

Abstract
The main aim of this study was to identify putative β-bends and the role of the N- and C-terminus in the CGRP receptor antagonist hα CGRP8–37, which was measured against hα CGRP inhibition of twitch responses in the rat prostatic vas deferens.
With a bend-biasing residue (proline) at position 16 in hα CGRP8–37 (10−5 M) an inactive compound was produced, while alanine at the same position retained antagonist activity (apparent pKB 5.6±0.1 at 10−5 M). Proline at position 19 within hα CGRP8–37 (10−5 M) was an antagonist (apparent pKB 5.8±0.1).
Incorporation of a bend-forcing structure (beta-turn dipeptide or BTD) at either positions 19,20 or 33,34 in hα CGRP8–37 (10−5 M) antagonized hα CGRP responses (apparent pKB 6.0±0.1 and 6.1±0.1, respectively). Replacement by BTD at both positions 19,20 and 33,34 within hα CGRP8–37 competitively antagonized responses to hα CGRP (pA2 6.2; Schild plot slope 1.0±0.1).
Hα CGRP8–37 analogues (10−5 M), substituted at the N-terminus by either glycine8, or des-NH2 valine8 or proline8 were all antagonists against hα CGRP (apparent pKB 6.1±0.1, 6.5±0.1 and 6.1±0.1, respectively), while hα CGRP8–37 (10−5 M) substituted in three places by proline8 and glutamic acid10,14 was inactive.
Replacement of the C-terminus by alanine amide37 in hα CGRP8–37 (10−5 M) failed to antagonize hα CGRP responses.
Peptidase inhibitors did not alter either the agonist potency of hα CGRP or the antagonist affinities of hα CGRP8–37 BTD19,20 and 33,34 and hα CGRP8–37 Gly8 (against hα CGRP responses).
In conclusion, two β-bends at positions 18–21 and 32–35 are compatible with high affinity by BTD and is the first approach of modelling the bioactive structure of hα CGRP8–37. Further, the N-terminus of hα CGRP8–37 is not essential for antagonism, while the C-terminus interacts directly with CGRP receptor binding sites of the rat vas deferens.
Keywords: BTD, hα CGRP8–37, hα CGRP, peptidase inhibitors, CGRP2 receptor, rat prostatic vas deferens
Introduction
CGRP is a single chain 37 amino acid neuropeptide with an N-terminal disulphide loop between Cys2 and Cys7, and an amidated C-terminus (Rosenfeld et al., 1983). The peptide is a potent vasodilator (Brain et al., 1985; Marshall et al., 1986a,1986b) and has a variety of biological actions on the heart, neuronal tissue, skeletal muscle, immune and inflammatory responses (see Poyner, 1992 for review). CGRP exerts its effects via specific receptors, CGRP1 and CGRP2 (Dennis et al., 1989; 1990; Mimeault et al., 1991; Quirion et al., 1992). Mainly, the distinction is based on the differing antagonist affinities for the C-terminal fragment hα CGRP8–37, which has a higher affinity for the former than the latter receptor.
A pre-requisite to understanding the relation between CGRP8–37 structure and activity, would be to establish the structural features which determine the interaction of the peptide with its receptor(s). The N-terminal amphipathic α-helix may be an important feature for the interaction of the peptide with its receptors (e.g. Lynch & Kaiser, 1988; Mimeault et al., 1992). Structural features downstream of the helix have not yet been identified. However, both conformational and modelling studies suggested a tendency for two β-bend formations, one terminating the α-helix around residues 17 and 21 (Lynch & Kaiser, 1988; Breeze et al., 1991; Hubbard et al., 1991) and another around the C-terminal region 29 to 35 (Hubbard et al., 1991, Hakala & Vinhinen, 1994). Beta-turn regions have been shown to be important features of several biologically active peptides, including enkephalin, angiotensin II and gramicidin S, and substantial evidence exists that many of these peptides adopt β-turns in their active receptor bound conformations (Smith & Pease, 1980). A β-bend is a reverse turn, involving four residues formed by an intramolecular hydrogen bond between the C=O of residue i (i.e., the first residue of a turn) and the N-H of residue i+3 (i.e., the residue located three residues towards the carboxyl terminus). One approach towards peptidomimetics is to replace these β-turn regions with structures that bias (proline) or force (BTD; Nagai & Sato, 1985) the conformation of the native peptide (Figure 1).
Figure 1.
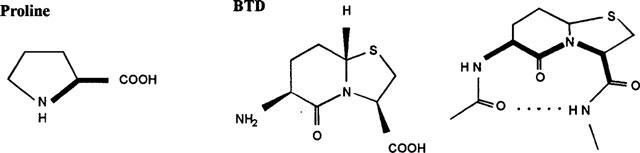
Chemical structure of the bend-biasing amino acid proline and the BTD (beta-turn dipeptide) peptidomimetic. Bold lines illustrate bend-biasing (proline) and bend-forcing (BTD) regions. The BTD mimic replaces the i+1 and i+2 amino acid residues of a four residue β-turn with its backbone conformation based on a 1-thioindolizine structure. Dotted lines represent hydrogen bonding between C=O of residue i and NH of residue i+3.
