

Abstract
C335Stop is a constitutively active mutant of the TRH receptor (TRH-R). To investigate the mechanism of the decreased responsiveness of C335Stop TRH-R, we studied cellular Ca2+ concentrations ([Ca2+]i) in AtT20 cells stably transfected with C335Stop TRH-R cDNA, or Ca2+-activated chloride currents in Xenopus laevis oocytes expressing this mutant receptor after injection of cRNA. The competitive TRH-R binding antagonist, chlorodiazepoxide (CDE), was used as an inverse agonist to study the contribution of constitutive activity to desensitization.
Acute treatment with CDE resulted in a rapid (within minutes) decrease in [Ca2+]i and an increase in the response amplitude to TRH with no measurable change in receptor density. Conversely, removal of chronically administered CDE caused a rapid increase in [Ca2+]i and a decrease in TRH response amplitude.
CDE abolished heterologous desensitization induced by C335Stop TRH-R on muscarinic m1-receptor (m1-R) co-expressed in Xenopus oocytes.
Chelation of extracellular calcium with EGTA caused a rapid decrease in [Ca2+]i and a concomitant increase in the response to TRH in AtT20 cells expressing C335Stop TRH-Rs.
Chelerythrine, a specific inhibitor of protein kinase C (PKC), reversed the heterologous desensitization of the response to acetylcholine (ACh). The phosphoserine/phosphothreonine phosphatase inhibitor, okadaic acid, abolished the effect of chelerythrine.
Down-regulation of PKC by chronic exposure to phorbol 12-myristate 13-acetate (PMA) or acute inhibition with chelerythrine caused a partial resensitization of the response to TRH.
Western analysis indicated that the α subtype of protein kinase C was down-regulated in cells expressing C335Stop TRH-Rs. Following a 5 min exposure to PMA, the residual αPKC translocated to the particular fraction.
We propose that cells expressing the constitutively active mutant TRH-R rapidly desensitize their response, utilizing a mechanism mediated by an increase in [Ca2+]i and PKC.
Keywords: Thyrotropin-releasing hormone, G-protein-coupled receptors, constitutive activity, inverse agonism, desensitization, cytosolic calcium, protein kinase C
Introduction
Several laboratories have reported that single amino acid substitutions in the sequences of G-proteins-coupled receptors (GPCRs) result in constitutive activity: namely, signalling activity independent of their cognate agonists (Kjelsberg et al., 1992; Ren et al., 1993; Samama et al., 1993; Shenker et al., 1993; Parma et al., 1993). Indeed, Kjelsberg et al. (1992) showed that at the site that confers constitutive activity upon its mutation in the α1B-adrenergic receptor, only the naturally occurring amino acid can maintain an inactive conformation in the absence of agonist. Many of the mutations shown to confer constitutive activity in GPCRs were located at the putative carboxyl-terminal domain of the third cytoplasmic loop, and in the sixth transmembrane segment (Kjelsberg et al., 1992; Ren et al., 1993; Samama et al., 1993; Shenker et al., 1993; Parma et al., 1993). The discovery of the involvement of constitutively active receptors in human diseases (Shenker et al., 1993; Parma et al., 1993) has increased the interest in these types of mutations.
We described a constitutively active mutant of the thyrotropin-releasing hormone receptor (TRH-R), which was truncated because the codon at position-335 in the carboxyl terminus was changed to a stop codon (C335Stop TRH-R) (Matus-Leibovitch et al., 1995). Until recently, this domain of GPCRs had not been linked to constitutive activity. Parker & Ross (1991) demonstrated that truncation of the carboxyl terminus of the avian β-adrenergic receptor resulted in 2–3 fold increased basal activity. This was, however, coupled with higher agonist-stimulated activity and represents a change in receptor regulation different from other GPCRs.
Using a protein kinase C (PKC)-responsive reporter gene, we recently demonstrated that wild type (WT) TRH-R exhibits low level constitutive activity, and competitive binding antagonists of TRH-R (e.g., chlorodiazepoxide (CDE), Drummond et al., 1989; Gershengorn & Paul, 1986) act as inverse agonists (Jinsi-Parimoo & Gershengorn, 1997). The truncation of TRH-R at residue 335 results in the loss of two neighbouring cysteine residues at positions 335 and 337 that are potentially subject to palmitoylation and may be involved in the coupling of TRH-R to G-proteins (Findlay & Eliopoulos, 1990; O'Dowd et al., 1989). We demonstrated the marked constitutive activity of this mutant by using CDE (Heinflink et al., 1995): in pituitary AtT20 cells stably expressing C335Stop TRH-Rs, CDE acts as an inverse agonist, and prolonged incubation with this agent results in enhanced TRH responsiveness and C335Stop TRH-R expression level.
In our previous studies (Matus-Leibovitch et al., 1995; Heinflink et al., 1995), cells expressing C335Stop TRH-Rs were found to exhibit higher basal cellular calcium concentration ([Ca2+]i), smaller amplitudes of TRH-induced [Ca2+]i elevation and electrophysiological responses, and substantially slower response kinetics than cells expressing WT TRH-Rs. These findings were compatible with a decrease in efficiency of coupling to G-proteins due to truncation of the receptor and/or desensitization induced by the constitutive activity.
Because cells chronically treated with CDE exhibited an increase in expression of C335Stop TRH-Rs, it was difficult to determine whether the increased response to TRH was due to resensitization or to increase in receptor density. In order to clarify this issue, we studied the effect of acute exposure to CDE, which does not cause an increase in C335Stop TRH-R density. Our results strongly suggest that the decreased responsiveness of AtT20 cells or Xenopus oocytes expressing C335Stop TRH-Rs is due in part to desensitization via rapid changes in [Ca2+]i and activation of PKC.
Methods
AtT20 pituitary cells
AtT20 cells stably expressing similar WT and C335Stop TRH-Rs densities (approximately 3×105 receptors/cell, Heinflink et al., 1995) were grown in Dulbecco's Modified Eagle's Medium supplemented with 5–7.5% whole foetal calf serum at 37° and 5% CO2, essentially as previously described (Matus-Leibovitch et al., 1995). Cultures were renewed every 10–15 passages from frozen stock. To ensure the selection of the transfected line, cells were grown in the presence of 200 μg/ml of Geneticin. For analysis of [Ca2+]i, cells were grown for 24–48 h on 22 mm diameter #1 round coverslips.
Xenopus laevis oocytes
Defolliculated oocytes were obtained from mature Xenopus females, essentially as previously described (Shapira et al., 1990). In vitro transcribed cRNA for WT TRH-R (1–5 ng/oocyte), C335Stop TRH-R (10–20 ng/oocyte) or the m1-R (1 ng/oocyte) was injected 24–48 h before assay, as described previously (Matus-Leibovitch et al., 1995; Oron et al., 1987; Lupu-Meiri et al., 1993). The larger amount of the mutant receptor mRNA was required in order to obtain measurable reponses (Matus-Leibovitch et al., 1995). Co-expression of different receptors (m1-Rs, gastrin-releasing peptide-Rs, neuromedin B-Rs and TRH-Rs), in the amounts of cRNAs used here, did not inhibit either response, suggesting that neither the expression mechanism nor the post-receptor signal transduction components were limiting (Matus-Leibovitch et al., 1995; Shapira & Oron, unpublished).
