

Abstract
12,14-dichlorodehydroabietic acid (12,14-Cl2DHA) reduced GABA-stimulated uptake of 36Cl− into mouse brain synaptoneurosomes suggesting inhibition of mammalian GABAA receptor function.
12,14-Cl2DHA did not affect the binding of [3H]-muscimol to brain membranes but displaced specifically bound [3H]-EBOB. The inhibitory effect on [3H]-EBOB binding was not reversible. 12,14-Cl2DHA reduced the availability of [3H]-EBOB binding sites (Bmax) without changing the KD of the radioligand for remaining sites. 12,14-Cl2DHA did not affect the rate of association of [3H]-EBOB with its chloride channel receptor, but increased the initial rate of [3H]-EBOB dissociation.
12,14-Cl2DHA enhanced the incidence of EPSCs when rapidly applied to cultured rat cortical neurones. Longer exposures produced block of IPSCs with marked increases in the frequency of EPSCs and min EPSCs. 12,14-Cl2DHA also irreversibly suppressed chloride currents evoked by pulses of exogenous GABA in these cells.
Ultimately, 12,14-Cl2DHA inhibited all synaptic traffic and action currents in current clamped cells indicating that, in contrast to picrotoxinin (which causes paroxysmal bursting), it is not fully selective for the GABAA receptor-chloride channel complex.
The depolarizing block seen with 12,14-Cl2DHA in amphotericin-perforated preparations implicates loss of Ca2+ buffering in the polarity change and this may account for inhibition of spontaneous action potentials.
Our investigation demonstrates that 12,14-Cl2DHA blocks GABA-dependent chloride entry in mammalian brain and operates as a non-competitive insurmountable GABAA antagonist. The mechanism likely involves either irreversible binding of 12,14-Cl2DHA to the trioxabicyclooctane recognition site or a site that is allosterically coupled to it. We cannot exclude, however, the possibility that 12,14-Cl2DHA causes localized proteolysis or more extensive conformational change within a critical subunit of the chloride channel.
Keywords: 36Cl− uptake; cortical neurones; 12,14-dichlorodehydroabietic acid; [3H]-EBOB binding; GABA receptor; mammalian brain; patch clamp; synaptoneurosomes
Introduction
Where the bleaching of wood pulp with chlorine is carried out, dehydroabietic acid, a tricyclic diterpene monocarboxylic acid commonly found in coniferous trees, undergoes chemical modification generating several chlorinated compounds including 12,14-dichlorodehydroabietic acid (12,14-Cl2DHA; Thakore & Oehlschlager, 1977). Both chlorinated and non-chlorinated compounds have the potential to cause significant toxicity to fish in situations where pulp mill effluents are discharged into water bodies (Leach & Thakore, 1976; 1977). Although it is known that the exposure of fish to tricyclic diterpene monocarboxylic (or resin) acids leads to significant accumulation in the brain (Kruzynski, 1979; Oikari et al., 1982) and symptoms of nervous system impairment occur (Oikari et al., 1982), little is known about how these compounds act at the cellular and molecular level in brain. We have recently found that resin acids are capable of activating the release of excitatory and inhibitory neurotransmitters from synaptosomes prepared from the brains of fish and mammals (Nicholson, 1994; Zheng & Nicholson, 1996). A rise in cytosolic free [Ca2+] is a critical event associated with neurotransmitter vesicle depletion, and resin acids appear to initiate this through intraterminal discharge of Ca2+, although significant dependence on extracellular Ca2+ was also observed (Zheng & Nicholson, 1998). The three resin acids we have studied to date include abietic acid, dehydroabietic acid (see Morales et al., 1992 for structures), and 12,14-Cl2DHA (see Figure 1 for structure), and of these, 12,14-Cl2DHA is the most potent, increasing cytosolic free [Ca2+] and stimulating transmitter release in the low micromolar range (Zheng & Nicholson, 1996; 1998).
Figure 1.
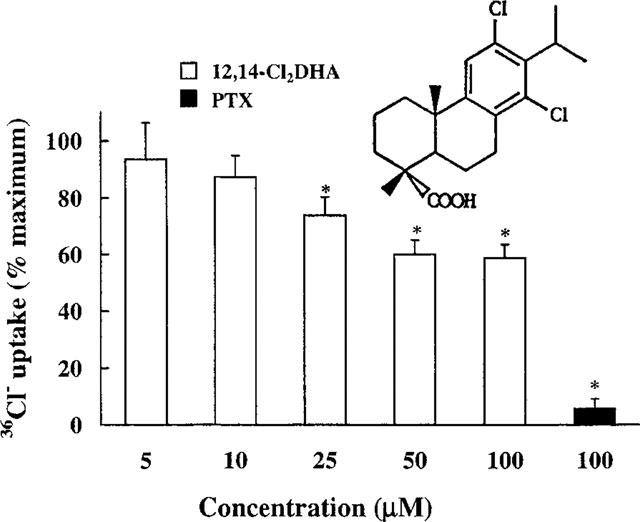
Inhibition by 12,14-Cl2DHA of GABA-dependent 36Cl− uptake into membrane vesicles prepared from mouse brain. The effect of picrotoxin (PTX) is also shown. Each value represents the mean±s.e.mean of 3–8 independent determinations. Asterisks indicate a statistically significant difference (P<0.05) from controls (Bonferroni's method after one way ANOVA). In these experiments, GABA-dependent uptake was 14.8±1.7 nmol chloride mg protein−1. The structure of 12,14-Cl2DHA is also displayed.
In connection with an unrelated investigation which required a γ-aminobutyric acid (GABA)-sensitive 36Cl− flux assay to be established, we unexpectedly found that 12,14-Cl2DHA also had the ability to inhibit GABA-stimulated entry of 36Cl− into mouse brain vesicles. Initial electrophysiological experiments conducted on cultured cortical neurons from embryonic rats indicated that 12,14-Cl2DHA enhanced the incidence of EPSCs with associated IPSC reduction, suggesting disinhibition of GABA. It was therefore of interest to characterize the mechanism of action of this chlorinated resin acid at the GABAA receptor-chloride channel complex in more depth, particularly since it shows no obvious structural resemblance to competitive GABAA receptor antagonists or the convulsant agents which act at the chloride channel.
Methods
Chemicals
The study compound 12,14-Cl2DHA was purchased at 99% purity from Helix Biotech Corporation (Richmond, BC, Canada). The compound was freshly formulated each day from frozen aliquotted dimethyl sulphoxide (DMSO) stock solutions. Radiochemicals were obtained from Dupont NEN and the other pharmacological agents used in this study were from Sigma.
Animals
This investigation was conducted using male CD1 mice (18–25 g; Charles River Laboratories, St. Constant, Quebec) and embryonic Sprague-Dawley rats were used for culture preparation. Animals were given continuous access to food and water and sacrificed by cervical dislocation and decapitation. All procedures relating to the housing and euthanasia of animals were carried out in compliance with Canadian Council on Animal Care guidelines or Schedule 1 Home Office Animals (Scientific Procedures) Act 1986, (U.K.).