Therefore, the major target of this study was to investigate the putative β-bend regions of hα CGRP8–37 at the CGRP receptor in the rat prostatic vas deferens, which contains CGRP2 receptors (Dennis et al., 1989; Wisskirchen et al., 1998). Using alanine (which conserves structure but removes functionality), proline (which can bias a bend), and BTD (which forces a bend) as surrogates in hα CGRP8–37, these were assayed against hα CGRP. Further, the role of the N-terminal region and the C-terminus of hα CGRP8–37 was investigated, using structural modifications at position 8 (glycine, proline, des-NH2 valine), in the helical region (proline8 and glutamic acid10,14), and at the C-terminus (alanine amide37), which were studied on hα CGRP responses. To check for possible peptide degradation, peptidase inhibitors were tested on hα CGRP and CGRP antagonists. Preliminary accounts for part of the present study have been published in abstract form (Wisskirchen et al., 1994).
Methods
Male Sprague Dawley rats (300–450 g) were stunned and killed by cervical dislocation. The vas deferens was isolated, and the prostatic portion was suspended under 0.5 g resting tension in Krebs solution containing (mM): Na+ 143, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, Cl− 128, HCO3− 25, HPO42− 1.2, SO42− 1.2 and glucose 11 at 37°C, oxygenated with 95%O2 and 5%CO2, and allowed to equilibrate for 75 min. Contractile responses were induced by electrical field stimulation at 60 V, 0.2 Hz, 1.0 ms through parallel platinum wire electrodes either side of the tissues. The isometric tone was recorded with Grass FT.03 transducers.
Twitch responses to field stimulation were tested for stability for 10 min, and 40 min later, a cumulative concentration response curve to hα CGRP was obtained. The effect of hα CGRP8–37 analogues (10−5 M; 20 min pretreatment) was studied on second curves to hα CGRP, 40 min later. The analogues were substituted in position 8 by glycine (hα CGRP8–37 Gly8), des-NH2 valine (hα CGRP8–37 des-NH2 Val8), or proline (hα CGRP8–37 Pro8), in position 8, 10 and 14 by proline and glutamic acid (hα CGRP8–37 Pro8, Glu10,14), in position 16 by alanine (hα CGRP8–37 Ala16), in position 16 or 19 by proline (hα CGRP8–37 Pro16, hα CGRP8–37 Pro19), and in position 19,20 and/or 33,34 by BTD (hα CGRP8–37 BTD19,20; hα CGRP8–37 37BTD33,34; hα CGRP8–37 BTD19,20 and 33,34). Hα CGRP8–37 BTD19,20 and 33,34 was also tested at 3×10−6 M (20 min pretreatment) against hα CGRP responses. All CGRP fragments were tested on basal tone (i.e. unstimulated preparation) and on twitch responses.
A mixture of the peptidase inhibitors (amastatin, bestatin, captopril, phosphoramidon and thiorphan; 10−6 M each; 30 min pretreatment) was studied on responses to hα CGRP in the absence and presence of hα CGRP8–37, hα CGRP8–37 BTD19,20 and 33,34 or hα CGRP8–37 Gly8 (10−5 M). The effect of peptidase inhibitors on CGRP8–37 analogues (measured against hα CGRP responses) was compared with results obtained in their absence. Peptidase inhibitors were tested on basal tone and on twitch responses.
Chemicals
Amastatin, bestatin, captopril, phosphoramidon and thiorphan were obtained from Sigma, U.K. Hα CGRP, hα CGRP8–37, hα CGRP8–37 analogues and fragments were donated by Glaxo Wellcome Research Laboratories (Beckenham, Kent, U.K.), and were synthesised as described below. Apart from hα CGRP8–37 BTD19,20 and 33,34 being dissolved and diluted in DMSO, all other peptides were dissolved and diluted in distilled water, to form a 10−2 M stock solution, and stored at −20°C. Peptidase inhibitors were dissolved and diluted in DMSO to form a stock solution of 10−4 M, and were stored at −20°C.
Peptide synthesis
Peptides were prepared using solid phase synthesis with an Applied Biosystems 432A (0.025 mmole) or 430A (0.1 mmole) synthesizer, using Fmoc (Fluorenyloxycarbonyl) chemistry and unmodified FastMoc cycles. All peptides were synthesized as the C-terminal amide using Rink amide resin (Novabiochem). Phthaloyl-BTD-OEt was synthesized with minor modifications from a literature procedure (Nagai & Sato, 1985), producing the compound in six steps from L-glutamic acid in 38% overall yield. This was deprotected using hydrazine and base hydrolysis under pH static conditions (pH 11.5), followed by reprotection using Fmoc chloride as previously described (Caprino & Han, 1972), to give Fmoc-BTD-OH.
After complete synthesis and final Fmoc deprotection, the peptide was cleaved from the resin using triflouroacetic acid with 5% triethylsilane (Pearson et al., 1989) as scavenger. The crude material was purified to homogeneity by preparative reverse phase chromatography (HPLC) using a Poros 20 R2 (Perseptive Biosystem) (21.2×250 mm) column using a linear gradient of 5–50% acetonitrile over 20 min at 4 ml min−1, or a Zorbax C8 (21.2×250 mm) column using a linear gradient of 5–50% acetonitrile (0.2% triflouroacetic acid) over 20 min at 20 ml min−1.