[Ca2+]i imaging in AtT20 cells
The analysis of [Ca2+]i was performed essentially as described in Matus-Leibovitch et al. (1995) and Heinflink et al. (1995). Briefly, for rapid kinetic analysis, cells grown on coverslips and loaded with Fura 2-AM were placed in a laminar flow perfusion chamber (Warner Instrument Corporation, U.S.A.). Applied Imaging (U.K.) Magical imaging system was used in a Tardis mode (Applied Imaging proprietary two-wavelength fluorescence acquisition software). The temporal resolution was 360 ms per pair of frames (340–380 nm excitation). A 0.85 numerical aperture ×20 fluorescence objective allowed simultaneous analysis of 20–50 individual cells. For low temporal resolution analysis, a Miracal imaging system (Life Science Resources Limited, U.K.) permitted acquisition of a pair of frames within 1.2–2.0 s. A 1.30 numerical aperture ×40 fluorescence oil-immersion objective allowed simultaneous analysis of 10–20 individual cells. Solutions were changed in the chamber. The response to TRH was a transient elevation of [Ca2+]i, with the maximum observed 6–12 s after addition of the agonist. The agonist was present throughout the response, which decayed to baseline values within 1–3 min. Peak amplitude, peak net amplitude (after subtracting baseline values) and peak amplitude/basal ratios were measured. The analysis of [Ca2+]i in both systems was done by averaging all the cells in the field. The two systems were calibrated according to the equation of Grynkiewicz et al. (1985). We used maximum/minimum values obtained by adding saturating CaCl2 or excess EGTA to either (in mM): KCl 100, NaCl 10, MgCl2 1 solution containing 2–5 μM Fura 2-FA, or to Fura 2-AM-loaded AtT20 cells pretreated with 1 μM ionomycin.
Electrophysiology
Two-electrode voltage clamp measurements were performed at a holding potential (VH)=−90 to −100 mV, as previously described (Lupu-Meiri et al., 1990, 1993). Chloride currents were continuously recorded. Responses to TRH or acetylcholine (ACh) were very rapid (0.5–3 s time-to-peak) after the initial latency of 1–4 s. Peak current amplitudes were measured.
TRH binding assay
The TRH-R density in AtT20 cells was estimated by the binding of [3H]Me-TRH, an analogue of higher affinity than TRH (Vale et al., 1971), according to a previously described protocol (Vale et al., 1971; Straub et al., 1990).
Detection of PKC subtypes
Four subtypes of PKC have been reported in AtT20 cells (McFerran et al., 1995): α, β, ε and ξ. We used the Western blot assay, essentially as described by Eto et al. (1995) for GH4C1 cells, with the following modifications: (1) Cells were grown on 60 mm plates and harvested close to confluence, 48 h post plating; (2) Homogenization was done with a Polytron (2×10 s, at setting 70, on ice) in 0.25 M sucrose, MgCl2 1 mM, Hepes 20 mM, pH 7.5; (3) Homogenates were centrifuged 36,000×g for 90 min. Cytosol and particulate fraction were separated on SDS–PAGE at 2–10 μg/lane, according to need. Protein was determined according to Bradford (1976) and on nitrocellulose transfers, with Ponceau S. Mouse brain homogenate served as a standard in all determinations.
Statistics
All experiments were performed several times with incorporated replicates. In experiments on AtT20 cells, 20–50 cells were analysed for each condition on each coverslip. n denotes the number of individual coverslips; N denotes the number of different experiments performed on separate days. A large number of oocytes (n) from a number of donors (N) was assayed because of possible variability of expression in oocytes of different frogs.
Results were presented as mean±s.e.mean. All results were analysed be either paired or unpaired Student's t-test. P values<0.05 were regarded significant.
Materials
Fura 2-AM and Fura 2-FA were obtained from either Molecular Probes (Eugene, OR, U.S.A.) or from Teflabs (Austin, TX, U.S.A.). Collagenase type IA, geneticin, leupeptin, soybean trypsin inhibitor, benzamidine and phenylmethylsulphonyl fluoride (PMSF), anti-PKC antibodies and goat-anti-rabbit IgG antibody conjugated to peroxidase were purchased from Sigma (Rechovot, Israel). Dulbecco's Modified Eagle's Medium and foetal calf serum were obtained from Biological Industries, Beit-Haemek, Israel. In vitro translation was carried out with a Promega RiboprobeR kit, modified to contain a higher concentration of T7 polymerase (Promega, Madison, WI, U.S.A.) and m7G(5′)ppp(5′)G cap (Boehringer-Manheim, Manheim, Germany). Bradford reagent was from BioRad (Hercules, CA, U.S.A.). All other chemicals were of analytical grade.
Drugs
Chelerythrine hydrochloride and okadaic acid were purchased from Alomone Labs (Jerusalem, Israel). ACh, TRH, CDE and phorbol 12-myristate, 13-acetate (PMA) were purchased from Sigma (Rechovot, Israel). [3H]Me-TRH was purchased from DuPont-NEN (Boston, MA, U.S.A.).
Results
The effects of acute exposure to or removal of CDE on basal [Ca2+]i.
To demonstrate the constitutive activity of C335Stop TRH-Rs, we used CDE, a representative competitive antagonist of the benzodiazepine family (Drummond et al., 1989). CDE was deemed an appropriate tool to study the constitutive activity and desensitization of the mutated receptor in view of our previous findings (Heinflink et al., 1995) that C335Stop and WT TRH-Rs in AtT20 cells have similar Me-TRH dissociation constants, EC50s for TRH, CDE dissociation constants and IC50s.
We previously reported that chronic exposure to CDE lowered [Ca2+]i, markedly increased the response to TRH in AtT20 cells expressing C335Stop TRH-Rs, and increased C335Stop TRH-Rs density (Heinflink et al., 1995). In order to dissociate changes in [Ca2+]i from those due to receptor density, we used short times of exposure to CDE while continuously monitoring [Ca2+]i. Adding 20 μM CDE to cells caused a rapid (2–4 min) decrease in basal [Ca2+]i from 137±12 to 109±11 nM (n=14, P<0.001). Subsequent removal of CDE resulted in an almost immediate increase in [Ca2+]i to 133±8 nM (n=4). A representative tracing of acute addition and removal of CDE is shown in Figure 1A.
Figure 1.
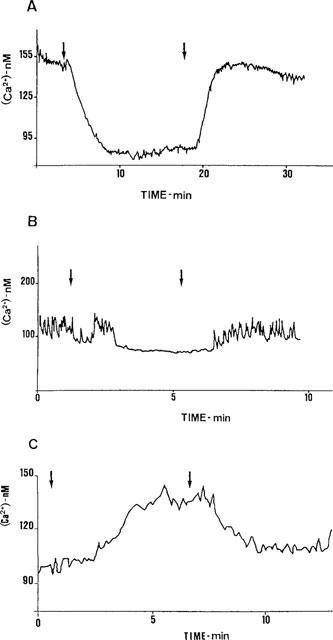
The effect of CDE on [Ca2+]i in AtT20 cells expressing C335Stop TRH-Rs. Cells were assayed for [Ca2+]i as described in Methods. CDE was added or removed by changing the perfusion medium. (A) Recording of average [Ca2+]i of several cells. The buffer was changed (first arrow) to one that included 20 μM CDE. After 14 min (second arrow), CDE was washed off. (B) Recording of a single cell that exhibited marked [Ca2+]i fluctuations. (C) Cells were incubated overnight with 20 μM CDE. All solutions during the Fura2 AM loading and washes included 20 μM CDE. The buffer was changed to one without CDE (first arrow), and after 6 min (second arrow) to a buffer that contained CDE. Representative recordings of 4–14 individual experiments.
In several experiments, many cells expressing C335Stop TRH-Rs exhibited spontaneous [Ca2+]i fluctuations. Addition of 20 μM CDE to these cells resulted in a decrease in [Ca2+]i and disappearance of the fluctuations. Fluctuations were re-initiated when CDE was washed off (Figure 1B). These results confirm the constitutively active character of this mutant, and suggest that the residual activity is manifested in some cells as variable [Ca2+]i fluctuations.