36Chloride uptake into synaptoneurosomes
36Cl− uptake assays were performed with synaptoneurosomes essentially as described by Harris & Allen (1985) with certain modifications (Bloomquist et al., 1986). Whole brains were removed from two CD1 mice and quickly cooled in ice-cold isolation buffer (in mM) NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, glucose 54 and HEPES 20 adjusted to pH 7.4 with Tris base. Brains were then coarsely chopped with a razor blade and homogenized by hand in 2.5 ml of isolation buffer (8 excursions). The homogenate was diluted with a further 12.5 ml of isolation buffer and then filtered through two layers of 100 μm nylon mesh. The filtrate was centrifuged at 1000×g for 15 min. After resuspending this pellet in 10 ml of isolation buffer and centrifuging (1000×g for 15 min), the synaptoneurosome pellet was resuspended in 2.5 ml of isolation buffer containing BSA (1 mg ml−1) and held on ice. Synaptoneurosomes (200 μl; approx. 2.6 mg protein) were incubated with test compounds or DMSO for 15 min at 30°C. 36Cl− uptake was started by addition of 200 μl assay buffer (in mM) NaCl 145, KCl 4.7, CaCl2 2.5, MgSO4 1.2, glucose 27 and HEPES 20 adjusted to pH 7.4 with Tris base, containing 0.6 μCi [36Cl]-HCl with or without 100 μM GABA as appropriate. Incubations were terminated after 4 s by rapid mixing with 4 ml ice-cold assay buffer, followed immediately by filtration (Whatman GF/C), and further washing (3×4 ml). Initial experiments confirmed that the levels of DMSO employed had no effect on the assay.
[3H]-muscimol binding
Crude synaptic membranes were isolated from the brains of four CD1 mice and stored frozen according to the procedure of Beaumont et al. (1978). Binding assays were conducted using the filtration protocol of Negro et al. (1995). Synaptic membranes (approx. 0.5 mg protein) were incubated in Tris-citrate buffer (pH 7.1; 0.5 ml) containing [3H]-muscimol (11.8 Ci mmol−1, 20 nM final concentration) with 12,14-Cl2DHA, drugs or control solvent (DMSO) for 30 min, at 4°C in darkness. Incubations were stopped by adding ice-cold Tris-citrate buffer (4 ml), followed by rapid filtration on Whatman GF/C filters. Filters were rinsed quickly with Tris-citrate buffer (3×4 ml), dried and then incubated with 7% SDS prior to quantitation of radioactivity. Assays were performed at least in duplicate. Non-specific binding was assessed in the presence of 100 μM GABA and averaged 4.03% of total.
[3H]-EBOB binding
Brain membranes were routinely isolated from 4 CD1 mice and stored frozen prior to assay. The preparation of membranes and the [3H]-EBOB binding assay were carried out according to methods published by Cole & Casida (1992). Brain membranes (400 μg protein) in 1 ml sodium phosphate buffer (10 mM, pH 7.5) containing 300 mM NaCl were incubated with [propyl-2,3-3H]-ethynyl bicycloorthobenzoate ([3H]-EBOB) (38 Ci mmol−1; 750 pM final concentration) together with study compound or control solvent (DMSO) as necessary, for 90 min at 37°C. Reactions were terminated by rapid filtration through Whatman GF/C filters and membranes were subjected to three washes with 4 ml ice-cold phosphate buffer. Assays were performed at least in duplicate and non-specific binding was determined with lindane at a saturating concentration (5 μM). Potential effects on the association kinetics of [3H]-EBOB were determined by incubating the membrane preparation for 10 min with 12,14-Cl2DHA or solvent control prior to adding radioligand and then monitoring specific binding of radioligand until a steady state situation was reached. To assess the effect of study compound on the dissociation kinetics of [3H]-EBOB, the membrane preparation was equilibrated with [3H]-EBOB, then challenged with 12,14-Cl2DHA (10 or 50 μM) plus displacer or solvent control plus displacer. Equilibrium binding assays employed concentrations of [3H]-EBOB ranging from 75–1500 pM in the presence of either 12,14-Cl2DHA (10 μM) or DMSO.
Cell culture
Neuronal cultures were prepared from cerebral cortices of 17–18 day old rat embryos (Lees & Leach, 1993). Cells were plated onto poly-D-lysine coated coverslips (25–50,000 cells ml−1) in Dulbecco's Modified Eagle Medium supplemented with 10% foetal calf serum and 100 u μg ml−1 penicillin/streptomycin. After 12–24 h the plating medium was replaced by a maintenance medium comprising, Neurobasal medium, with 2% B27 supplement, 1% glutamax (Gibco) and 100 u μg ml−1 penicillin/streptomycin. Cells were used in experiments after 14–35 days in vitro.
Electrophysiology
Cultured networks on coverslips were placed in a 5 mm perspex trench on the mechanical stage of a Nikon Diaphot inverted microscope equipped with phase contrast optics and Narishige hydraulic micromanipulators. Cells were vigorously perfused (circa 2 ml min−1) with saline (in mM) NaCl 142, KCl 5, CaCl2 2, MgCl2 2, HEPES 5, glucose 10 adjusted to pH 7.4. 50 nM tetrodotoxin (TTX) was added (as indicated in the text) to block action potentials and evoked synaptic activity in cultured networks. Patch clamp recording micropipettes were fabricated from thin-walled borosilicate glass capillary tubes (1.7 mm outer diameter) using a Mecanex BBCH programmable puller. Pipette saline consisted of (mM) KCl or potassium gluconate 132, MgCl2 2, CaCl2 1, HEPES 10, EGTA 11 adjusted to pH 7.4. All potentials cited are those based on the preamplifier null potential and take no account of the liquid junction offset (circa −14 mV by direct measurement: see Neher, 1992) inherent in the use of asymmetrical, (gluconate-based), pipette solutions which were used for the majority of experiments. To achieve non-invasive recordings, without the use of strong intracellular Ca2+ buffering, the gluconate based pipette saline was supplemented with 130 μM amphotericin B (freshly prepared from DMSO stocks within 2–3 h of the experiment) to obtain perforated patch recordings. Borosilicate patch pipettes (3–5 MΩ) were used. 60–80% series resistance compensation was applied at the List EP7 pre-amplifier. Pyramidal neurones with input resistances of ⩾100–200 MΩ were studied. Whole cell currents were filtered at 0.5–3 KHz prior to digitization and monitored on a Gould chart recorder or analysed using WCP (John Dempster, Strathclyde University) or CED software.