All peptides were fully characterized by high field (600 MHz) 1H-NMR using a Bruker AM×600 spectrometer (initial studies were made using a Bruker AMX 500 spectrometer), and electrospray mass spectrometry using a VG Bio-Q (VG Instruments) at a needle voltage of 3.75 kV, cone voltage of 30 v tuned to 100 resolving power, scanned over mass range 500–1700. The samples for mass spectrometry were prepared (approximately 5–10 pmol μl−1) in acetonitrile/water (50–50 v v−1 with 1% formic acid) and introduced by flow injection into a mobile phase of the same composition at a flow rate of 3 μl min−1: The molecular weight of the peptide was determined using the +5 to +2 charge states.
Data analysis
The reduction in twitch tension of the field-stimulated prostatic vas deferens in response to applied drugs is expressed as percentage inhibition of twitch responses. All values are given as mean±s.e.mean. Differences were tested for significance using one-way ANOVA, Dunnett's test and Student's t-test (for paired and unpaired groups), as appropriate, accepting significance at P<0.05.
The IC50 values (molar concentration of the agonist that produced 50% of the maximal response) were calculated by non-linear regression curve fitting, using Graphpad Prism 2.0 (Graphpad Software, U.S.A.), and were used to determine the pIC50 values (−log of IC50). The Hill slope of each non-linear regression curve was determined (using Graphpad Prism 2.0), to check whether curves in the absence and presence of antagonists were parallel. In the presence of an antagonist used at a single concentration, an apparent pKB value was calculated, given by the equation:
where CR is the concentration ratio of the IC50 values in the presence and absence of the antagonist and [B] is the molar concentration of the antagonist. By this method, apparent pKB values for antagonists were determined from experiments where the agonist maximum response was unaltered. Where multiple concentrations of antagonist were used, a Schild plot of log (CR-1) against log [B] was plotted, and a linear regression carried out to derive the pA2 value and the Schild plot slope, using Graphpad Prism 2.0. The pA2 values were calculated from the individual control concentration response curves and the respective curves obtained in the presence of hα CGRP8–37 and analogues.
Results
Twitch responses evoked by field stimulation in the prostatic vas deferens resulted in reproducible uniform phasic contractions with a tension of 1.1±0.2 g (n=7). Hα CGRP inhibited twitch responses with a pIC50 of 8.0±0.1 (Hill slope 1.0±0.1; n=4). Hα CGRP8–37 analogues (up to 10−5 M) did not affect either basal tone or twitch responses, i.e. they were devoid of agonist activity.
Effect of proline and alanine replacement around the predicted central bend region of hα CGRP8–37
Hα CGRP8–37 Pro16 (10−5 M), with proline in position 16, failed to alter responses to hα CGRP (Figure 2a; Table 1), i.e. incorporation of a bend-biasing residue16 in hα CGRP8–37 abolished antagonism. Hα CGRP8–37 Ala16 (10−5 M) and hα CGRP8–37 Pro19 (10−5 M), with proline further downstream at position 19, antagonized hα CGRP responses (Figure 2b and c), and gave apparent affinities similar to that of hα CGRP8–37 (Table 1). These data indicated that incorporation of a bend-biasing residue (proline) retained antagonism of hα CGRP8–37 in position 19 but not in position 16.
Figure 2.
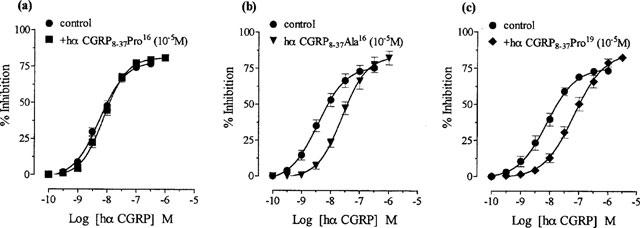
Effect of replacement by either proline16, alanine16 or proline19 in hα CGRP8–37 on hα CGRP responses in the rat prostatic vas deferens. Concentration response curves to hα CGRP on twitch responses, and in the presence of 10−5M of (a) hα CGRP8–37 Pro16 (b) hα CGRP8–37 Ala16 and (c) hα CGRP8–37 Pro19. Results are expressed as percentage inhibition of twitch responses. Points and error bars represent the mean±s.e.mean of four or five separate experiments.
Table 1.
Antagonist affinities of hα CGRP8-37 and analogues on hα CGRP-induced twitch inhibition in rat prostatic vas deferens

Effect of BTD replacement in the predicted bend regions of hα CGRP8–37
Hα CGRP8–37 BTD19,20 (10−5 M), with BTD at positions 19 and 20, antagonized hα CGRP responses (Figure 3a; Table 1). Similarly, hα CGRP8–37 BTD33,34 (10−5 M), with replacement by BTD at positions 33 and 34, antagonized responses to hα CGRP (Figure 3b; Table 1). Hα CGRP8–37 BTD19,20 and 33,34 (3×10−6 and 10−5 M), with two BTD molecules in positions 19,20 and 33,34 concentration-dependently antagonized hα CGRP consistent with competitive antagonism (Figure 4; Table 1). These results demonstrated that enforcement of a β-bend with BTD in positions 19,20 and 33,34, preserved the antagonist activity of hα CGRP8–37.