In a complementary experiment on cells expressing C335Stop TRH-Rs and exposed to 20 μM CDE for 18–24 h, removal of CDE resulted in an immediate increase in basal [Ca2+]i from 79±15 to 149±25 nM (n=4, P<0.05). Re-addition of CDE caused [Ca2+]i decrease to 97±7 nM (P<0.1, Figure 1C). Although overnight treatment with CDE upregulates C335Stop TRH-Rs density (Heinflink et al., 1995), the subsequent removal or addition of the antagonist was too rapid to affect receptor density and most likely represents direct inhibition of receptor function.
Acute addition or removal of CDE in AtT20 cells expressing the WT TRH-Rs yielded no changes in [Ca2+]i (n=6, not shown). By comparison, removal of the antagonist after 24 h incubation appeared to evoke small increases in basal [Ca2+]i, from 75±21 to 96±30 nM, n=3 while its subsequent re-addition decreased in [Ca2+]i, to 79±15 nM, n=3; these changes were not statistically significant. Hence, the inverse agonist CDE had a significant effect on basal [Ca2+]i in cells expressing constitutively active mutant TRH-Rs, and a small effect in cells expressing WT TRH-Rs. This was compatible with our finding that CDE acts as an inverse agonist (Heinflink et al., 1995), and with the reported limited constitutive activity of WT TRH-Rs (Jinsi-Parimoo & Gershengorn, 1997). The effects of CDE were mediated by TRH-Rs, since the addition or removal of the antagonist had no effect on [Ca2+]i in untransfected AtT20 cells (not shown).
The effects of exposure to or removal of CDE on the response to TRH
The use of acutely administered CDE is complicated by the potential for two opposing effects. On the one hand, CDE is a competitive antagonist of TRH binding to TRH-Rs. The response to TRH reflects therefore the kinetics of dissociation of CDE rather than the kinetics of association of TRH, even at supramaximal concentrations. Hence, the response to TRH in the presence of CDE should be partially inhibited. Indeed, in AtT20 cells expressing WT TRH-Rs, the [Ca2+]i increase due to stimulation by TRH was inhibited by 54% (Figure 2A). On the other hand, chronic exposure to CDE inhibits the constitutive activity of C335Stop TRH-Rs and raises receptor density (Heinflink et al., 1995). We proposed that the increased response in AtT20 cells expressing C335Stop TRH-Rs chronically exposed to CDE could be attributed not only to increased receptor density, but also to resensitization (Heinflink et al., 1995). Acute exposure to 20 μM CDE did not affect C335Stop TRH-R density for up to 1 h (not shown). Unlike the inhibition observed in cells expressing WT TRH-Rs, the response to TRH in cells expressing C335Stop TRH-Rs was the same after a 10 min exposure to CDE as in cells not exposed to CDE (Figure 2A). This is consistent with the idea that the binding inhibitory effect of the antagonist was balanced by resensitization.
Figure 2.
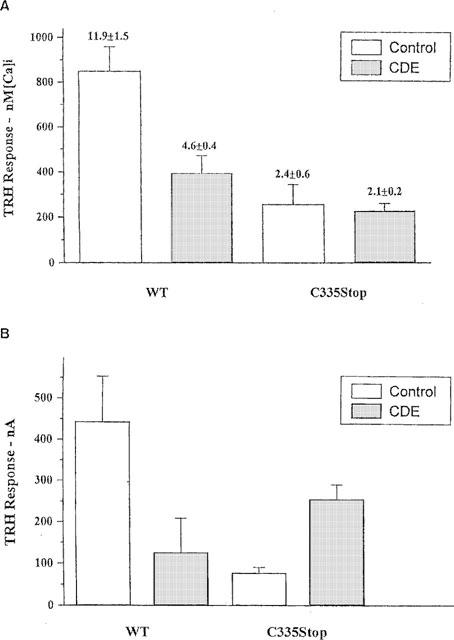
The effect of CDE on the response to TRH. (A) AtT20 cells expressing WT or C335Stop TRH-Rs and continuously monitored for [Ca2+]i were challenged with 5 μM TRH and the peak [Ca2+]i response to the agonist (6–12 s after the addition of the hormone) was recorded. The results are presented as mean±s.e.mean. CDE (20 μM), when present, was added 10 min before the challenge with TRH. The numbers over the columns represent the fold increase of the TRH response over basal [Ca2+]i. (B) Xenopus oocytes were injected with the appropriate in vitro transcribed RNAs. Chloride current responses to 10 μM TRH (time-to-peak approximately 2–3 s) were recorded under two-electrode voltage clamp (VH=−90 to −100 mV, see Methods) after approximately 24 h. CDE (20 μM) was added 120 min before the challenge with the hormone.
The potentiating effect of either chronic or acute exposure to CDE on the response to TRH was greater in Xenopus oocytes than in AtT20 cells expressing C335Stop TRH-Rs. Oocytes injected with 10–20 ng of C335Stop TRH-R cRNA and challenged with 1 μM TRH exhibited small responses (78±13 nA, n=272, N=25) or current fluctuations of 5–20 nA, despite the significantly higher expression level of the mutant receptor (Matus-Leibovitch et al., 1995). One hundred and twenty min exposure to 20 μM CDE resulted in a large increase in TRH response amplitude (to 256±35 nA, n=226, N=14, see Figure 2B). A similar effect was seen upon shorter (10–15 min) incubation with CDE (not shown). In oocytes expressing WT TRH-Rs, on the other hand, the presence of CDE caused pronounced inhibition, as expected from a competitive binding antagonist (for representative experiment see Figure 2B).
To assess the effect of acute removal of CDE, oocytes were exposed to 20 μM CDE overnight and the response to 1 μM TRH was measured before and after the removal of the antagonist. Chronic exposure to CDE resulted in a dramatic increase in the response amplitude (905±320 nA, n=57, N=5). The removal of the antagonist for 30–60 min resulted in a partial return to a desensitized state. In three experiments, the TRH response amplitude decreased by 50±16% (n=40, N=3). Hence, similar to the effect of the removal of CDE from AtT20 cells (see Figure 1C), the effect of CDE in oocytes was rapidly reversible.
The effect of CDE on heterologous desensitization in oocytes
We previously described the heterologous desensitization induced by C335Stop TRH-Rs when co-expressed with other GPCRs (Matus-Leibovitch et al., 1995). To confirm that C335Stop TRH-Rs cause rapidly reversible desensitization of other GPCRs, we co-expressed C335Stop TRH-Rs and m1-Rs in Xenopus oocytes. As previously reported, the mutant receptor caused a major decrease in the response to ACh (by 64±10%, n=29–49, N=6) in oocytes expressing both receptors. Incubation with 20 μM CDE caused not only a homologous resensitization of the C335Stop TRH-Rs-mediated response to TRH (Figure 3A), but a complete reversal of heterologous desensitization of the muscarinic response (Figure 3B). CDE had no effect on responses to ACh in oocytes expressing this receptor alone, even upon chronic exposure (24 h, not shown). This experiment demonstrates that CDE acts as an inverse agonist at C335Stop TRH-Rs, leading to resensitization of the heterologously desensitized m1-Rs. Unlike C335Stop TRH-Rs, co-expression of WT TRH-Rs with m1-Rs did not inhibit the response to ACh (see also Matus-Leibovitch et al., 1995), while incubation with CDE did not potentiate it (not shown).
Figure 3.