For electrophysiological studies the stock solutions of 12,14-Cl2DHA were diluted 1000 times into saline. 0.1% DMSO has previously been shown to produce no effect on the parameters reported here and was routinely added to drug-free salines and perfused from glass reservoirs via teflon lines. The compound was rapidly and quantitatively delivered to cultured cells using the Y-tube technique (Murase et al., 1989) which allows data to be derived on the effect of brief pulsing. In other experiments, the study compound was superfused through the recording chamber enabling the net effects on cellular excitation at equilibrium to be determined. GABA was applied at a saturating concentration of 1 mM or at 10 μM (<EC50: Lees et al., 1998) as 0.5–1 s pulses through the Y-tube. The failure of GABA to overcome the inhibitory effects of 12,14-Cl2DHA at equilibrium after extensive washing was taken to indicate irreversibility. Unless otherwise stated, all observations reported were replicated in a minimum of three cells.
Data analysis
Results are expressed as mean±standard error of the mean (s.e.m.). Where required, data sets were analysed using the statistical test indicated in figure legends. A P value <0.05 was taken as significant.
Other methods
Radioactivity associated with mouse brain synaptoneurosomes and membrane fractions was determined by liquid scintillation counting (Beckman LS 3801). The amount of protein in samples was determined using the procedure of Peterson (1977).
Results
Inhibition of GABA-dependent 36Chloride uptake by 12,14-Cl2DHA
12,14-Cl2DHA inhibited GABA-stimulated uptake of 36Cl− into brain membrane vesicles in a concentration-related manner. At 50 μM and above the inhibition plateaued to approximately 40% (Figure 1). 12,14-Cl2DHA was not soluble in saline above 100 μM and this precluded experiments at higher concentrations. The IC50 of 12,14-Cl2DHA was calculated at 16.4 μM and this compound had no effect on basal accumulation of 36Cl− up to 100 μM (data not shown).
Effects of 12,14-Cl2DHA on [3H]-EBOB binding
12,14-Cl2DHA (50 μM) had no effect on high affinity specific binding of [3H]-muscimol in situations where bicuculline (50 μM) but not picrotoxin (100 μM) produced significant inhibition (data not shown), indicating that 12,14-Cl2DHA does not bind to the GABA recognition site.
The specific component of [3H]-EBOB binding was inhibited by 12,14-Cl2DHA between approximately 1 and 100 μM (Figure 2) and the IC50 was established at 9.38±0.73 μM. Equilibrium binding experiments with [3H]-EBOB over increasing radioligand concentrations, indicated high affinity binding to a single class of recognition site in agreement with Cole & Casida (1992). At 10 μM, 12,14-Cl2DHA significantly reduced the apparent concentration of [3H]-EBOB binding sites from 1220±98 to 775±106 fmol mg−1 protein, representing a 36.5% decrease (Figure 3). The affinity of chloride channels for this radioligand (KD=2.6± 0.29 nM) was not significantly changed by 12,14-Cl2DHA. The effect of 12,14-Cl2DHA on the rate of association of [3H]-EBOB is shown in Figure 4a. The semilogarithmic plots of association data show clearly that 10 μM 12,14-Cl2DHA does not affect the rate at which [3H]-EBOB associates with its binding site. In sharp contrast, an identical concentration of 12,14-Cl2DHA increased displacer-dependent dissociation of [3H]-EBOB from the steady state receptor : ligand complex by 1.7 fold (Figure 4b). 50 μM 12,14-Cl2DHA, a concentration approximately five times higher than the IC50, caused a more rapid (2.8 fold) increase in radioligand dissociation. To obtain information on the extent to which 12,14-Cl2DHA-related inhibition of [3H]-EBOB binding can be reversed, the membrane preparation was incubated for 10 min with 15 μM 12,14-Cl2DHA, then centrifuged and resuspended in fresh binding medium a further one to three times. At each stage, the effect of the initial 12,14-Cl2DHA exposure on [3H]-EBOB binding was assessed. No statistically significant reduction in the ability of 12,14-Cl2DHA to inhibit [3H]-EBOB binding was detected following sequential washing (Figure 5). Parallel experiments confirmed that the absolute level of specific [3H]-EBOB binding remained unchanged in the membranes throughout this procedure (data not shown).
Figure 2.
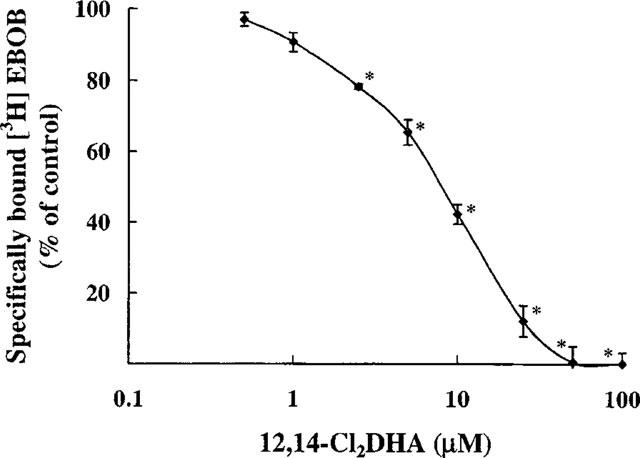
Displacement by 12,14-Cl2DHA of specific binding of [3H]-EBOB to membrane fragments prepared from mouse brain. Values represent means±s.e.mean of 3–5 experiments. Asterisks indicate a statistically significant difference (P<0.05) from solvent controls (Bonferroni's method after one way ANOVA).
Figure 3.
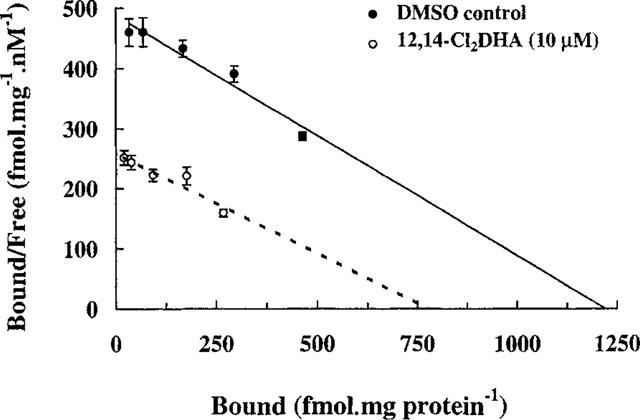
Scatchard plots of [3H]-EBOB binding to mouse brain membranes in the absence and presence of 10 μM 12,14-Cl2DHA. The data show means±s.e.mean of three separate experiments. Lines were generated by linear regression analysis. The difference in mean Bmax values is statistically significant (P<0.05; Bonferroni's method after one way ANOVA). Differences in KD values are not statistically significant.
Figure 4.