Figure 3.
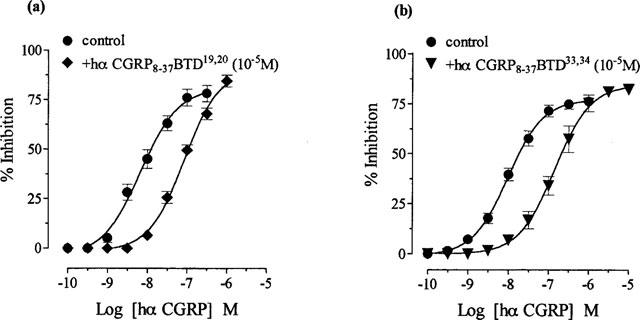
Effect of replacement by BTD at either positions 19,20 or 33,34 in hα CGRP8–37 on hα CGRP responses in the rat prostatic vas deferens. Concentration response curves to hα CGRP on twitch responses, and in the presence of 10−5M of (a) hα CGRP8–37 BTD19,20 and (b) hα CGRP8–37 BTD33,34. Results are expressed as percentage inhibition of twitch responses. Points and error bars represent the mean±s.e.mean of four or five separate experiments.
Figure 4.
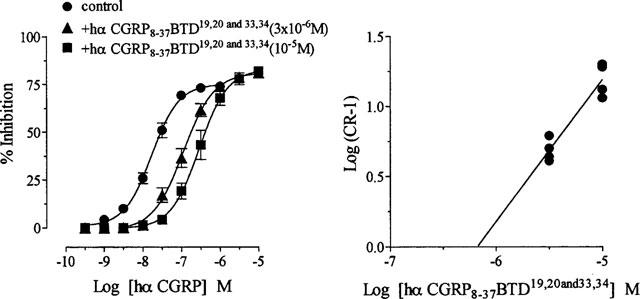
Effect of replacement by BTD at positions 19,20 and 33,34 in hα CGRP8–37 on hα CGRP responses in rat prostatic vas deferens. Graph (left) showing a concentration response curve to hα CGRP on twitch responses, and in the presence of hα CGRP8–37 BTD19,20 and 33,34 at 3×10−6M and 10−5M. The Schild plot (right) for hα CGRP8–37 BTD19,20 and 33,34 against hα CGRP. Results on the graph are expressed as percentage inhibition of twitch responses. Points and error bars represent the mean±s.e.mean of four separate experiments. On the Schild plot, points represent individual data of eight separate experiments.
Effect of structural modifications at the N-terminal region of hα CGRP8–37
Hα CGRP8–37 analogues (10−5 M), replaced by either glycine8 (hα CGRP8–37 Gly8), des-NH2 valine8 (hα CGRP8–37 des-NH2 Val8) or proline8 (hα CGRP8–37 Pro8) antagonized hα CGRP responses with similar affinities (Figure 5a,b,c; Table 1), i.e. structural changes at the N-terminus did not significantly alter antagonism of hα CGRP8–37 (P>0.05). Hα CGRP8–37 Pro8 Glu10,14 (10−5 M) substituted by proline8 and glutamic acid10,14 did not antagonize hα CGRP responses (Figure 5d; Table 1).
Figure 5.
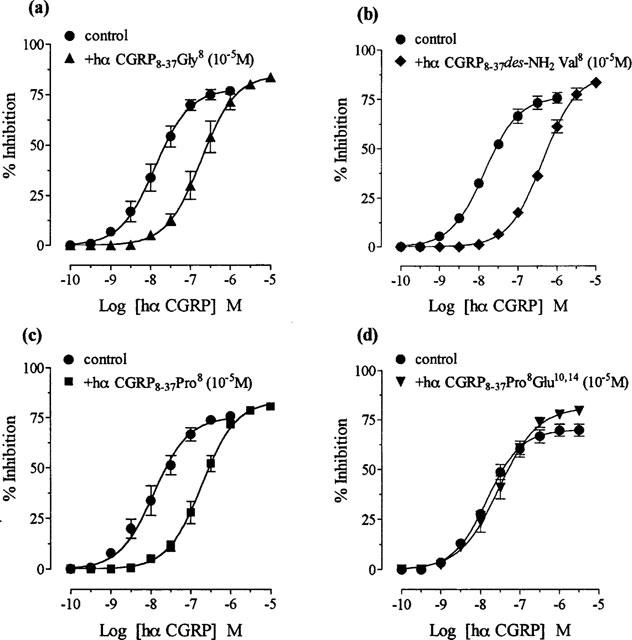
Effect of replacement by either glycine8, des-NH2 valine8, proline8 or proline8 and glutamic acid10,14 in hα CGRP8–37 on hα CGRP responses in the rat prostatic vas deferens. Concentration response curves to hα CGRP on twitch responses, and in the presence of 10−5M of (a) hα CGRP8–37 Gly8, (b) hα CGRP8–37 des-NH2 Val8, (c) hα CGRP8–37 Pro8 and (d) hα CGRP8–37 Pro8 Glu10,14. Results are expressed as percentage inhibition of twitch responses. Points and error bars represent the mean±s.e.mean of four or five separate experiments.