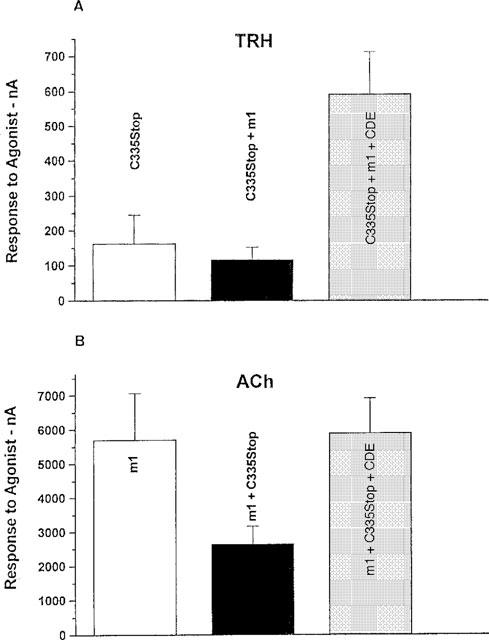
The effect of CDE on the heterologous desensitization of m1-Rs. Xenopus oocytes were injected with cRNAs for C335Stop TRH-Rs (10 ng/oocyte) alone, m1-Rs (1 ng/oocyte) alone, or a mixture of both. After 24 h, the cells were tested for responses to 10 μM TRH (A) or 1 μM ACh (B) (time-to-peak 0.5–2 s), alone or after 4 h exposure to 20 μM CDE. A representative experiment of the three performed is shown.
The effects of basal [Ca2+]i on the response to TRH
In the previous section, we showed that CDE rapidly lowered [Ca2+]i in AtT20 cells, and appeared to resensitize both AtT20 cells and Xenopus oocytes expressing C335Stop TRH-Rs to a challenge with the agonist. To test whether changes in [Ca2+]i are involved in the desensitization mechanism, we chelated extracellular calcium by adding an excess of EGTA to the incubation buffer. AtT20 cells were very sensitive to chelation of extracellular calcium. Upon addition of EGTA, [Ca2+]i rapidly decreased in cells expressing WT (from 90±9 to 56±7 nM, n=13, P<0.001, Figure 4A) or C335Stop TRH-Rs (from 97±10 to 72±9 nM, n=19, P<0.01, Figure 4B). Re-addition of calcium to the buffer in doses of 1.8–2.0 mM above the concentration of EGTA resulted in a rapid return of [Ca2+]i to pre-EGTA baseline levels (Figure 4).
Figure 4.
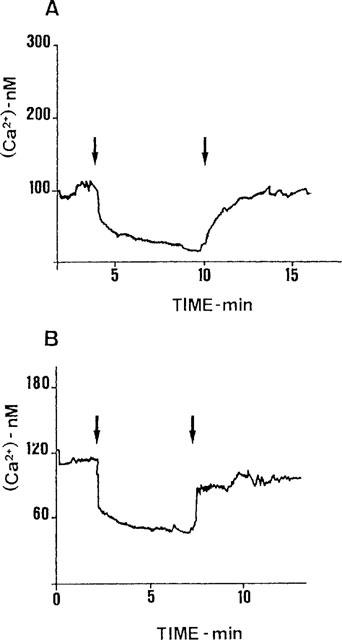
The effect of EGTA on basal [Ca2+]i. AtT20 cells expressing WT (A) or C335Stop TRH-Rs (B) were assayed for [Ca2+]i as described in Methods. The two representative tracings show the changes in [Ca2+]i upon addition (first arrow) and removal (second arrow) of EGTA (approximately 2 mM over [Ca2+]i in the buffer), averaged over all the cells in the field.
Chelation of buffer calcium for 1 min not only significantly lowered [Ca2+]i but also markedly potentiated the response to TRH in AtT20 cells expressing C335Stop TRH-Rs. The amplitude of the [Ca2+]i transient increased from 213±13 to 412±79 nM (P<0.0001), the net amplitude of the transient (after subtracting the baseline values) increased from 120±12 to 306±65 nM (P<0.0001), and the fold response from 2.6±0.2 to 4.1±0.4 (P<0.005, n=60 and 10, respectively, Figure 5). Despite a similar decrease in [Ca2+]i, in cells expressing WT TRH-Rs, there was no effect on the TRH response. After treatment with EGTA, the peak amplitude was 738±190 nM, the net amplitude 682±185 nM, and the fold increase 11.3±3.0 vs 716±216, 636±202 nM and 8.5±1.8, respectively, in cells not exposed to EGTA (n=6–7, Figure 5). These data were consistent with the idea that the desensitization resulting from the constitutive activity of C335Stop TRH-Rs was mediated to a large extent by the persistent increase in [Ca2+]i.
Figure 5.
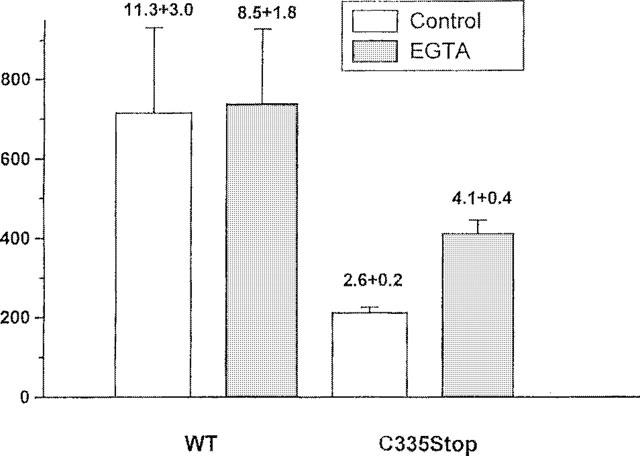
The effect of EGTA on the response to TRH. AtT20 cells expressing WT or C335Stop TRH-Rs were treated with EGTA for 1 min and then challenged with 5 μM TRH. The numbers over the columns denote the fold increase of the peak [Ca2+]i response over basal [Ca2+]i.
The involvement of PKC in desensitization of cells expressing C335Stop TRH-Rs
The involvement of [Ca2+]i in desensitization suggested that one of the calcium-sensitive steps in the signalling pathway might be mediating this effect. Since our previous results suggested that rapid desensitization of the response to TRH in Xenopus oocytes was partially mediated by PKC (Lipinsky et al., 1995a,1995b), we tested the role of PKC in desensitization of cells expressing C335Stop TRH-Rs. Two different approaches were employed: down-regulation of PKC by chronic exposure to PMA (Glikes et al., 1994), and inhibition by chelerythrine, a specific inhibitor of PKC (McFerran & Guild, 1994; McFerran et al., 1995).
Exposure for 24 h to 0.1 μM PMA caused a marked decrease in the response to TRH of AtT20 cells expressing WT TRH-Rs. The amplitude of the TRH-evoked [Ca2+]i transient decreased from 851±109 in control cells, to 402±68 nM in cells maintained in the presence of PMA (n=29 and 5, respectively). In contrast, in cells expressing C335Stop TRH-Rs, the amplitude of the transient increased from 195±52 to 311±49 nM (P<0.01, N=5, Figure 6), a value close to that obtained in cells expressing WT TRH-Rs and chronically exposed to PMA. These results were consistent with the idea that down-regulation of PKC results in resensitization of the response to TRH.
Figure 6.
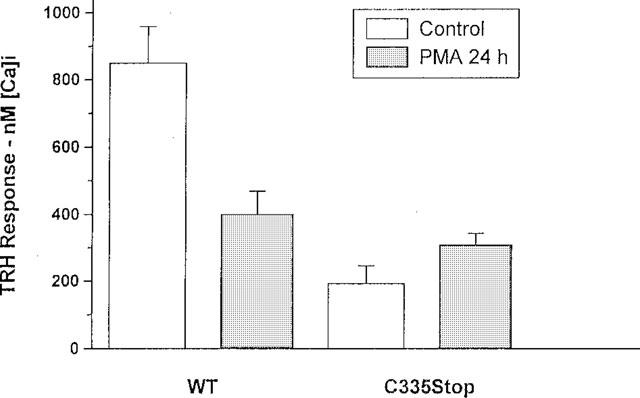
The effect of down-regulation of PKC on the response to TRH. AtT20 cells expressing WT or C335Stop TRH-Rs were exposed to 0.1 μM PMA for 24 h and assayed for the [Ca2+]i response to 5 μM TRH.