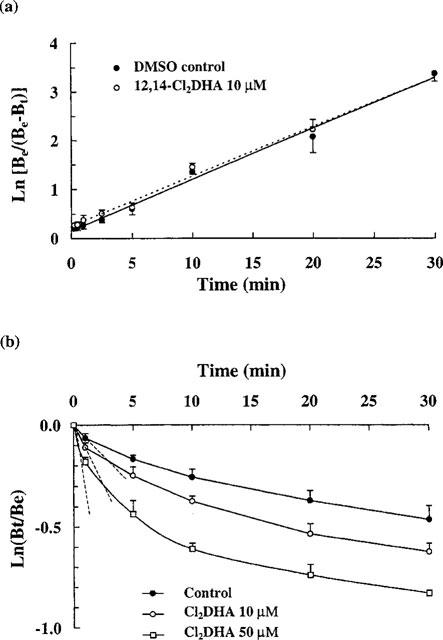
(a) Kinetics of association of [3H]-EBOB with chloride channels in the absence and presence of 10 μM 12,14-Cl2DHA. Semilogarithmic plots are displayed and lines were generated by linear regression analysis. Data points represent means and bars s.e.means of 3–5 preparations. No statistically significant differences in rate are present. (b) Time course for dissociation of [3H]-EBOB from the chloride channel complex. Note: 12,14-Cl2DHA produced significant (P<0.05) concentration-related increases in the initial rate of radioligand dissociation compared to the control (Bonferroni's method after one way ANOVA). The data show means±s.e.mean of three independent experiments.
Figure 5.
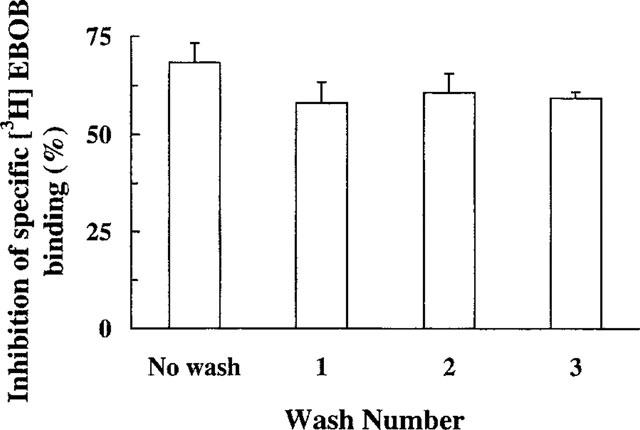
The failure of sequential washing of brain membranes to reverse the inhibitory effect of 15 μM 12,14-Cl2DHA on [3H]-EBOB binding. Values are means±s.e.mean of three independent experiments. No statistically significant differences were observed in multiple comparisons between unwashed membranes and membranes receiving up to three washes.
Rapid application of 12,14-Cl2DHA via the Y-tube
1 or 10 μM 12,14-Cl2DHA evoked increases in the incidence of post-synaptic currents in pyramidal neurones whole cell clamped at −45 mV (n=4). When applied as a brief pulse (20–30 s) at 1 μM this action was apparently reversible (Figure 6a). At 1 or 10 μM EPSCs were so frequent that they appeared almost co-incident (Figure 6b), although 12,14-Cl2DHA did not elicit marked/consistent changes in input conductance or holding current consistent with a direct post-synaptic effect or effective summation of the phasic currents (in this recording configuration). As shown in Figure 6c, more prolonged application of 10 μM 12,14-Cl2DHA via the Y-tube, appeared to selectively block IPSCs (after a transient increase in their incidence) whilst enhancing markedly the rate of EPSC/minEPSC incidence in three of the three cells examined (consistent with biochemical effects on the GABAA receptor). Continued exposure to 10 μM 12,14-Cl2DHA eventually resulted in depression of incidence of both IPSCs and EPSCs (possibly due to transmitter depletion and/or receptor desensitization).
Figure 6.
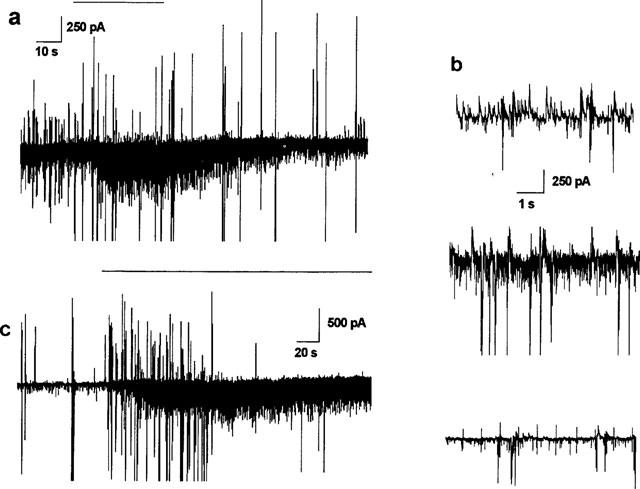
(a) A pulse of 1 μM 12,14-Cl2DHA (horizontal bar) via the Y-tube results in a marked increase in EPSC incidence after circa 10 s exposure. The response was reversed by washing with physiological saline. Cell clamped at −45 mV using gluconate in the recording pipette. (b) Higher resolution images showing similar responses to 10 μM 12,14-Cl2DHA in a different cell. Top, pretreatment saline blank from the Y-tube; middle, peak response to 12,14-Cl2DHA showing the marked increase in the incidence of EPSCs; bottom, continued exposure results in a net depression of spontaneous activity (the regular bipolar spikes are residual uncompensated capacative transients and current responses to voltage jumps driven through the patch electrode to gauge membrane input conductance). Vh: −45 mV. (c) At 10 μM in the same cell as (a), 12,14-Cl2DHA transiently elevated the incidence of EPSCs and IPSCs. Note the sustained effect on the inward currents (downward deflections). The IPSCs appear to be selectively blocked within 2 min.
Isolation of GABAA currents in TTX saline
Bath superfusion of 25 μM 12,14-Cl2DHA in TTX saline fully blocked inward chloride currents, evoked by 500 ms pulses of exogenous GABA (10 μM), within 10 min of superfusion (mean time to full block 7.25±1.2 min, n=4). Even with extensive washing (>10 min) with control saline, this effect was completely irreversible (Figure 7). Again, no consistent or marked changes in cellular holding current or input resistance (not quantified) were associated with 12,14-Cl2DHA application in these experiments (not shown).
Figure 7.