Effect of an alanine replacement at the C-terminus of hα CGRP8–37
Hα CGRP8–37 Ala37 (10−5 M), with alanine amide at position 37, failed to alter hα CGRP responses (pIC50 8.2±0.1 and 8.1±0.1 (n=4 each) for hα CGRP in the absence and presence of hα CGRP8–37 Ala37, respectively), i.e. incorporation of alanine at the C-terminus abolished antagonism.
Effect of peptidase inhibitors
A mixture of peptidase inhibitors (amastatin, bestatin, captopril, phosphoramidon, thiorphan; 10−6 M each) in DMSO or DMSO alone had no effect on basal tone or twitch responses. The antagonist activity of either hα CGRP8–37 BTD19,20 and 33,34 or hα CGRP8–37 Gly8 on hα CGRP responses was not significantly altered in the presence of peptidase inhibitors (P>0.05; Figure 6a and b). The apparent pKB values were 6.2±0.1 and 6.5±0.1 for hα CGRP8–37 BTD19,20 and 33,34 and 6.1±0.1 and 5.9±0.1 for hα CGRP8–37 Gly8, before and after treatment with peptidase inhibitors, respectively. Incubation (for 30 min) with 10−5 M thiorphan alone did not alter the pIC50 value for hα CGRP or the pKB value for hα CGRP8–37 (10−5 M against hα CGRP; P>0.05; data not shown).
Figure 6.
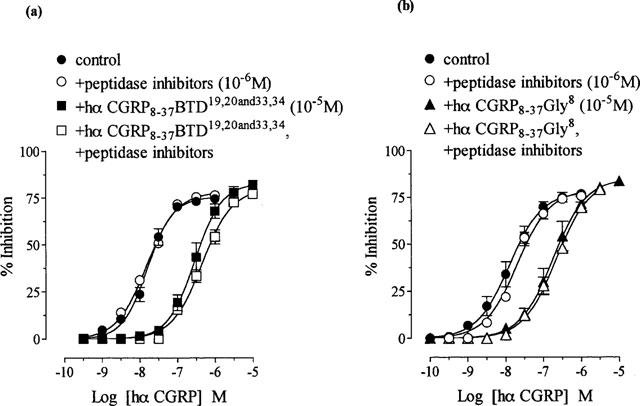
Effect of peptidase inhibitors (amastatin, bestatin, captopril, phosphoramidon, thiorphan; 10−6M) on antagonism by hα CGRP8–37 BTD19,20 and 33,34 and hα CGRP8–37 Gly8 against hα CGRP responses in the rat prostatic vas deferens. Concentration response curves to hα CGRP on twitch responses, and in the presence of 10−5M of (a) hα CGRP8–37 BTD19,20 and 33,34 and (b) hα CGRP8–37 Gly8, before and after treatment with peptidase inhibitors. Results are expressed as percentage inhibition of twitch responses. Points and error bars represent the mean±s.e.mean of four separate experiments.
Discussion
This study extends the known pharmacology of the bioactive conformation of hα CGRP8–37 at the CGRP receptor, by providing evidence for two β-bend regions and their exact location (18–21 and 32–35) by the use of BTD as a conformational probe. Further, the present data confirm that the N-terminus is not important for antagonism by hα CGRP8–37, while the C-terminus is essential for interaction with CGRP receptor binding sites in the rat vas deferens.
Both CD and two-dimensional 1H-NMR spectroscopy suggested that following the α-helix in CGRP, residue 18 (arginine) may be involved in a β-bend formation (Lynch & Kaiser, 1988; Breeze et al., 1991). However, in terms of the helical content and its termination reports from the use of CD (Manning, 1989; Hubbard et al., 1991) and NMR (Boulanger et al., 1995) have reached different conclusions. Therefore, the present study investigated whether region 15–18 or 18–21 in hα CGRP8–37 could be involved in a β-turn. For region 15–18 (leucine15, leucine16, serine17, arginine18), a bend formation is unlikely since incorporation of a bend-biasing structure16 (proline) abolished antagonism, while a residue with structure-conserving character (alanine16) retained weak antagonist activity, indicating that leucine16 side chains are not essential for receptor interaction. The loss of antagonism by proline16 may be due to disruption of the ordered helical folding, which would agree with the proposal that the α-helix is a critical factor for interaction with the receptor in the rat vas deferens (Lynch & Kaiser, 1988; Dennis et al., 1989; Mimeault et al., 1991; 1992). For region 18–21, a bioactive bend structure appears likely, as incorporation of a bend-biasing residue19 (proline) retained antagonism, indicating that serine19 side chains are not essential for receptor interaction. From examination of the sequence, an 18–21 bend terminating the helix is reasonable, as the residues within this region are arginine18, serine19, glycine20, glycine21, and glycine residues are ‘helix breakers' (Chou & Fasman, 1977) and being flexible, favour turns.