To complement these results, we exposed AtT20 cells expressing C335Stop TRH-Rs to 5 μM chelerythrine for 5–10 min, which resulted in a modest potentiation of the TRH response. The net amplitude of the [Ca2+]i transient increased from 90±16 to 130±23 nM (P<0.2, NS), while the fold response increased from 1.71±0.09 to 2.20±0.16 (P<0.02, n=11–14). Thus, the contribution of PKC to the desensitization of the response appears limited. To investigate this further, we performed similar experiments in Xenopus oocytes expressing C335Stop TRH-Rs. In four experiments performed on four different donors, 20 min exposure to 10–20 μM chelerythrine resulted in a major potentiation of the response to TRH: from 107±26 nA in untreated controls to 301±55 nA in oocytes treated with chelerythrine, n=50 and 47, respectively, P<0.002. Similarly, in oocytes co-expressing C335Stop TRH-Rs and m1-Rs, chelerythrine almost completely abolished the heterologous desensitization. This effect of chelerythrine was inhibited by a phosphoserine/phosphothreonine phosphatase inhibitor, okadaic acid (Figure 7). In oocytes co-expressing WT TRH-Rs and m1-Rs, okadaic acid caused a modest inhibition of the muscarinic response (Figure 7). These down-regulation and inhibition experiments suggest that PKC is involved in the homologous and heterologous desensitization caused by the constitutive activity of C335Stop TRH-Rs.
Figure 7.
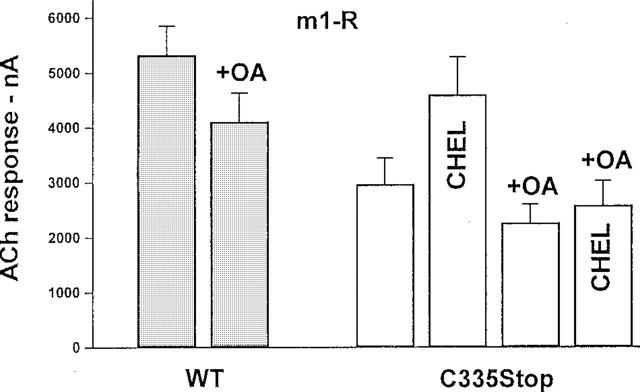
The effects of chelerythrine and okadaic acid on heterologous desensitization of m1-Rs. Xenopus oocytes were injected with cRNAs coding for m1-Rs (1 ng/oocyte) together with cRNAs coding for either WT (1 ng/oocyte) or C335Stop TRH-Rs (10 ng/oocyte). After 24 h, the cells were tested for responses to 10 μM ACh. Where indicated, oocytes were preincubated with chelerythrine (CHEL, 20 μM, for 40 min) and/or okadaic acid (OA, 1 μM, 2 h). The bars represent means±s.e.mean of 3–6 experiments on different donors.
The role of PKC in the desensitization resulting from constitutive activity was studied further by comparing the expression of PKC in AtT20 cells transfected with either WT or C335Stop TRH-Rs. We, like McFerran et al. (1995), were able to detect PKC α, β1, ε and ζ. The β1 subtype was barely detectable, even when 10 μg of cytosol or particulate fraction protein were analysed. Since only the α subtype is regulated by both Ca2+ and diacylglycerol, we studied this PKC isozyme.
In AtT20 cells expressing C335Stop TRH-Rs, the amount of PKC-α decreased in both fractions, suggesting that the constitutive activity down-regulates this enzyme subtype by approximately 40%. Short-term (1 h) incubation of cells with 20 μM CDE caused a moderate decrease in the proportion of the enzyme in the particulate fraction (41 vs 53%, with or without CDE, respectively). CDE had no effect on the distribution of the enzyme in cells expressing the WT TRH-Rs. In AtT20 cells expressing either the WT or the mutant TRH-Rs, acute incubation (5 min) with 0.1 μM PMA caused major translocation of PKC to the particulate fraction (87 and 91% of total in particulate fraction, respectively), while overnight incubation with PMA caused extensive down-regulation. The latter was sometimes accompanied by the appearance of immunoreactive, lower molecular mass degradation products. Representative experiments are shown in Figure 8.
Figure 8.
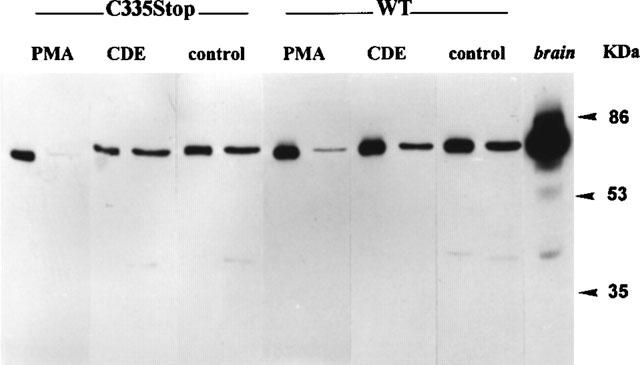
The effects of constitutive activity and drugs on the expression of PKC-α. Cytosolic and particulate fractions of AtT20 cells expressing either WT or C335Stop TRH-Rs were assayed for the expression of PKC-α, as described in Methods. The Figure is a representative Western analysis. The right and left lanes of each pair were loaded with 5 and 8 μg cytosolic and particulate protein, respectively. In the right-most lane, reference mouse brain homogenate was loaded. Cells were incubated with 20 μM CDE for 1 h or with 0.1 μM PMA for 5 min.
Discussion
Based on our previous characterization of the constitutively active C335Stop TRH-R mutant, we suggested that the decreased signalling activity of this receptor in response to TRH could be attributed to both a decrease in its coupling efficiency, and to its constitutive activity resulting in pronounced desensitization (Matus-Leibovitch et al., 1995). Although constitutively-active GPCRs have been described previously (Kjelsberg et al., 1992; Ren et al., 1993; Samama et al., 1993; Shenker et al., 1993; Parma et al., 1993), to our knowledge this was the first demonstration that changes in the carboxyl terminus of GPCRs activating the PLC signalling pathway could lead to increased agonist-independent activity. Since then, additional GPCRs have been shown to be constitutively active due to alteration of their carboxyl termini. Prezeau et al. (1996) showed that an alternatively spliced variant of the metabotropic glutamate receptor is constitutively active. Hasegawa et al. (1996) reported that an alternatively spliced variant and a C-terminus-truncated mutant of the prostaglandin E3 receptor possess agonist-independent activity.
We showed previously that the competitive antagonist of TRH-R binding, CDE, behaves as an inverse agonist and causes increases in the responses to TRH both in AtT20 cells and Xenopus oocytes expressing C335Stop TRH-Rs (Heinflink et al., 1995). In these studies, the increased responses to TRH observed with CDE could be accounted for by inhibition of the constitutive activity of the mutant receptor that allowed for resensitization of the signal transduction pathway, by a CDE-induced increase in receptor density, or by both mechanisms (Heinflink et al., 1995). The present work was aimed at clarifying this issue.
To avoid changes in receptor density, we exposed AtT20 cells or Xenopus oocytes expressing C335Stop TRH-Rs to CDE for a very short period, which allowed no measurable change in receptor binding. Under these conditions, exposure to CDE inhibited responses to TRH in AtT20 cells and Xenopus oocytes expressing WT TRH-Rs, consistent with its acting as a competitive binding antagonist of the TRH-Rs. In contrast, CDE did not inhibit TRH responses in AtT20 cells expressing C335Stop TRH-Rs, most likely because the inhibition of TRH binding was offset by resensitization. More importantly, acute administration of CDE potentiated the responses to TRH in oocytes expressing the mutant TRH-Rs. Hence, competitive inhibition of TRH binding to its receptor not only did not affect the activity mediated by this receptor in AtT20 cells, it even increased it in Xenopus oocytes. This phenomenon was compatible with a rapidly reversible desensitization induced by the constitutive activity of the mutant receptor. Perhaps the most convincing evidence for desensitization resulting from the constitutive activity of C335Stop TRH-R was the ability of CDE, acting as an inverse agonist of TRH-Rs, to resensitize the response of m1-Rs to ACh when C335Stop TRH-Rs and m1-Rs were co-expressed in the same oocyte.