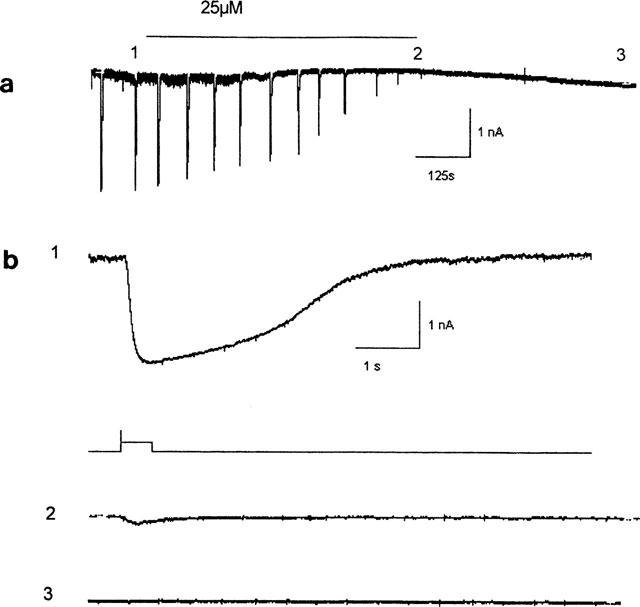
(a) Current responses to regular 500 ms pulses of 10 μM GABA on a compressed time base. The cell was clamped at −45 mV in 50 nM TTX and the inward currents reflect the use of chloride in the recording pipette. The horizontal bar indicates the period of superfusion with 25 μM 12,14-Cl2DHA. Note that the GABA response was still completely blocked even after extensive washing (3). (b) Higher resolution sweeps of the events marked numerically above. The unlabelled sweep indicates the timing/duration of the GABA pulse from the Y-tube.
In a further four cells, without TTX, outward currents evoked by 10 μM or 1 mM GABA (using gluconate-based pipette salines) were also completely blocked by 10 μM 12,14-Cl2DHA (Figure 8a). Before and after equilibration with 12,14-Cl2DHA and again after washing, a 500 ms pulse of 1 mM GABA (a saturating concentration to gauge the maximal response) was applied (Figure 8b). Even this high concentration of agonist did not produce a detectable response in treated preparations indicating that the blocking action of 12,14-Cl2DHA was insurmountable/non-competitive. In these experiments, it was apparent that the blocking action was not selective for inhibitory synaptic currents. Inward EPSCs and action currents were also blocked by the molecule with similar onset kinetics. It is noteworthy that the non-selective depressant effects were much faster in onset by superfusion than by Y-tube application.
Figure 8.
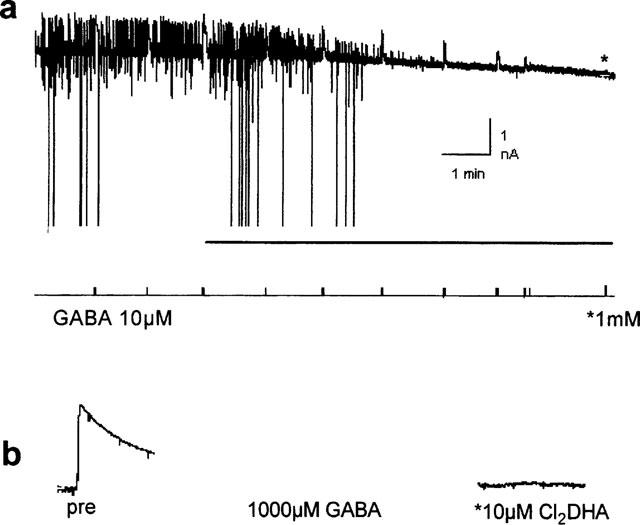
(a) Spontaneous activity in a cell whole-cell clamped at −45 mV using gluconate in the patch electrode. Inhibitory (GABAergic) events are upward deflections. The large downward events are ‘action currents'. Exogenous GABA was applied as indicated in the lowest trace. 10 μM 12,14-Cl2DHA was bath applied (horizontal bar). (b) Current evoked by a saturating concentration of GABA in a different cell before and after equilibration with the resin acid in an identical experiment.
Comparison of 12,14-Cl2DHA with picrotoxinin in current clamped cells
The patch amplifier was used in current clamp mode to make comparisons with 10 μM picrotoxinin (bath perfusion). The alkaloid depressed IPSP amplitude and elicited protracted hyperexcitation (nine of nine treated cells) and paroxysmal depolarizing shifts in eight of nine cells examined (Figure 9a and b). These effects were at least partially reversible in two of two neurones (not shown). In contrast, 12,14-Cl2DHA at 10 μM appeared to indiscriminately block synaptic traffic and action currents in the cells within a few minutes of superfusing the bath (four of four cells examined; Figure 9b and c). To examine the possibility that this reflected an effect on the viability of the recording configuration or interference with excitable properties of the cells, we applied depolarizing currents through the recording electrode. After spontaneous synaptic potentials had been fully blocked by 12,14-Cl2DHA, only one of the four treated cells was able to generate an action potential which was very broad and was not followed by any after hyperpolarization (AHP). At least at the somatic recording site, no consistent polarity shifts were associated with resin acid action. It was noticeable that 12,14-Cl2DHA (but not picrotoxinin) appeared to widen spontaneous action potentials and erode the associated AHP in the early phases of treatment prior to induction of electrical silence (three of five treated cells; Figure 10a and b).
Figure 9.
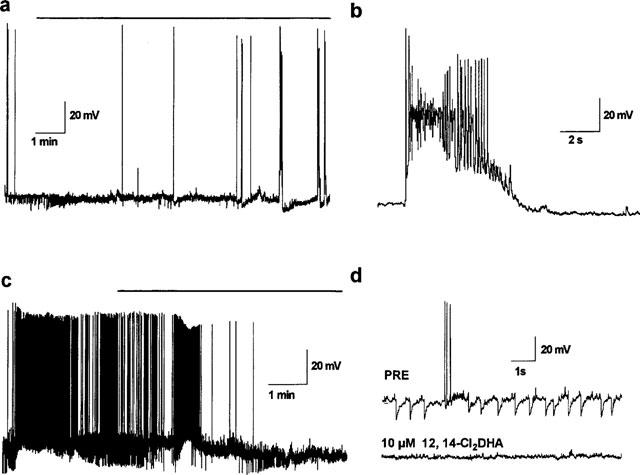
(a) Spontaneous activity in a current clamped cell superfused with 10 μM picrotoxinin (horizontal bar). Note the selective, progressive block of IPSPs (downward deflections) and the increased incidence of spiking/burst firing in the treated cell. (b) One of these bursts at higher resolution: these paroxysmal depolarizing shifts are typical features of disinhibition in the cultured cortical cells. (c) 10 μM12,14-Cl2DHA (bar) did not evoke this type of epileptiform activity. In all cells examined the compound blocked all synaptic currents and spontaneous spiking (after transient periods of hyperexcitation). (d) Faster sweeps (from the cell depicted in c) demonstrating spontaneous synaptic events and action potentials pretreatment (upper trace) and the lack of physiological activity after prolonged exposure to 12,14-Cl2DHA.
Figure 10.