The present data confirm that a β-turn at position 18–21 is compatible with high affinity binding, as enforcement by BTD at positions 19,20 produced no loss of antagonist activity of hα CGRP8–37. The nature of the semi-rigid BTD structure is such that it forces a bend at the point of substitution (i+1 and i+2; Figure 1), and therefore mimics a four-residue β-bend structure. The retention of antagonism by this bend-forcing structure19,20 in hα CGRP8–37 indicates that serine19 and glycine20 residues are not essential for receptor interaction and are not involved in the helical folding. Therefore, the helix may be terminated at residue 17 by an 18–21 β-turn.
Initial CD spectroscopic studies predicted another bend in the C-terminal region around residues 29 to 34 of CGRP (Hubbard et al., 1991), which would be favoured by the presence of glycine33. Molecular modelling studies suggested either a four-residue 32–35 β-turn or a three residue 32–34 γ-turn (Hakala & Vihinen, 1994), while a model CGRP peptide (CGRP6–37 Pro7 Pro8 Cys31 Cys36) with a stabilized β-bend (cysteine-induced) between 32 and 35, showed binding affinity and biological activity (adenylate cyclase activation) in a rabbit lung assay (Hakala et al., 1994). A recent structure affinity relationship study on CGRP27–37 analogues pointed to the possibility of either a 32–35 or a 33–36 β-turn (Rist et al., 1998). The current study shows that a 32–35 β-bend (valine32, glycine33, serine34, lysine35) is a tolerated conformation in hα CGRP8–37, as enforcement of this bend by BTD33,34 (i.e. in the i+1 and i+2 position) produced the same antagonist affinity as hα CGRP8–37, indicating that glycine33 and serine34 residues are not essential for receptor recognition. Therefore, a β-turn at position 32–35 is a possible bioactive structure in hα CGRP8–37.
Evidence that both turns 18–21 and 32–35 are biologically relevant is provided by the findings that constraint by BTD at 19,20 and 33,34 preserved hα CGRP8–37 antagonism, in the vas deferens. Therefore, the current study is consistent with the idea that the bioactive conformation of hα CGRP8–37 is characterized by two chain reversals, which by their virtue of folding the peptide chain would ensure a more compact formation of hα CGRP8–37 upon interaction with its receptors.
The incorporation of BTD has been shown to be a useful method to identify two potential bend regions in hα CGRP8–37, and confirms that the motifs are accessible and consistent with β-turns at position 18–21 and 32–35. The successful mimicry of these turns by BTD whilst retaining antagonism of hα CGRP8–37, is the first approach towards a structural model of the peptide's bioactive conformation. However, whether hα CGRP8–37 may adopt alternative conformations in these regions (e.g. 19–21 γ-turn, Boulanger et al., 1995; 32–34 γ-turn, Hakala & Vihinen, 1994; 33–36 β-turn, Rist et al., 1998), cannot be excluded by the current data.
For the N-terminus of hα CGRP8–37, both CD spectroscopy and structure activity studies suggested that valine8 does not participate in the formation of the α-helix or in the binding affinity at CGRP receptors (Mimeault et al., 1991; 1992). The current study indicates that valine8 is not essential for receptor interaction, since deletion of both the N-terminal charge (des-NH2 valine) and the iso-propyl side chain (glycine) is tolerated without loss of antagonism. Further, valine8 is probably not important for maintaining the integrity of the α-helix, since neither a helix breaker8 (glycine) nor a helix promoting residue8 (proline, which can bias the folding of a helix if placed at its N-terminus (Presta & Rose, 1988)) altered the antagonist affinity of hα CGRP8–37. Therefore, in agreement with the literature, this study concludes that the N-terminus does not play a role in antagonism by hα CGRP8–37 at the CGRP receptor in the rat vas deferens.
For the N-terminal α-helical segment in hα CGRP8–37, it has been suggested that its length and amphipathic structure is critical for interaction with CGRP receptors (Mimeault et al., 1991; 1992). However, it has not been possible to show an obvious relationship between the aqueous solution structure of this region and antagonist potency in a variety of alanine-substituted analogues (e.g. Boulanger et al., 1996). In the current study, it was hoped to increase the helical stability of hα CGRP8–37 by introducing helix promoting (proline8) and hydrophilic residues (glutamic acid10,14), but this resulted in loss of affinity. While there is no direct evidence about the effect of these substitutions on the α-helix one explanation could be that the triple substitution abolished its amphipathic character. However, what remains unclear is whether the helix in hα CGRP8–37 is important in presenting the correct orientation or not.
It has been suggested that an intact C-terminus (F-NH2) is essential for biological activity and receptor binding of CGRP (Thiebaud et al., 1991; Poyner et al., 1992), and conformational studies indicated no significant difference between the solution structure of CGRP and des-F-NH2 CGRP (O'Connell et al., 1993). The current investigation on the C-terminus of hα CGRP8–37 indicates that phenylalanine37 (F-NH2) is directly involved in the interaction with receptor binding sites, since deletion of the hydrophobic phenyl group (alanine amide37) abolished antagonism. This would agree with previous findings by Rist et al., (1998) who reported that even the substitution of F-NH2 by Y-NH2, which has only one additive hydroxyl group, destroyed receptor recognition of hα CGRP27–37, in human SK-N-MC cells. Therefore, the present study confirms that the C-terminus interacts directly with CGRP receptor binding sites, and that the phenyl group is essential for receptor recognition.
