

Abstract
We previously established that the formation of both α- and β/γ-secretase-derived products generated by human embryonic kidney 293 cells (HEK293) expressing either wild type or mutant βAPP could be stimulated by agonists of the cyclic AMP/protein kinase A pathways. This cyclic AMP-dependent effect modulates post-translational events since it is not prevented by actinomycin D or cycloheximide.
We show here that two protein kinase A inhibitors, H89 and PKI, both trigger dose-dependent inhibition of the basal constitutive production of Aβ40 and Aβ42 by HEK293 cells expressing wild type βAPP751.
H89 also potently inhibits the total Aβ produced by the neocortical neuronal cell line TSM1.
These two inhibitors also drastically reduce the recovery of Aβ40 and Aβ42 produced by HEK293 cells expressing the Swedish (Sw) βAPP and M146V-presenilin 1 (PS1) mutations responsible for cases of the early-onset forms of Familial Alzheimer's disease (FAD).
By contrast, H89 and PKI do not significantly affect the recovery of the physiological α-secretase-derived fragment APPα.
Our study indicates that protein kinase A inhibitors selectively lower the formation of Aβ40 and Aβ42 in human cells expressing normal and mutant βAPP and PS1 without affecting the physiological α-secretase pathway in these cells. Selective inhibitors of protein kinase A may be of therapeutic value in both sporadic and Familial Alzheimer's disease, since they may decrease the production of Aβ that is thought to be responsible for the neurodegenerative process.
Keywords: Alzheimer's disease, amyloid β peptides, APPα, protein kinase A, PKI, H89, HEK293 cells, neurons, mutant βAPP, mutant presenilins
Introduction
Sporadic and familial forms of Alzheimer's disease (FAD) are characterized by similar extracellular proteinaceous deposits called senile plaques that invade the cortical and subcortical areas of affected brains (Hardy & Allsop, 1991). These neuropathological lesions are mainly composed of amyloid β peptide (Aβ), a 39–43 amino-acid poorly soluble peptide (Selkoe, 1991). The onset of genetic forms of Alzheimer's disease generally precedes that of the sporadic cases by several decades. This is thought to be due to the drastic overproduction of Aβ and, particularly that of the readily aggregable 42 aminoacid form of Aβ (for review see Checler, 1995). The acceleration of the Aβ production has been demonstrated to be due to the presence of missense mutations in the β amyloid precursor protein (βAPP, Citron et al., 1992; Cai et al., 1993; Felsenstein et al., 1994) and more recently, in two homologous proteins named presenilins 1 and 2 (PS1, PS2) (Borchelt et al., 1996; Duff et al., 1996; Citron et al., 1997; Tomita et al., 1997; Xia et al., 1997; Ancolio et al., 1997; Marambaud et al., 1998b). The fact that distinct proteins, all responsible for aggressive forms of Alzheimer's disease, could trigger similar phenotypic overproduction of Aβ argues in favour of a therapeutic strategy aimed at slowing down the production of this peptide. In this context, putative therapeutic targets could be β- and γ-secretases, (the proteolytic activities responsible for the release of Aβ from its precursor) or other mechanisms responsible for the regulation of βAPP processing. Effectors of the protein kinase C have been shown to decrease Aβ production and increase secretion of the α-secretase-derived physiological product APPα in various cell lines (Caporaso et al., 1992; Gillespie et al., 1992; Buxbaum et al., 1993; Hung et al., 1993). Furthermore, in gene-targeted mice overproducing Aβ, the administration of the PKC stimulator phorbol 12,13-dibutyrate (PDBu) led to drastic inhibition of the production of Aβ (Savage et al., 1998).
We recently showed that the maturation of βAPP appears to be under control of the protein kinase A (PKA) pathway in human cells and neurons overexpressing normal and FAD-linked βAPP (Marambaud et al., 1998a). However, unlike modulators of PKC, effectors of the PKA pathway stimulated production of both Aβ and APPα (Marambaud et al., 1998a) suggesting that the target of PKA was probably located upstream of both α- and β/γ- secretases cleavages. Here we show that two distinct PKA inhibitors drastically reduce the constitutive production of both Aβ40 and Aβ42 in stably transfected HEK293 cells expressing wild type (wt) and Swedish mutated (Sw) βAPP751. We also establish that PKA inhibitors almost completely prevent the formation of Aβs by HEK293 cells overexpressing wt- and M146V-PS1. Interestingly, the inhibitors do not significantly affect the recoveries of APPα or its α-secretase-derived C-terminal stub, p10. Our data indicate that PKA inhibitors selectively affect the β/γ-secretase pathway in human cells and are potential pharmacological which may be able to reduce Aβ formation in both sporadic and FAD-linked Alzheimer's disease.