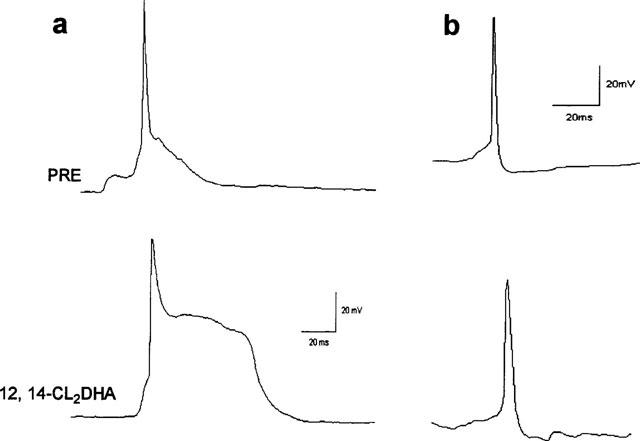
Action potential profiles in control saline (top sweeps) then in the early phase of treatment with 10 μM 12,14-Cl2DHA (bottom sweeps). (a) A relatively broad spike with a presumptive Ca2+ mediated plateau phase. The latter component was drastically enlarged and prolonged by 12,14-Cl2DHA. (b) A fast action potential with a pronounced AHP in a different cell: note the 12,14-Cl2DHA enhanced the spike duration and partially suppressed the after potential.
Effect of 12,14-Cl2DHA in amphotericin-perforated cells
Since our previous experiments (Zheng & Nicholson, 1998) have demonstrated that the resin acids elevate cytoplasmic free Ca2+, we studied the effects of 12,14-Cl2DHA on cells using the amphotericin-perforated patch technique (which does not rely on high cytoplasmic EGTA concentrations and strong artificial buffering of Ca2+ in the soma and proximal neurites of the cells selected). Again, in every cell tested, 12,14-Cl2DHA (10 μM) elicited a dramatic enhancement in EPSP/C incidence in impaled cells (five of five) following a transient (20–120 s) hyperpolarization/outward current (Figure 11a). In this recording configuration relatively large and consistent (quantified for all four current-clamped cells) net depolarizing shifts in transmembrane polarity were observed compared to the whole cell data (Figure 11b). Full-blown action potential propagation was confined to the very early period of exposure to 12,14-Cl2DHA.
Figure 11.
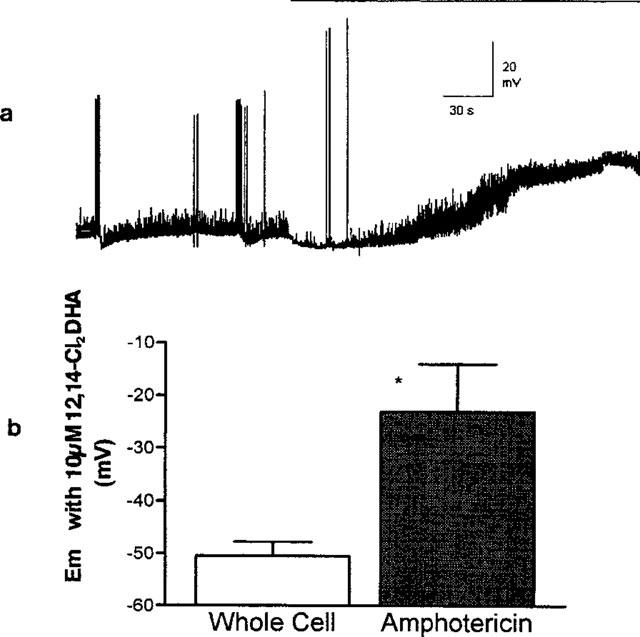
(a) Perforated patch recording from a current clamped neurone resting at circa −55 mV. 10 μM 12,14-Cl2DHA (horizontal bar) evoked relatively large and sustained depolarizing response using this none-invasive technique, accounting for the depressant effects on synaptic traffic noted in earlier experiments. Note the typical pronounced transient hyperpolarization and the brief period of spiking prior to the sustained enhancement in incidence of EPSPs. (b) Net effect of 12,14-Cl2DHA at 10 μM on cellular resting membrane potential in the indicated recording configurations. A marked and consistent somatic depolarization was only seen using non-invasive recording. The datasets (n=4) were significantly different (non-parametric Mann-Whitney test, P<0.05).
Discussion
The finding by Harris & Allen (1985) that stimulation of 36chloride uptake into mouse brain vesicles by GABA is (a) mimicked by muscimol and 3-aminopropane sulphonic acid but not baclofen, (b) blocked by bicuculline and picrotoxin and (c) enhanced by pentobarbital, demonstrated functional coupling of GABAA receptors to chloride channels. Since we have shown that 12,14-Cl2DHA inhibits GABA-dependent 36chloride accumulation in this in vitro system and has no effect on basal accumulation of 36chloride, it can be concluded that the study compound interferes specifically with GABAA receptor-mediated opening of chloride ion channels. To our knowledge, this is the first report of a tricyclic diterpene monocarboxylic acid acting as a GABAA antagonist.
Our fluxing experiments were carried out with 100 μM GABA to activate 36chloride entry. Under these conditions, inhibition of GABA-dependent chloride influx by 12,14-Cl2DHA is incomplete and, for reasons unknown, this contrasts to the complete block observed in electrophysiological experiments. However, the fluxing profile is similar to that found for lindane which, in more elaborately designed fluxing experiments, was shown to inhibit GABA-stimulated 36chloride uptake non-competitively (Wafford et al., 1989). To address the question of whether 12,14-Cl2DHA was acting competitively or non-competitively and to obtain concurrent information on the mechanism of blockade at the GABAA receptor-chloride channel complex, we conducted radioligand binding studies with [3H]-muscimol and [3H]-EBOB, which selectively label the GABAA binding site (Agey & Dunn, 1989) and the non-competitive blocker site (Cole & Casida, 1992) on this complex respectively.
The high affinity binding of [3H]-muscimol to brain membranes is inhibited by GABAA receptor agonists and also competitive, but not non-competitive antagonists (Wang et al., 1979). At its maximum effective concentration for inhibition of GABAA receptor-activated 36chloride uptake, 12,14-Cl2DHA had no statistically significant effect on the high affinity binding of [3H]-muscimol to brain membranes. Our binding experiments show that this effect is markedly different from that of bicuculline and GABA, but that a similarity exists between 12,14-Cl2DHA and the non-competitive GABAA receptor blocker picrotoxin. The lack of any displacing effect of 12,14-Cl2DHA on [3H]-muscimol binding, makes it unlikely that a competitive mechanism for inhibition is involved.
A key biochemical finding is that 12,14-Cl2DHA acts as an effective inhibitor of the binding of [3H]-EBOB to mouse brain membranes demonstrating its ability to exert a major influence on the non-competitive blocker site. Indeed the IC50s for 12,14-Cl2DHA in both the 36chloride flux and [3H]-EBOB binding assays show good agreement, and when compared to values for other non-competitive antagonists, place 12,14-Cl2DHA at similar potency to a number of polychlorocycloalkane insecticides (Bloomquist et al., 1986), but at lower potency compared with t-butylbicyclophosphorothionate and picrotoxin (Cole & Casida, 1992; Gant et al., 1987). In contrast to the competitive GABAA receptor agonists, compounds containing acidic functions like 12,14-Cl2DHA have not been widely reported as non-competitive blockers of the GABAA-gated chloride channel, although precedents are established for both carboxylic and phosphonic acids (Li & Casida, 1994).