A number of hα CGRP8–37 analogues have been identified as useful tools to determine the presence of bend structures and the role of the N- and C-terminus in hα CGRP8–37. The affinity determinants produced by the active hα CGRP8–37 analogues closely match those to hα CGRP8–37, supporting the presence of a CGRP2 receptor in the rat vas deferens. Furthermore, the observation that antagonist affinities were not increased in the presence of various peptidase inhibitors is consistent with previous reports (Wisskirchen et al., 1998), and agrees with the conclusion that peptidergic degradation is not a factor that affects the affinity of hα CGRP8–37 in the rat vas deferens.
In conclusion, the present findings provide evidence for two possible bioactive β-bend regions at positions 18–21 and 32–35 in hα CGRP8–37. The successful incorporation of BTD as a mimic of these β-turns is the first approach towards a structural model for hα CGRP8–37 at its receptor(s). Furthermore, these studies confirm that the N-terminus does not play a role in antagonism of hα CGRP8–37, while the C-terminus is essential for receptor interaction. The discovery of equally potent hα CGRP8–37 analogues may be helpful in characterizing CGRP receptors further.
Acknowledgments
We thank David Cooper (MS) and Paul Sanderson (NMR) for their analytical expertise.
Abbreviations
- BTD
beta-turn dipeptide
References
- BOULANGER Y., KHIAT A., CHEN Y., SENECAL L., TU Y., ST-PIERRE S., FOURNIER A. Structure of human calcitonin gene-related peptide and of its antagonist human calcitonin gene-related peptide 8-37 by nmr and molecular modelling. Peptide Res. 1995;8:206–213. [PubMed] [Google Scholar]
- BOULANGER Y., KHIAT A., LAROCQUE A., FOURNIER A., ST-PIERRE S. Structural comparison of alanine-substituted analogues of the calcitonin gene-related peptide 8-37. Int. J. Peptide Protein Res. 1996;47:477–483. doi: 10.1111/j.1399-3011.1996.tb01098.x. [DOI] [PubMed] [Google Scholar]
- BRAIN D., WILLIAMS T.J., TIPPINS J.R., MORRIS H.R., MACYNTYRE I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- BREEZE A.L., HARVEY T.S., BAZZO R., CAMPBELL I.D. Solution structure of human calcitonin gene-related peptide by 1H-NMR and distance geometry with restrained molecular dynamics. Biochemistry. 1991;30:575–582. doi: 10.1021/bi00216a036. [DOI] [PubMed] [Google Scholar]
- CAPRINO L.A., HAN G.Y. The 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972;37:3404–3409. [Google Scholar]
- CHOU P.Y., FASMAN G.D. β-turns in peptides. J. Mol. Biol. 1977;115:135–175. doi: 10.1016/0022-2836(77)90094-8. [DOI] [PubMed] [Google Scholar]
- DENNIS T., FOURNIER A., CADIEUX A., POMERLEAU F., JOLICEUR F.B., ST-PIERRE S., QUIRION R. hCGRP8-37, a calcitonin gene-related peptide antagonist revealing calcitonin gene- related peptide receptor heterogeneity in brain and periphery. J. Pharmacol. Exp. Ther. 1990;254:123–128. [PubMed] [Google Scholar]
- DENNIS T., FOURNIER A., ST-PIERRE S., QUIRION R. Structure-activity profile of calcitonin gene-related peptide in peripheral and brain tissues. Evidence for receptor multiplicity. J. Pharmacol. Exp. Ther. 1989;251:718–725. [PubMed] [Google Scholar]
- HAKALA J.M.L., VALO T., VIVAINEN S., HERMONEN J., HEINO P., HALME M., KOSKINEN A.M. Constrained analogues of the calcitonin gene-related peptide. Biochem. Biophys. Res. Commun. 1994;202:497–503. doi: 10.1006/bbrc.1994.1956. [DOI] [PubMed] [Google Scholar]
- HAKALA J.M.L., VIHINEN M. Modelling the structure of the calcitonin gene-related peptide. Protein Eng. 1994;7:1069–1075. doi: 10.1093/protein/7.9.1069. [DOI] [PubMed] [Google Scholar]
- HUBBARD A.M., MARTIN S.R., CHAPLIN L.C., BOSE C., KELLY S.M., PRICE N.C. Solution structures of calcitonin gene-related peptide analogues of calcitonin gene-related peptide and amylin. Biochem. J. 1991;275:785–788. doi: 10.1042/bj2750785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYNCH B., KAISER E.T. Biological properties of two models of calcitonin gene related peptide with idealized amphiphilic α-helices of different lengths. Biochemistry. 1988;27:7600–7607. doi: 10.1021/bi00420a005. [DOI] [PubMed] [Google Scholar]