Methods
Antibodies
FCA3340 and FCA3542 specifically recognize the C- termini of Aβ40 and Aβ42, respectively (Barelli et al., 1997). FCA18 (Barelli et al., 1997) recognizes the N-terminus of Aβ. WO2 (Ida et al., 1996) recognizes the N-termini of Aβ and βAPP. The 207 antibody (Cephalon, West Chester, U.S.A.) interacts with the N-termini of βAPP and APPα. BR188 recognizes the C-termini of both βAPP and p10. A scheme illustrating the various antibody specificities is presented in Figure 1.
Figure 1.
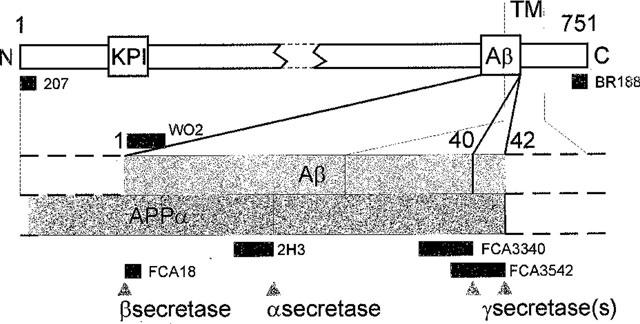
Organization of βAPP and epitopes recognition by various antibodies. The organization of βAPP751 is shown. Epitopes recognized by the various antibodies are indicated by black bars. KPI, Kunitz Protease Inhibitor domain; TM, Transmembrane Domain.
HEK293 and TSM1 cell culture and stable transfections in HEK293
HEK293 cells and the TSM1 neocortical neuronal cell line (Chun & Jaenisch, 1996) were grown in 5% CO2 in F12/DMEM (50/50) supplemented with 10% foetal calf serum containing penicillin (100 u ml−1), streptomycin (50 μg ml−1) and geneticin (1 mg ml−1). HEK293 cells were stably transfected by calcium phosphate precipitation with 1 μg of pcDNA3-containing either wtβAPP751, Sw-βAPP751, wt-PS1 or M146V-PS1 and transfectants were identified as described (Marambaud et al., 1997a; Chevallier et al., 1997; Ancolio et al., 1997).
Cells treatment with inhibitors of PKA and detection of βAPP, APPα and p10
Cells were incubated at 37°C for 7 h in the presence or in the absence of various concentrations of myristoylated PKI (Quality Control Biochemicals, Hopkinton, U.S.A.) or H89 (Calbiochem, Meudon, France). Media were immunoprecipitated with a 3000 fold dilution of 207 antibody and APPα was revealed on Western blots with mAb2H3 as previously described (Marambaud et al., 1997a). Intracellular βAPP and p10 were analysed with WO2 and BR188 antibodies, respectively, as previously described (Marambaud et al., 1998).
Metabolic labelling and detection of secreted Aβ 40 and Aβ 42
Cells were preincubated for 1 h in the presence or absence of the indicated concentrations of PKA inhibitors then metabolically labelled (50 μCi ml−1 [35S]-methionine/cysteine (Tran35S label; ICN) for 6 h in the continued presence of the same concentrations of protein kinase A inhibitors. Conditioned media were collected, diluted in a one tenth volume of RIPA 10× (NaCl 150 mM, Tris 50 mM, pH 8) buffer then Aβ42 and Aβ40 were analysed by sequential immunoprecipitation with FCA3542 and FCA3340, respectively as described (Marambaud et al., 1998a,1998b).
Western blotting of total Aβ
HEK293 cells overexpressing wt-PS1 and M146V-PS1 or neuronal cells were incubated without or with PKA inhibitors then conditioned media were recovered as described above. Total Aβ was immunoprecipitated overnight in RIPA buffer with a 350 fold dilution of FCA18, in presence of protein A-Sepharose. Pellets were resuspended with loading buffer then submitted to a 16.5% Tris-tricine SDS–PAGE and Western blotted for 45 min. Nitrocellulose sheets were incubated in boiled 1× PBS− for 5 min then for 30 min in PBS−-Tween (1× PBS−, 0.05% Tween) containing 5% skim milk then membranes were exposed overnight to a 425 fold dilution of mAbWO2 antibody in PBS−-Tween (containing 1% skim milk). Nitrocellulose sheets were rinsed in PBS−-Tween then incubated with a goat anti-mouse IgGs coupled to peroxidase, revealed and quantified by enhanced chemiluminescence as previously described (Marambaud et al., 1997b).