Saturation analysis shows very clearly that 12,14-Cl2DHA reduces the Bmax for [3H]-EBOB without any effect on KD, indicating that it interacts with the trioxabicyclooctane receptor through a non-competitive mechanism. Our results also demonstrate that 12,14-Cl2DHA, at a concentration close to the IC50, is unable to influence the rate of formation of the [3H]-EBOB:chloride channel receptor complex reinforcing the idea that simple competitive inhibition is not involved. This conclusion is further supported by the observation that 12,14-Cl2DHA produces a concentration-related increase in the rate of dissociation of the radioligand : trioxabicyclooctane receptor complex over and above that of an excess concentration of displacer. When these results are considered together with the fact that attempts to reverse the inhibitory influence of 12,14-Cl2DHA on [3H]-EBOB binding by extensive washing were not successful, the most reasonable conclusion is that 12,14-Cl2DHA binds irreversibly to the trioxabicyclooctane recognition site or an adjacent site that is allosterically linked to it. Being weak acids, tricyclic diterpenene monocarboxylic acids are predominantly ionized at physiological pH (McLeay et al., 1979), and this gives molecules such as 12,14-Cl2DHA amphiphilic properties. Thus although the effects of 12,14-Cl2DHA on [3H]-EBOB binding appear quite specific, we do not rule out the possibility that this compound may induce localized proteolysis of the trioxabicyclooctane recognition site, or more global conformational change to critical transmembrane or extracellular domains of the chloride channel complex.
The electrophysiological results presented here are consistent with the biochemical data in that 12,14-Cl2DHA appears to be a potent and irreversible antagonist of the GABAA receptor in mammalian brain. The action of the GABAA receptor is much slower in onset than the transient facilitation of transmitter release which is presumably due to the presynaptic cytoplasmic-Ca2+ elevation characterized in an earlier study (Zheng & Nicholson, 1998). The slow onset of the GABA blockade, its irreversible nature and the effects on spontaneous traffic are not observed with the well characterized non-competitive GABAA antagonist picrotoxinin (Lees & Leach, 1993). The irreversible nature of the block may simply reflect physicochemical differences and the need to diffuse to a hydrophobic blocking site coupled to a high-affinity receptor interaction. However, the net effect of 12,14-Cl2DHA on synaptic traffic confirms that the interaction with the picrotoxinin site in the GABAA chloride channel is not the sole site of action. As we have previously reported the paroxysmal depolarizing shift and fulminant hyperexcitation is the hallmark of disinhibition in these cultured circuits (Lees & Leach 1993, Lees & Calder, 1996) regardless of whether bicuculline picrotoxin congeners, or cage-convulsants are used (Lees, unpublished). The lack of selectivity for inhibitory currents and potentials indicates that 12,14-Cl2DHA may also cause loss of Ca2+ buffering in the treated cells. This has been shown to result in a significant depletion of neurotransmitter vesicular pools in nerve terminals (Zheng & Nicholson, 1998) and may explain the eventual loss of excitatory drive in the circuits. Similar net effects on spontaneous synaptic currents and action currents were previously noted in the same cells exposed to the respiratory uncoupler surangin B which precipitates a comparatively small but sustained increase in cytoplasmic Ca2+. A sustained loss of Ca2+ buffering is almost invariably a terminal phase in cell death and results in electrical silence in our neuronal networks (Zheng et al., 1998).
Using amphotericin-perforated cells we unmasked a profound net depolarization which was sufficient to block spontaneous action potentials despite the marked enhancement in EPSP incidence. EGTA or intracellular dialysis clearly limits this effect. The depolarization may reflect the influence of cytoplasmic Ca2+ on membrane polarity or decreased washout of excitatory amino acid receptors but the detailed mechanism was not sought in this study. This depolarizing block (due to steady state inactivation of the voltage-gated Na+ channel) would explain the elimination of synaptic activity seen in the whole cell experiments. Clearly, 12,14-Cl2DHA and picrotoxinin have the capacity to disinhibit via GABAA receptors, but only the former blocks conduction and hence excitatory drive concurrently. 12,14-Cl2DHA was also transiently able to modify action potential kinetics in the cultured circuits. It is noteworthy that molecules which mediate Ca2+ entry such as dihydroavermectin B1 (Lees & Beadle, 1986) or regulate Ca2+ release for example the ryanoids (Sham et al., 1995), have a similar plethora of effects on cell surface signalling proteins which are not directly related to their acknowledged site of action.
12,14-Cl2DHA, at concentrations which are achieved in the brains of intoxicated fish, clearly has the potential to disrupt signalling in the CNS of mammals. Further studies will now be required to confirm the primary site of action of the compound. Our results to date suggest an important role for Ca2+ homeostasis but since the GABAA receptor is so widespread and both sites show similar sensitivity, the latter may be an important alternative target underpinning transient neuroexcitation.
We believe that the general pharmacological profile described here for 12,14-Cl2DHA likely extends to other structurally related resin acids (such as abietic acid and dehydroabietic acid) since recent experiments have confirmed that these compounds also have the ability to inhibit GABA-dependent 36chloride uptake and [3H]-EBOB binding (data not shown).
Acknowledgments
This research was supported in part by Natural Sciences and Engineering Research Council of Canada grants (OGP 0042113, EQP 0092242 and EQP 0123003) to RAN. GL thanks the Wellcome Trust for financial support and Helen Jackson for technical support with cell culture.