To determine the pathway of desensitization induced by the constitutively active TRH-R mutant, we monitored the basal [Ca2+]i in AtT20 cells. We previously reported that basal [Ca2+]i was substantially higher in cells transfected with the mutant receptor (Matus-Leibovitch et al., 1995). In the present series of experiments, we observed a 100% increase in some batches and only a 10% difference in others. Analysis of all the experiments yielded a value of 74.1±4.9 nM in cells expressing the WT TRH-Rs (n=61), and 90.1±4.9 nM in cells transfected with C335Stop TRH-Rs (n=116, P<0.05)–an increase of 23%. This level of [Ca2+]i was significantly above the control value, but apparently below toxic values.
Addition of CDE caused a rapid decrease in [Ca2+]i in cells expressing C335Stop TRH-Rs, but not in cells expressing WT TRH-Rs. This was direct proof of the constitutive activity of C335Stop TRH-R, which affected an event downstream to the initial steps of the signal transduction pathway. To show that the receptor-induced changes in basal [Ca2+]i are involved in the desensitization process, we lowered the basal [Ca2+]i by chelating the Ca2+ in the incubation buffer. AtT20 cells responded to the chelation of extracellular calcium with a rapid decrease in [Ca2+]i, which could be rapidly reversed by increasing calcium in the buffer. The decrease in [Ca2+]i did not affect the response to TRH in AtT20 cells expressing WT TRH-Rs, but markedly potentiated the response in cells expressing the mutant receptors. We concluded that the rapid desensitization induced by C335Stop TRH-Rs is at least partly mediated by the persistent increase in [Ca2+]i.
Our previous observations in oocytes led us to postulate that the very rapid desensitization caused by limited occupancy of TRH-R is mediated, at least in part, by PKC (Lipinsky et al., 1995a,1995b). In cells expressing WT TRH-Rs, acute activation of PKC appears to dramatically inhibit TRH responses: in AtT20 cells by 75–96% (Perlman & Gershengorn, 1991), and in Xenopus oocytes by more than 90% (Lipinsky et al., 1995b). These effects are consistent with the idea that PKC serves as a feedback mechanism to limit the extent and duration of the response: that is, it causes desensitization. On the other hand, down-regulation of PKC by chronic exposure to PMA inhibited the response to the agonist in AtT20 cells expressing WT TRH-Rs. These apparently contradictory results can be attributed to the multiple effects of PKC activation on virtually any component of the cellular signal transduction pathway. For example, we have previously shown (Lupu-Meiri et al., 1989) that in Xenopus oocytes, PMA inihibits muscarinic responses, but markedly potentiates responses to the injection of inositol trisphosphate. Nevertheless, down-regulation of PKC potentiated the response to TRH in AtT20 cells expressing C335Stop TRH-Rs. Acute inhibition of PKC also modestly increased the response to TRH in AtT20 cells expressing C335Stop TRH-Rs, and to a greater extent in oocytes expressing this receptor. In a complementary experiment, heterologous desensitization of the muscarinic response by co-expressed C335Stop TRH-Rs was abolished by chelerythrine. The effect of chelerythrine was inhibited by okadaic acid, confirming that desensitization was linked to serine/threonine phosphorylation. We concluded, therefore, that the desensitization induced by the constitutively active C335Stop TRH-R mutant is mediated in part by the Ca-PKC pathway.
Indeed, constitutive activity of C335Stop TRH-Rs was reflected in partial down-regulation of PKC-α. This phenomenon was similar, though less extensive, to the down-regulation of the enzyme by its chronic activation by PMA. CDE decreased the proportion of the enzyme detected in the particulate fraction (i.e., the activated enzyme) in cells expressing either the WT or the mutant TRH-Rs. Since short-term activation of PKC by PMA translocates the enzyme to the particulate fraction and inhibits TRH responses (Lipinsky et al., 1995b), the opposite effect of CDE is compatible with the resensitization of the TRH response in cells expressing C335Stop TRH-Rs.
The involvement of PKC in agonist-induced desensitization has been described in many systems. Two recent examples are the heterologous desensitization of angiotensin II and vasopressin receptors in cardiomyocytes (Zhang et al., 1996), and the homologous desensitization of the δ-opioid receptor expressed in Xenopus oocytes (Ueda et al., 1995). Recently, Ancellin & Morel, (1998) reported a PKC-mediated homologous and heterologous desensitization in Xenopus oocytes expressing vasopressin and muscarinic receptors. In other cases, desensitization could not be fully ascribed to the activation of PKC alone. Cai et al. (1996) reported that acute desensitization of the δ-opioid receptor in NG108-15 cells was fully antagonized by a non-specific kinase inhibitor, staurosporine, but only partially by the specific PKC inhibitor, calphostin C. These results suggest that different protein kinases may be involved in rapid desensitization. Similarly, down-regulation of PKC antagonized desensitization of the 5-HT2A receptor only during the early stage of agonist exposure (Roth et al., 1995). Richardson et al. (1995) have shown that the desensitization of the IL-8A receptor is only partially mediated by PKC, and proceeds via phosphorylation of residues located at the distal part of carboxyl terminus of the receptor. These few examples demonstrate that desensitization may be mediated by PKC alone or in conjunction with other kinases, both other second messenger-dependent kinases as well as receptor-specific protein kinases (Premont et al., 1995; Chuang et al., 1996). Thus, less than complete resensitization of the TRH response by inhibition of PKC could be interpreted as caused by multiple mechanisms of desensitization induced by the prolonged constitutive activity of the C335Stop mutant.
The molecular mechanisms of desensitization are very complex due to the above mentioned variety of kinases and their targets within the signal transduction cascade. A further complication stems from the functional definition of desensitization, which ranges from a decrease of the slope of the onset of the response, to effects of prolonged (sometimes hours) or multiple sequential exposures to agonists. Another factor is the relationship between the fractional occupancy of the receptor and desensitization. In oocytes, we have been investigating rapid desensitization caused by limited fractional occupancy of receptors. But, even at supramaximal agonist concentrations, the very rapid kinetics and the transient nature of the electrophysiological response preclude equilibrium binding and reflect low receptor occupancy. The [Ca2+]i response in AtT20 cells, which is both rapid and transient, also suggests limited occupancy. Our results here refer to this type of desensitization, probably important from a physiological point of view and less relevant to drug therapy. Indeed, we previously demonstrated that prolonged challenge of cells expressing TRH-Rs with TRH causes desensitization unrelated to the activation of PKC (Perlman & Gershengorn, 1991).
We could not demonstrate full recovery of the TRH response in cells expressing C335Stop TRH-Rs (compared to responses elicited in cells expressing WT TRH-Rs) by adding CDE or by decreasing [Ca2+]i (57 and 58% recovery, respectively, in AtT20 cells). But, treatment with CDE fully restored muscarinic response in oocytes (mediated by the WT m1-Rs) in heterologous desensitization (Figure 3B), supporting the idea that C335Stop may be partially uncoupled. If this is correct, the response to TRH of cells expressing C335Stop TRH-Rs cannot reach the values seen in the WT TRH-R, even if desensitization is fully prevented. This hypothesis must be studied directly by examining the importance of the carboxyl terminus and the two cysteine residues in receptor-G-protein coupling.
When the desensitization and subsequent resensitization of the response mediated by C335Stop TRH-R in our two model systems are compared, it is striking that oocytes and mammalian cells exhibit qualitatively identical phenomena, despite differences of species, cell size and assay techniques. Oocytes, however, appear to resensitize to a greater extent than AtT20 cells, both with CDE and chelerythrine, probably reflecting the different assays employed in the two systems. While in AtT20 cells the transient (but relatively slow, time-to-peak of approximately 8 s, not shown) accumulation of [Ca2+]i is measured, in oocytes the very rapid Cl− current transient reflects the earliest part of [Ca2+]i rise at the inner surface of the membrane (Lipinsky et al. 1995b), and may be a much more sensitive measure of [Ca2+]i dynamics.