Results and discussion
In basal constitutive conditions, HEK293 cells stably transfected with wtβAPP751 produced quantitable amounts of Aβ40 and Aβ42 that could be immunoprecipitated with FCA3340 and FCA3542 (Figure 2B and C), two polyclonal antibodies specifically directed towards the Aβ40 and Aβ42 C-termini, respectively (Barelli et al., 1997). As previously described (Marambaud et al., 1998), these cells also produced a doublet of low molecular weight (referred to as ×40/42 in the Figure 2) that are not recognized by FCA18 (not shown), an antibody reacting with the N-terminus of Aβ (Barelli et al., 1997). The smallest of these two N-terminally truncated Aβ probably corresponds to a recently described α′/γ-secretase derived product (Xu et al., 1998). Two distinct inhibitors of PKA, H89 (Chijiwa et al., 1990) and the fully selective blocking agent PKI (Cheng et al., 1986), inhibited the formation of both Aβ40 and Aβ42 (Figure 2B and C) with a similar dose-dependent effect (Figure 2F). The maximal inhibition achieved by PKI and H89 corresponded to about 80 and 90% of Aβ production (Figure 2F), respectively. These two inhibitors also prevented the formation of ×-40 and ×-42 (Figure 2B and C) but did not affect the recovery of secreted APPα (Figure 2D), its C-terminal counterpart p10 (Figure 2E) or βAPP (Figure 2A).
Figure 2.
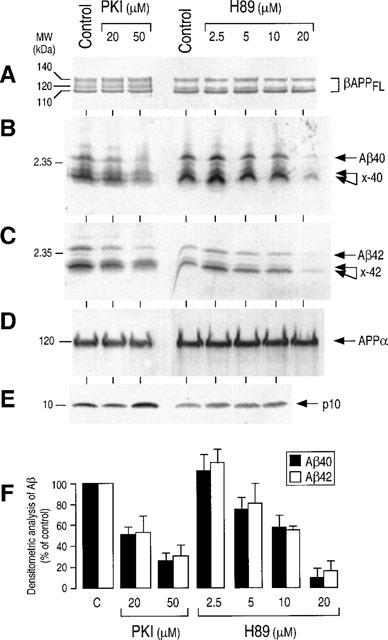
Effect of PKA inhibitors on the βAPP maturation by wtβAPP-expressing HEK293 cells. Stably transfected HEK293 cells overexpressing wild type βAPP were incubated for 7 h at 37°C in the absence (control) or in the presence of the indicated concentrations of H89 or myristoylated PKI. Secreted APPα (D) was immunoprecipitated with 207 antibody and then was analysed by SDS–PAGE, Western blotted and revealed with mAb2H3 as described in the Methods. Intracellular full-length βAPP (βAPPFL, A) and p10 (E) were identified by Western blot analysis with WO2 and BR188 antibodies, respectively. For Aβ detection, cells were preincubated with PKA inhibitors as above for 1 h then metabolically labelled for 6 h in the presence of the inhibitors. Aβ42 and x-42 (C) and Aβ40 and x-40 (B) were sequentially immunoprecipitated as described in the Methods, with FCA3542 and FCA3340, respectively. (F) Illustrates the quantitative densitometric analyses expressed as the per cent of control obtained in absence of inhibitors. Values are the means of four independent experiments.
We previously showed that the protein kinase A pathway modulated wtβAPP maturation in HEK293 cells at a post-translational level since dBut-cyclic AMP- and forskolin-stimulated Aβ and APPα production were not affected by actinomycin D and cycloheximide (Marambaud et al., 1998a). The fact that both physiological and potentially pathogenic βAPP maturation products were affected argued in favour of PKA phosphoprotein target(s) located upstream of α- and β/γ secretases. Our data indicate that PKI discriminates between the PKA-mediated effect on Aβ and APPα productions. This could be due to the involvement of distinct PKA isoforms (Parvathenani et al., 1998), differently susceptible to PKI and H89, and responsible for the phosphorylation of proteins specifically involved in the α- or β/γ-secretase pathways.
