J. Biol. Chem. 1998;273:1309–1315. doi: 10.1074/jbc.273.3.1309. [DOI] [PubMed] [Google Scholar]
- RANSOM R.W., GOODMAN C.B., YOUNG G.S. Bradykinin stimulation of phosphoinositoside hydrolysis in guinea-pig ileum longitudinal muscle. Br. J. Pharmacol. 1992;105:919–924. doi: 10.1111/j.1476-5381.1992.tb09078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIZZI A., GOBEIL F., BOGONI G., CALO C., INAMURA N., REGOLI D. FR173657: a new, potent, non-peptide kinin B2 receptor antagonist. An in vitro study. Hypertension. 1997;29:951–956. doi: 10.1161/01.hyp.29.4.951. [DOI] [PubMed] [Google Scholar]
- SAWUTZ D.G., SALVINO J.M., DOLLE R.E., CASIANO F., WARD S.J., HOUCK W.T., FAUNCE D.M., DOUTY B.D., BAIZMAN E., AWAD M.M.A., MARCEAU F., SEOANE P.R. The non-peptide WIN 64338 is a bradykinin B2 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4693–4697. doi: 10.1073/pnas.91.11.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEWART J.M. Bradykinin antagonists: development and applications. Biopolymers. 1995;37:143–155. doi: 10.1002/bip.360370208. [DOI] [PubMed] [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of Pharmacologic Calculations with Computer Programs. New York: Springer-Verlag; 1981. [Google Scholar]