Understanding constitutive signalling by GPCRs is becoming increasingly important. Subtle changes in primary structure of GPCRs result in a major change in their coupling properties. In this respect, C335Stop TRH-R represents GPCRs which are altered in the carboxyl terminus and until recently were not associated with constitutive activity. It is interesting that a competitive antagonist still acts as an inverse agonist, suggesting that the binding of ligands can affect the conformation of the truncated carboxyl terminus and change its availability for interacting with the G-protein.
A number of WT (or native) GPCRs (Hasegawa et al., 1996; Samama et al., 1994; Tiberi & Caron, 1994), as well as GPCRs found to be mutated in certain disease states (Shenker et al., 1993; Parma et al., 1993; Lefkowitz, 1993), have been shown to exhibit agonist-independent activity. The majority of these GPCRs retain the ability to respond to agonists. Our data on rapid desensitization-resensitization of the signalling cascade show that the level of responsiveness of these receptors can be modulated by counter-regulatory mechanisms elicited by activation of the signalling cascade. A change in [Ca2+]i or a change in the activity of cellular protein kinases can switch the constitutively active (and therefore persistently desensitized) receptor to a more or less responsive one. Thus, lower response to an agonist may be secondary to desensitization of a constitutively active receptor, rather than loss of receptor function. This phenomenon may produce paradoxical effects under certain circumstances. For example, an agonist may cause further desensitization, while an antagonist may produce inverse-agonistic effects on a constitutively active receptor and resensitize the signal transduction pathway. The significance of these theoretic possibilities will be assessed in the future in in vitro systems, and may prove to have important implications for certain diseases caused by constitutively active receptors in humans.
In conclusion, C335Stop TRH-R mutant exhibits decreased responsiveness to TRH due to persistent desensitization resulting from its constitutive activity. This desensitization is both homologous and heterologous, is rapidly reversible by treatment with CDE (which acts as an inverse agonist), by lowering [Ca2+]i, by down-regulation of PKC, or by treatment with chelerythrine. These last two phenomena suggest the involvement of PKC in the desensitization mechanism.
Acknowledgments
This work was supported by a Bi-National Science Foundation grant to Y.O. and M.C.G. The work described in this paper partially fulfills the requirements of a PhD thesis of H.G. at the Sackler Faculty of Medicine, Tel Aviv University.
Abbreviations
- ACh
acetylcholine
- [Ca2+]i
cytosolic calcium concentration
- CDE
chlorodiazepoxide
- C335Stop TRH-R
a constitutively active mutant of the thyrotropin-releasing hormone receptor truncated at cys335
- GPCRs
membrane receptors coupled to guanine nucleotide regulatory proteins
- G-protein
guanine nucleotide-binding regulatory protein
- m1-R
muscarinic m1-receptor
- NS
not significant
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- WT
wild type
References
- ANCELLIN N., MOREL A. Homologous and heterologous acute desensitization of vasopressin V1a receptor in Xenopus oocytes. Cell. Signal. 1998;10:217–223. doi: 10.1016/s0898-6568(97)00124-1. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;71:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- CAI Y., ZHANG Y., WU Y., PIE G. δ opioid receptor in neuronal cells undergoes acute and homologous desensitization. Biochem. Biophys. Res. Commun. 1996;219:342–347. doi: 10.1006/bbrc.1996.0235. [DOI] [PubMed] [Google Scholar]
- CHUANG T.T., LACOVELLI L., SALLSES M., DE BLASI A. G protein-coupled receptors: Heterologous regulation of homologous desensitization and its implications. Trends Pharmacol. Sci. 1996;17:416–421. doi: 10.1016/s0165-6147(96)10048-1. [DOI] [PubMed] [Google Scholar]
- DRUMMOND A.H., HUGHES P.J., RUIZ-LARREA F., JOELS L.A. Use of receptor antagonists in elucidating the mechanism of action of TRH in GH3 cells. Ann. NY. Acad. Sci. 1989;553:197–204. doi: 10.1111/j.1749-6632.1989.tb46642.x. [DOI] [PubMed] [Google Scholar]
- ETO E., AKITA Y., SAIDO T.C., SUZUKI K., KAWASHIMA S. The role of calpain-calpastatin system in thyrotropin-releasing hormone-induced selective down-regulation of a protein kinase C isozyme, nPKCs, in rat pituitary GH4C1 cells. J. Biol. Chem. 1995;270:25115–25120. doi: 10.1074/jbc.270.42.25115. [DOI] [PubMed] [Google Scholar]
- FINDLAY J., ELIOPOULOS E. Three-dimensional modeling of G-protein-linked receptors. TIPS. 1990;11:492–499. doi: 10.1016/0165-6147(90)90050-i. [DOI] [PubMed] [Google Scholar]
- GERSHENGORN M.C., PAUL M.E. Evidence for tight coupling of receptor occupancy by thyrotropin-releasing hormone to phospholipase C-mediated phosphoinositide hydrolysis in rat pituitary cells: Use of chlorodiazepoxide as a competitive antagonist. Endocrinology. 1986;119:833–839. doi: 10.1210/endo-119-2-833. [DOI] [PubMed] [Google Scholar]
- GILKES A.F., GUILD S.B., CRAMB G. Phorbol ester activation of protein kinase C inhibits CNP-stimulated cyclic GMP production in the mouse AtT-20 pituitary tumor cell line. Biochem. Biophys. Res. Commun. 1994;204:1318–1324. doi: 10.1006/bbrc.1994.2607. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- HASEGAWA H., NEGISHI M., ICHIKAWA A. Two isoforms of the prostaglandin E receptor EP3 subtype different in agonist-independent constitutive activity. J. Biol. Chem. 1996;271:1857–1860. doi: 10.1074/jbc.271.4.1857. [DOI] [PubMed] [Google Scholar]
- HEINFLINK M., NUSSENZVEIG D.R., GRIMBERG H., LUPU-MEIRI M., ORON Y., GERSHENGORN M.C. A constitutively active mutant thyrotropin-releasing hormone receptor is chronically down-regulated in pituitary cells: Evidence using chlorodiazepoxide as a negative antagonist. Mol. Endocrinol. 1995;9:1455–1460. doi: 10.1210/mend.9.11.8584022. [DOI] [PubMed] [Google Scholar]
- JINSI-PARIMOO A., GERSHENGORN M.C. Constitutive activity of native thyrotropin-releasing hormone receptors revealed using a protein kinase C-responsive reporter gene. Endocrinology. 1997;138:1471–1475. doi: 10.1210/endo.138.4.5059. [DOI] [PubMed] [Google Scholar]
- KJELSBERG M.A., COTECCHIA S., OSTROWSKI J., CARON M.G., LEFKOWITZ R.J. Constitutive activation of the α1B- adrenergic receptor by all amino acid substitutions at a single site. Evidence for a region which constrains receptor activation. J. Biol. Chem. 1992;267:1430–1433. [PubMed] [Google Scholar]
- LEFKOWITZ R.J. G-protein-coupled receptors: Turned on to ill effect. Nature. 1993;365:603–604. doi: 10.1038/365603a0. [DOI] [PubMed] [Google Scholar]