We previously established that the α- and β/γ- secretase pathways in HEK293 cells overexpressing FAD-linked mutated SwβAPP displayed similar responsiveness to PKA agonists (Marambaud et al., 1998a). These cells also responded similarly to PKI and H89 treatments. Thus, these two inhibitors drastically inhibited the secretion of Aβ40, Aβ42 and their ×40/42 related products (Figure 3B and C) with similar dose-response curves (Figure 3F) without affecting secreted APPα (Figure 3D), intracellular p10 (Figure 3E) or βAPP expression (Figure 3A). Although the sites of Aβ production appear distinct for SwβAPP and wt-βAPP (Haass et al., 1995), our data indicate that PKI and H89 exhibit identical effects in the two cell systems. This could indicate that PKA targets similar phosphoprotein(s) involved in common early steps of the secretory/routing processes occurring for wt- and Sw-βAPP maturation.
Figure 3.
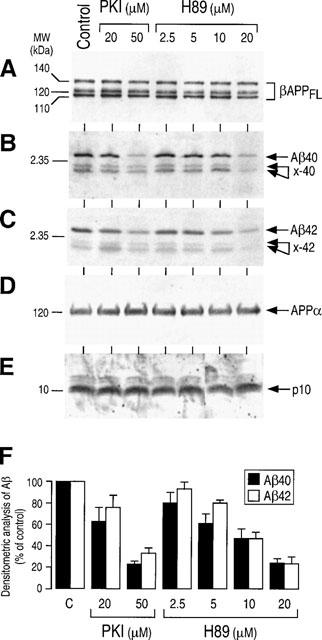
Effect of PKA inhibitors on the βAPP maturation by Swedish mutated βAPP-expressing HEK293 cells. Stably transfected HEK293 cells overexpressing Swedish mutated βAPP were incubated as in Figure 2 and analysed for βAPP (A), Aβ40 and x-40 (B), Aβ42 and x-42 (C), APPα (D) and p10 (E) as described in the Figure 2. Densitometric analyses of Aβ40 and Aβ42 recoveries were quantified as in the Figure 2 and are the means±s.e.mean of 4–5 independent experiments.
HEK293 cells expressing wild type and M146V-PS1 display low endogenous amounts of βAPP, leading to poorly detectable Aβ42 secretion. Therefore, we have used FCA18 to immunoprecipitate total secreted Aβ species (Figure 4B). It is interesting to note that, as previously described (Borchelt et al., 1996; Duff et al., 1996; Citron et al., 1997; Tomita et al., 1997; Xia et al., 1997; Ancolio et al., 1997; Marambaud et al., 1998b), the total amount of secreted Aβ (Figure 4B) was increased in control conditions by the presence of the FAD-linked mutation. As stated above, the specificity of FCA18 does not allow the recovery of the N-terminally truncated ×-40/42 fragments (Figure 4B). Aβ secretion by both wt- and M146V-PS1-expressing HEK293 cells is virtually completely abolished by PKI and to a lesser extent by H89 (Figure 4B). APPα (Figure 4C), p10 (Figure 4D) and βAPP expression (Figure 4A) were not affected by PKA inhibitors. Therefore, the PKA contribution to βAPP maturation is: (1) identical for endogenous βAPP and therefore, independent of the extent of βAPP expression; (2) can be observed in cells expressing mutant βAPP and PS1 proteins linked to familial forms of Alzheimer's disease. The effect of PKA inhibitors did not appear to be cell-type specific since the secretion of total Aβ by neocortical neurons was also drastically reduced by H89 (Figure 5).
Figure 4.