Abbreviations
- 12,14-Cl2DHA
12,14-dichlorodehydroabietic acid
- DMSO
dimethyl sulphoxide
- EGTA
ethylene glycol-bis (β-aminoethyl ether) N,N,N′N′-tetraacetic acid
- GABA
γ-aminobutyric acid
- [3H]-EBOB
[propyl-2,3-3H]-ethynyl bicycloorthobenzoate
- TTX
tetrodotoxin
References
- AGEY M.W., DUNN S. Kinetics of [3H]muscimol binding to the GABAA receptor in bovine brain membranes. Biochemistry. 1989;28:4200–4208. doi: 10.1021/bi00436a012. [DOI] [PubMed] [Google Scholar]
- BEAUMONT K., CHILTON W.S., YAMAMURA H.I., ENNA S.J. Muscimol binding in rat brain : association with synaptic GABA receptors. Brain Res. 1978;148:153–162. doi: 10.1016/0006-8993(78)90385-2. [DOI] [PubMed] [Google Scholar]
- BLOOMQUIST J.R., ADAMS P.M., SODERLUND D.M. Inhibition of γ-aminobutyric acid-stimulated chloride flux in mouse brain vesicles by polychlorocycloalkane and pyrethroid insecticides. NeuroToxicol. 1986;7:11–20. [PubMed] [Google Scholar]
- COLE L.M., CASIDA J.E. GABA-gated chloride channel: Binding site for 4′′-ethynyl-4-n-[2,3-3H2]propylbicycloorthobenzoate ([3H]EBOB) in vertebrate brain and insect head. Pestic. Biochem. Physiol. 1992;44:1–8. [Google Scholar]
- GANT D., ELDEFRAWI M.E., ELDEFRAWI A.T. Cyclodiene insecticides inhibit GABAA receptor-regulated chloride transport. Toxicol. Appl. Pharmacol. 1987;88:313–321. doi: 10.1016/0041-008x(87)90206-7. [DOI] [PubMed] [Google Scholar]
- HARRIS R.A., ALLEN A.M. Functional coupling of γ-aminobutyric acid receptors to chloride channels in brain membranes. Science. 1985;228:1108–1110. doi: 10.1126/science.2581319. [DOI] [PubMed] [Google Scholar]
- KRUZYNSKI G.M.Some effects of dehydroabietic acid (DHA) on hydromineral balance and other physiological parameters in juvenile sockeye salmon Oncorhynchus nerka 1979. PhD thesis. University of British Columbia, Vancouver, BC, Canada
- LEACH J.M., THAKORE A.N. Toxic constituents in mechanical pulping effluents. Tappi. 1976;59:129–132. [Google Scholar]
- LEACH J.M., THAKORE A.N. Compounds toxic to fish in pulp mill waste streams. Prog. Water Technol. 1977;9:787–798. [Google Scholar]
- LEES G., BEADLE D.J. Dihydroavermectin B1: actions on cultured neurones from the insect central nervous system. Brain Res. 1986;366:369–372. doi: 10.1016/0006-8993(86)91321-1. [DOI] [PubMed] [Google Scholar]
- LEES G., CALDER J. Interaction of lindane isomers with chloride currents in insect membranes–steric requirements for channel modulation and block. Pestic. Biochem. Physiol. 1996;55:40–48. doi: 10.1006/pest.1996.0033. [DOI] [PubMed] [Google Scholar]
- LEES G., EDWARDS M.D., HASSONI A.A., GANELLIN C.R., GALANAKIS D. Modulation of GABAA Receptors and Inhibitory Synaptic Currents by the Endogenous CNS Sleep Regulator cis-9,10 octadecenoamide (cOA) Br. J. Pharmacol. 1998;124:873–882. doi: 10.1038/sj.bjp.0701918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEES G., LEACH M.J. Studies on the mechanism of action of the novel anticonvulsant lamotrigine (Lamictal) using primary neuroglial cultures from rat cortex. Brain Res. 1993;612:190–199. doi: 10.1016/0006-8993(93)91660-k. [DOI] [PubMed] [Google Scholar]
- LI Q.X., CASIDA J.E. Structure-activity studies leading to potent chloride channel blockers: 5e-tert-butyl-2-[4-(substituted-ethynyl)phenyl]-1,3-dithianes. Bioorg. Med. Chem. 1994;2:1423–1434. doi: 10.1016/s0968-0896(00)82095-7. [DOI] [PubMed] [Google Scholar]
- MCLEAY D.J., WALDEN C.C., MUNRO J.R. Influence of dilution water on the toxicity of kraft pulp and paper mill effluent, including mechanisms of effect. Water Res. 1979;13:151–158. [Google Scholar]
- MORALES A., BIRKHOLZ D.A., HRUDLEY S.E. Analysis of pulp mill effluent contaminants in water, sediment and fish bile-fatty and resin acids. Water Environ. Res. 1992;64:660–668. [Google Scholar]
- MURASE K., RYU P.D., RANDIC M. Excitatory and inhibitory amino acids and peptide-induced responses in acutely dissociated dorsal horn neurons. Neurosci. Lett. 1989;103:56–63. doi: 10.1016/0304-3940(89)90485-0. [DOI] [PubMed] [Google Scholar]
- NEGRO M., CHINCHETRU M.A., FERNANDEZ A., CALVO P. Effect of ethanol treatment on rate and equilibrium constants for [3H]muscimol binding to rat brain membranes: alteration of two affinity states of the GABAA receptor. J. Neurochem. 1995;64:1379–1389. doi: 10.1046/j.1471-4159.1995.64031379.x. [DOI] [PubMed] [Google Scholar]
- NEHER E. Correction for liquid junction potentials in patch clamp experiments. Meths. Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- NICHOLSON R.A. Excitatory actions of dehydroabietic acid on mammalian synaptosomes. Pharmacol. Toxicol. 1994;75:274–279. doi: 10.1111/j.1600-0773.1994.tb00360.x. [DOI] [PubMed] [Google Scholar]
- OIKARI A., HOLMBOM B., BISTER H. Uptake of resin acids into tissues of the trout (Salmo gairdneri Richardson) Ann. Zool. Fenn. 1982;19:61–64. [Google Scholar]
- PETERSON G.L. A simplification of the protein assay of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- SHAM J.K., CLEEMAN L., MORAD M. Functional coupling of Ca++ channels and ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 1995;92:121–125. doi: 10.1073/pnas.92.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THAKORE A.N., OESCHLAGER A.C. Structures of toxic constituents in kraft mill caustic extraction effluents from 13C and 1H nuclear magnetic resonance. Can. J. Chem. 1977;55:3298–3303. [Google Scholar]
- WAFFORD K.A., SATTELLE D.B., GANT D.B., ELDEFRAWI A.T., ELDEFRAWI M.E. Noncompetitive inhibition of GABA receptors in insect and vertebrate CNS by endrin and lindane. Pestic. Biochem. Physiol. 1989;33:213–219. [Google Scholar]
- WANG Y.-J., SALVATERRA P., ROBERTS E. Characterization of [3H]muscimol binding to mouse brain membranes. Biochem. Pharmacol. 1979;28:1123–1128. doi: 10.1016/0006-2952(79)90316-2. [DOI] [PubMed] [Google Scholar]
- ZHENG J., LEONG D., LEES G., NICHOLSON R.A. Studies on the interaction of surangin B with insect mitochondria, insect synaptosomes and rat cortical neurones in primary Culture. Pestic. Biochem. Physiol. 1998;61:1–13. [Google Scholar]
- ZHENG J., NICHOLSON R.A. Influence of two naturally occurring abietane monocarboxylic acids (resin acids) and a chlorinated derivative on release of the inhibitory neurotransmitter γ-aminobutyric acid from trout brain synaptosomes. Bull. Env. Contam. Toxicol. 1996;56:114–120. doi: 10.1007/s001289900017. [DOI] [PubMed] [Google Scholar]
- ZHENG J., NICHOLSON R.A. Action of resin acids in nerve ending fractions isolated from fish central nervous system. Environmental Toxicol. Chem. 1998;17:1852–1859. [Google Scholar]