- LIPINSKY D., GERSHENGORN M.C., ORON Y. Contribution of response kinetics to the response pattern: Studies of responses to thyrotropin-releasing hormone in Xenopus oocytes. J. Cell. Physiol. 1995a;162:284–289. doi: 10.1002/jcp.1041620214. [DOI] [PubMed] [Google Scholar]
- LIPINSKY D., NUSSENZVEIG D.R., GERSHENGORN M.C., ORON Y. Desensitization of the response to thyrotropin-releasing hormone in Xenopus oocytes is an amplified process that precedes calcium mobilization. Pflugers Arch. 1995b;429:419–425. doi: 10.1007/BF00374158. [DOI] [PubMed] [Google Scholar]
- LUPU-MEIRI M., BEIT-OR A., CHRISTENSEN S.B., ORON Y. Calcium entry in Xenopus oocytes: Effects of inositol trisphosphate, thapsigargin and DMSO. Cell Calcium. 1993;14:101–110.2. doi: 10.1016/0143-4160(93)90080-p. [DOI] [PubMed] [Google Scholar]
- LUPU-MEIRI M., SHAPIRA H., ORON Y. Dual regulation by protein kinase C of the muscarinic response in Xenopus oocytes. Pflugers Arch. 1989;413:498–504. doi: 10.1007/BF00594180. [DOI] [PubMed] [Google Scholar]
- LUPU-MEIRI M., SHAPIRA H., ORON Y. Extracellular calcium participates in responses to acetylcholine in Xenopus oocytes. FEBS Lett. 1990;262:165–169. doi: 10.1016/0014-5793(90)80180-q. [DOI] [PubMed] [Google Scholar]
- MATUS-LEIBOVITCH N., NUSSENZVEIG D.R., GERSHENGORN M.C., ORON Y. Truncation of the thyrotropin-releasing hormone receptor carboxyl tail causes constitutive activity and leads to impaired responsiveness in Xenopus oocytes and AtT20 cells. J. Biol. Chem. 1995;270:1041–1047. doi: 10.1074/jbc.270.3.1041. [DOI] [PubMed] [Google Scholar]
- MCFERRAN B.W., GUILD S.B. Effects of protein kinase C activators upon the late stages of the ACTH secretory pathway of AtT-20 cells. Br. J. Pharmacol. 1994;113:171–178. doi: 10.1111/j.1476-5381.1994.tb16190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCFERRAN B.W., MACEWAN D.J., GUILD S.B. Involvement of multiple protein kinase C isozymes in the ACTH secretory pathway of AtT-20 cells. Br. J. Pharmacol. 1995;115:307–315. doi: 10.1111/j.1476-5381.1995.tb15878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'DOWD B.F., HNATOWICH M., CARON M.G., LEFKOWITZ R.J., BOUVIER M. Palmitoylation of the human beta 2-adrenergic receptor. Mutation of cys341 in the carboxyl tail leads to an uncoupled non-palmitoylated form of the receptor. J. Biol. Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- ORON Y., GILLO B., STRAUB R.E., GERSHENGORN M.C. Mechanism of membrane-electrical response to thyrotropin-releasing hormone in Xenopus oocytes injected with GH3 pituitary cell messenger ribonucleic acid. Mol. Endocrin. 1987;1:918–925. doi: 10.1210/mend-1-12-918. [DOI] [PubMed] [Google Scholar]
- PARKER E.M., ROSS E.M. Truncation of the extended carboxyl-terminal domain increases the expression and regulatory activity of the avian β-adrenergic receptor. J. Biol. Chem. 1991;266:9987–9996. [PubMed] [Google Scholar]
- PARMA J., DUPREZ L., VAN-SANDE J., COCHAUX P., GERVY C., MOCKEL J., DUMONT J., VASSART G. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;356:649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- PERLMAN J.H., GERSHENGORN M.C. Thyrotropin-releasing hormone stimulation of phosphoinositide hydrolysis desensitizes. Evidence against mediation by protein kinase C or calcium. Endocrin. 1991;129:2679–2686. doi: 10.1210/endo-129-5-2679. [DOI] [PubMed] [Google Scholar]
- PREMONT R.T., INGLESE J., LEFKOWITZ R.J. Protein kinases 3: Protein kinases that phosphorylate activated G protein-coupled receptors. FASEB J. 1995;9:175–182. doi: 10.1096/fasebj.9.2.7781920. [DOI] [PubMed] [Google Scholar]
- PREZEAU L., GOMEZA J., AHERN S., MARY S., GALVEZ T., BOCKAERT J., PIN J.P. Changes in the carboxyl-terminal domain of metabotropic glutamate receptor 1 by alternative splicing generate receptors with differing agonist-independent activity. Mol. Pharmacol. 1996;49:422–429. [PubMed] [Google Scholar]
- REN Q., KUROSE H., LEFKOWITZ R.J., COTECCHIA S. Constitutively active mutants of the α2-adrenergic receptor. J. Biol. Chem. 1993;268:16483–16487. [PubMed] [Google Scholar]
- RICHARDSON R.M., DUBOSE R.A., ALI H., TOMHAVE E.D., HARIBABU B., SNYDERMAN R. Regulation of human interleukin-8 receptor A: Identification of a phosphorylation site involved in modulating receptor functions. Biochemistry. 1995;34:14193–14201. doi: 10.1021/bi00043a025. [DOI] [PubMed] [Google Scholar]
- ROTH B.L., PALVIMAKI E., BERRY S., KHAN N., SACHS N., ULUER A., CHOUDHARY M.S. 5-hydroxytryptamine2A(5-HT2A) receptor desensitization can occur without down-regulation. J. Pharmacol. Exp. Ther. 1995;275:1638–1646. [PubMed] [Google Scholar]
- SAMAMA P., COTECCHIA S., COSTA T., LEFKOWITZ R.J. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- SAMAMA P., PEI G., COSTA T., COTECCHIA S., LEFKOWITZ R.J. Negative antagonists promote an inactive conformation of the β2-adrenergic receptor. Mol. Pharmacol. 1994;45:390–394. [PubMed] [Google Scholar]
- SHAPIRA H., LUPU-MEIRI M., GERSHENGORN M.C., ORON Y. Activation of two different receptors mobilizes calcium from distinct stores in Xenopus oocytes. Biophys. J. 1990;57:1281–1285. doi: 10.1016/S0006-3495(90)82646-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHENKER A., LAUE L., KOSUGI S., MERENDINO J.J., Jr, MINEGISHI T., CUTLER G.B., JR A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365:652–654. doi: 10.1038/365652a0. [DOI] [PubMed] [Google Scholar]
- STRAUB R.E., FRECH G.C., JOHO R.H., GERSHENGORN M.C. Expression cloning of a cDNA encoding the mouse pituitary thyrotropin-releasing hormone receptor. Proc. Natl. Acad. Sci U.S.A. 1990;87:9514–9518. doi: 10.1073/pnas.87.24.9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIBERI M., CARON M.G. High agonist-independent activity is a distinguishing feature of the dopamine D1B receptor subtype. J. Biol. Chem. 1994;269:27925–27931. [PubMed] [Google Scholar]
- UEDA H., MIYAMAE T., HAYASHI C., WATANABE S., FUKUSHIMA N., SASAKI Y., IWAMURA T., MISU Y. Protein kinase C involvement in homologous desensitization of δ-opioid receptor coupled to Gi1-phospholipase C activation in Xenopus oocytes. J. Neurosci. 1995;15:7485–7499. doi: 10.1523/JNEUROSCI.15-11-07485.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALE W., RIVIER J., BURGUS R. Synthetic TRF (thyrotropin releasing factor) analogues: II pGlu-M3imMe-His-Pro-NH2: A synthetic analogue with specific activity greater than that of TRF. Endocrin. 1971;89:1485–1488. doi: 10.1210/endo-89-6-1485. [DOI] [PubMed] [Google Scholar]
- ZHANG M., TURNBAUGH D., COFIE D., DOGAN S., KOSHIDA H., FUGATE R., KEM D.C. Protein kinase C modulation of cardiomyocyte angiotensin II and vasopressin receptor desensitization. Hyperten. 1996;27:269–275. doi: 10.1161/01.hyp.27.2.269. [DOI] [PubMed] [Google Scholar]