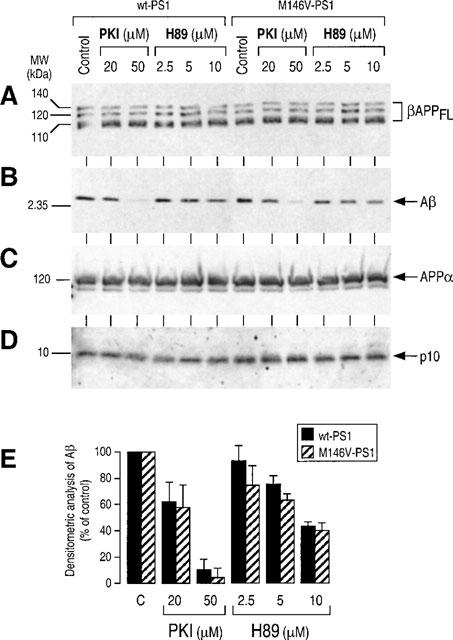
Effect of PKA inhibitors on βAPP maturation in wild type and M146V-presenilin1-expressing HEK293 cells. Stably transfected HEK293 cells overexpressing wt-PS1 or M146V-PS1 were incubated without (control) or in the presence of the indicated concentrations of H89 and PKI then analysed for βAPP (A), APPα (C) and p10 (D) as described in Figure 2. Total Aβ (B) was detected after immunoprecipitation with FCA18, electrophoresis and Western blot with mAbWO2 as described in Methods. Densitometric analyses of total secreted Aβ was quantified as in Figure 2 and are the means ±s.e.mean of three independent experiments.
Figure 5.
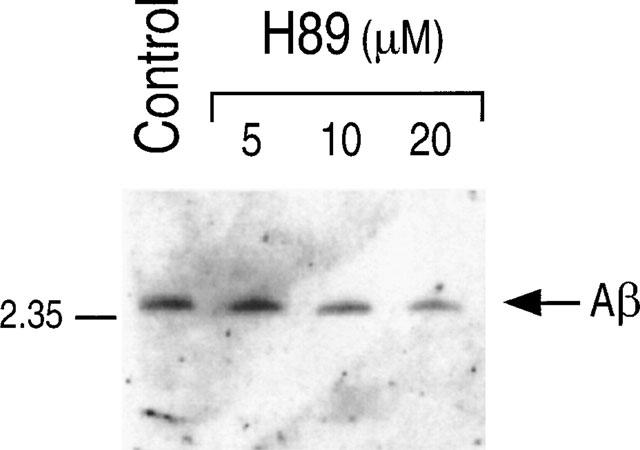
Effect of H89 on the secretion of Aβ by TSM1 neocortical neuronal cell line. TSM1 cells were incubated as described in Methods without (control) or in the presence of the indicated concentrations of H89. Total Aβ was detected after immunoprecipitation with FCA18, electrophoresis and Western blot with mAbWO2 as in Figure 4.
Several lines of evidence have indicated that βAPP maturation can be modulated by agents targeting the protein kinase C (PKC) pathway. Unlike agents modulating PKA, PKC effectors elicit increased APPα secretion and reduce Aβ recovery from various cell lines (for review see Checler, 1995). Savage et al. (1998) reported on the ability of PDBu, a PKC stimulator, to lower Aβ recovery in vivo, in gene-targeted mice. Here, we identify another therapeutic strategy aimed at diminishing Aβ production. Of most interest is the observation that this strategy could be effective in Alzheimer's disease of both sporadic and genetic origin. Work is in progress in the laboratory to assess whether this approach can be validated in animal models of Aβ overproduction.
Acknowledgments
We are very grateful to Dr K. Beyreuther (Heidelberg, Germany) for providing us with WO2. We thank Drs B. Greenberg and M. Savage for the kind supply of the 207 antibody. mAb2H3 was generously provided by Dr D. Schenk (Athena Neuroscience). BR188 was kindy given by Dr M. Goedert (MRC Cambridge). Dr Chun and Allelix Biopharmaceutical Inc (Missisauga, Canada) are thanked for providing the TSM1 cell line. We thank J. Kervella for secretarial assistance. AdC is recipient of a grant from the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP, Brazil). This work was supported by the Centre National de la Recherche Scientifique and the Institut National de la Santé et de la Recherche Médicale.
Abbreviations
- Aβ
amyloid βpeptide
- APPα
amyloid precursor protein α
- βAPP
βamyloid precursor protein
- FAD
familial Alzheimer's disease
- HEK
human embryonic kidney
- PDBu
phorbol 12,13-dibutyrate
- PKA
protein kinase A
- PKC
protein kinase C
- PS1-PS2
presenilins 1 and 2
- Sw
Swedish
- wt
wild type
References
- ANCOLIO K., MARAMBAUD P., DAUCH P., CHECLER F. α-secretase-derived product of β-amyloid precursor protein is decreased by presenilin 1 mutations linked to familial Alzheimer's disease. J. Neurochem. 1997;69:2494–2499. doi: 10.1046/j.1471-4159.1997.69062494.x. [DOI] [PubMed] [Google Scholar]
- BARELLI H., LEBEAU A., VIZZAVONA J., DELAERE P., CHEVALLIER N., DROUOT C., MARAMBAUD P., ANCOLIO K., BUXBAUM J.D., KHORKOVA O., HEROUX J., SAHASRABUDHE S., MARTINEZ J., WARTER J.-M., MOHR M., CHECLER F. Characterization of new polyclonal antibodies specific for 40 and 42 aminoacid-long amyloid β peptides: their use to examine the cell biology of presenilins and the immunohistochemistry of sporadic Alzheimer's disease and cerebral amyloid angiopathy cases. Mol. Med. 1997;3:695–707. [PMC free article] [PubMed] [Google Scholar]
- BORCHELT D.R., THINAKARAN G., ECKMAN C.B., LEE M.K., DAVENPORT F., RATOVITSKY T., PRADA C.-M., KIM G., SEEKINS S., YAGER D., SLUNT H.H., WANG R., SEEGER M., LEVEY A.I., GANDY S.E., COPELAND N.G., JENKINS N.A., PRICE D.L., YOUNKIN S.G., SISODIA S.S. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- BUXBAUM J.D., KOO E.H., GREENGARD P. Protein phosphorylation inhibits production of Alzheimer amyloid β/A4 peptide. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAI X.-D., GOLDE T.E., YOUNKIN S.G. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- CAPORASO L., GANDY S.E., BUXBAUM J.D., RAMABHADRAN T.V., GREENGARD P. Protein phosphorylation regulates secretion of Alzheimer β/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3055–3059. doi: 10.1073/pnas.89.7.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHECLER F. Processing of the β-amyloid precursor protein and its regulation in Alzheimer's disease. J. Neurochem. 1995;65:1431–1444. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- CHENG H-C., KEMP B.E., PEARSON R.B., SMITH A.J., MISCONI L., VAN PATTEN S.M., WALSH D.A. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J. Biol. Chem. 1986;261:989–992. [PubMed] [Google Scholar]
- CHEVALLIER N., JIRACEK J., VINCENT B., BAUR C.P., SPILLANTINI M.G., GOEDERT M., DIVE V., CHECLER F. Examination of the role of endopeptidase 3.4.24.15 in Aβ secretion by human transfected cells. Brit. J. Pharmacol. 1997;121:556–562. doi: 10.1038/sj.bjp.0701151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIJIWA T., MISHIMA A., HAGIWARA M., SANO M., HAYASHI K., INOUE T., NAITO K., TOSHIOKA T., HIDAKA H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinol:inesulfonamide (H89), of PC12D pheochromocytoma cells. J. Biol. Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- CHUN J., JAENISCH R. Clonal cell lines produced by infection of neocortical neuroblasts using multiple oncogenes transduced by retroviruses. Mol. Cell. Neurosci. 1996;7:304–321. doi: 10.1006/mcne.1996.0023. [DOI] [PubMed] [Google Scholar]
- CITRON M., OLTERSDORF T., HAASS C., MCCONLOGUE L., HUNG A.Y., SEUBERT P., VIGO-PELFREY C., LIEBERBURG I., SELKOE D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- CITRON M., WESTAWAY D., XIA W., CARLSON G., DIEHL T., LEVESQUE G., JOHNSON-WOOD K., LEE M., SEUBERT P., DAVIS A., KHOLODENKO D., MOTTER R., SHERRINGTON R., PERRY B., YAO H., STROME R., LIEBERBURG I., ROMMENS J., KIM S., SCHENK D., FRASER P., ST GEORGE HYSLOP P., SELKOE D. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nature Medicine. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- DUFF K., ECKMAN C., ZEHR C., YU X., PRADA C.-M., PEREZ-TUR J., HUTTON M., BUEE L., HARIGAYA Y., YAGER D., MORGAN D., GORDON M.N., HOLCOMB L., REFOLO L., ZENK B., HARDY J., YOUNKIN S. Increased amyloid-β42(43) in brains expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- FELSENSTEIN K.M., HUNIHAN L.W., ROBERTS S.B. Altered cleavage and secretion of a recombinant β-APP bearing the Swedish familial Alzheimer's disease mutation. Nature Genetics. 1994;6:251–256. doi: 10.1038/ng0394-251. [DOI] [PubMed] [Google Scholar]
- GILLESPIE S., GOLDE T.E., YOUNKIN S.G. Secretory processing of the Alzheimer amyloid β/A4 protein precursor is increased by protein phosphorylation. Biochem. Biophys. Res. Commun. 1992;187:1285–1290. doi: 10.1016/0006-291x(92)90442-n. [DOI] [PubMed] [Google Scholar]
- HAASS C., LEMERE C.A., CAPELL A., CITRON M., SEUBERT P., SCHENK D., LANNFELT L., SELKOE D. The Swedish mutation causes early-onst Alzheimer's disease by β-secretase cleavage within the secretory pathway. Nature Medicine. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- HARDY J., ALLSOP D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends in Pharmacol. Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- HUNG A.Y., HAASS C., NITSCH R.M., QIU W.Q., CITRON M., WURTMAN R.J., GROWDON J.H., SELKOE D.J. Activation of protein kinase C inhibits cellular production of the amyloid β-protein. J. Biol. Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- IDA N., JOHANNES H., PANTEL J., SCHRÖDER J., ZERFASS R., FÖRSTL H., SANDBRINK R., MASTERS C.L., BEYREUTHER K. Analysis of heterogeneous βA4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J. Biol. Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- MARAMBAUD P., ANCOLIO K., CHECLER F. Post-transcriptional contribution of a cAMP-dependent pathway to the formation of α-and β/γ-secretases-derived products of βAPP maturation in human cells expressing wild type and Swedish mutated βAPP. Mol. Medicine. 1998a;4:715–723. [PMC free article] [PubMed] [Google Scholar]
- MARAMBAUD P., ANCOLIO K., LOPEZ-PEREZ E., CHECLER F. Proteasome inhibitors prevent the degradation of familial Alzheimer's disease-linked presenilin 1 and trigger increased Aβ42 secretion by human cells. Mol. Medicine. 1998b;4:146–156. [PMC free article] [PubMed] [Google Scholar]
- MARAMBAUD P., CHEVALLIER N., BARELLI H., WILK S., CHECLER F. Proteasome contributes to the α-secretase pathway of amyloid precursor protein in human cells. J. Neurochem. 1997a;68:698–703. doi: 10.1046/j.1471-4159.1997.68020698.x. [DOI] [PubMed] [Google Scholar]
- MARAMBAUD P., LOPEZ-PEREZ E., WILK S., CHECLER F. Constitutive and protein kinase C-regulated secretory cleavage of Alzheimer's β amyloid precursor protein: different control of early and late events by the proteasome. J. Neurochem. 1997b;69:2500–2505. doi: 10.1046/j.1471-4159.1997.69062500.x. [DOI] [PubMed] [Google Scholar]
- PARVATHENANI L.K., BUESCHER E.S., CHACON-CRUZ E., BEEBE S.J. Type-I cAMP-dependent protein kinase delays apoptosis in human neutrophils at a site upstream of caspase-3. J. Biol. Chem. 1998;273:6736–6743. doi: 10.1074/jbc.273.12.6736. [DOI] [PubMed] [Google Scholar]
- SAVAGE M., TRUSKO S.P., HOWLAND D.S., PINSKER L.R., MISTRETTA S., REAUME A.G., GREENBERG B.D., SIMAN R., SCOTT R.W. Turnover of amyloid β-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci. 1998;18:1743–1752. doi: 10.1523/JNEUROSCI.18-05-01743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SELKOE D.J. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- TOMITA T., MARUYAMA K., SAIDO T.C., KUME H., SHINOZAKI K.T., S., CAPELL A., WALTER J., GRÜNBERG J., HAASS C., IWATSUBO T., OBATA K. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc. Natl. Acad. Sci. U.S.A. 1997;94:2025–2030. doi: 10.1073/pnas.94.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIA W., ZHANG J., KHOLODENKO D., CITRON M., PODLISNY M.B., TEPLOW D.B., HAASS C., SEUBERT P., KOO E.H., SELKOE D.J. Enhanced production and oligomerization of the 42-residue amyloid β-protein by chinese hamster ovary cells stably expressing mutant presenilins. J. Biol. Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- XU H., GOURAS G.K., GREENFIELD J.P., VINCENT B., NASLUND J., MAZZARELLI L., FRIED G., JOVANOVIC J.N., SEEGER M., RELKIN N.R., LIAO F., CHECLER F., BUXBAUM J.D., CHAIT B.T., THINAKARAN G., SISODIA S.S., WANG R., GREENGARD P., GANDY S. Estrogen reduces neuronal generation of Alzheimer β-amyloid peptides. Nature Medicine. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]