- MANNING M. Conformation of the alpha form of human calcitonin gene-related peptide (CGRP) in aqueous solution as determined by circular dicroism spectroscopy. Biochem. Biophys. Res. Commun. 1989;160:388–392. doi: 10.1016/0006-291x(89)91668-9. [DOI] [PubMed] [Google Scholar]
- MARSHALL I., AL-KAZWINI S.J., HOLMAN J.J., CRAIG R.K. Human and rat α-CGRP but not calcitonin cause mesenteric vasodilatation in rats. Eur. J. Pharmacol. 1986a;123:217–222. doi: 10.1016/0014-2999(86)90662-x. [DOI] [PubMed] [Google Scholar]
- MARSHALL I., AL-KAZWINI S.J., ROBERTS P.M., SHEPPERSON N.B., ADAMS A., CRAIG R.K. Cardiovascular effects of human and rat CGRP compared in the rat and other species. Eur. J. Pharmacol. 1986b;123:207–216. doi: 10.1016/0014-2999(86)90661-8. [DOI] [PubMed] [Google Scholar]
- MIMEAULT M., FOURNIER A., DUMONT Y., ST-PIERRE S., QUIRION R. Comparative affinities and antagonistic potencies of various human calcitonin gene-related peptide fragments on calcitonin gene-related peptide receptors in brain and periphery. J. Pharm. Exp. Ther. 1991;258:1084–1090. [PubMed] [Google Scholar]
- MIMEAULT M., QUIRION R., DUMONT Y., ST-PIERRE S., FOURNIER A. Structure-activity study of hCGRP8-37, a calcitonin gene-related peptide receptor antagonist. J. Med. Chem. 1992;35:2163–2168. doi: 10.1021/jm00090a003. [DOI] [PubMed] [Google Scholar]
- NAGAI U., SATO K. Synthesis of a bicyclic dipeptide with the shape of β-turn central part. Tetrahedron Lett. 1985;26:647–650. [Google Scholar]
- O'CONNELL J.P., KELLY S.M., RALEIGH D.P., HUBBARD J.A.M., PRICE N.C., DOBSON C.M., SMITH B.J. On the role of the C-terminus of α-calcitonin-gene-related peptide (αCGRP) Biochem. J. 1993;291:205–210. doi: 10.1042/bj2910205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEARSON D.A., BLANCHETTE M.L., GUINDON C.A. Trialkylsilanes as scavenger for the triflouroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
- POYNER D.R. Calcitonin gene-related peptide: Multiple actions, multiple receptors. Pharmacol. Ther. 1992;56:23–51. doi: 10.1016/0163-7258(92)90036-y. [DOI] [PubMed] [Google Scholar]
- POYNER D.R., ANDREW D.P., BROWN D., BOSE C., HANLEY M.R. Pharmacological characterisation of a receptor for calcitonin gene related peptide on rat, L6 myocytes. Br. J. Pharmacol. 1992;105:441–447. doi: 10.1111/j.1476-5381.1992.tb14272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRESTA L.G., ROSE G.D. Helix signals in proteins. Science. 1988;240:1632–1641. doi: 10.1126/science.2837824. [DOI] [PubMed] [Google Scholar]
- QUIRION R., VAN ROSSUM D., DUMONT Y., ST-PIERRE S., FOURNIER A. Characterization of CGRP1 and CGRP2 receptor subtypes. Ann. N.Y. Acad. Sci. U.S.A. 1992;657:88–105. doi: 10.1111/j.1749-6632.1992.tb22759.x. [DOI] [PubMed] [Google Scholar]
- RIST B., ENTZEROTH M., BECK-SICKINGER A.G. From micromolar to nanomolar affinity: A systematic approach to identify the binding site of CGRP at the human calcitonin gene-related peptide 1 receptor. J. Med. Chem. 1998;41:117–123. doi: 10.1021/jm970533r. [DOI] [PubMed] [Google Scholar]
- ROSENFELD M.G., MERMOD S.G., AMARA L.W., SAWCHENKO P.E., RIVIER J., VALE W.W., EVANS R.M. Production of a novel neuropeptide encoded by the calcitonin gene via tissue-specific RNA processing. Nature. 1983;304:129–135. doi: 10.1038/304129a0. [DOI] [PubMed] [Google Scholar]
- SMITH J.A., PEASE L.G. Reverse turn in peptides and proteins. CRC Crit. Rev. Biochem. 1980;8:315–399. doi: 10.3109/10409238009105470. [DOI] [PubMed] [Google Scholar]
- THIEBAUD D., AKATSU T., YAMASHITA T., SUDA T., NODA T., MARTIN R.E., FLETCHER A.E., MARTIN T.J. Structure-activity relationships in calcitonin gene-related peptide: cyclic AMP response in a preosteoblast cell line (KS-4) J. Bone Min. Res. 1991;6:1137–1142. doi: 10.1002/jbmr.5650061016. [DOI] [PubMed] [Google Scholar]
- WISSKIRCHEN F.M., BURT R.P., MARSHALL I. Pharmacological characterization of CGRP receptors mediating relaxation of the rat pulmonary artery and inhibition of twitch responses of the rat vas deferens. Br. J. Pharmacol. 1998;123:1673–1683. doi: 10.1038/sj.bjp.0701783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WISSKIRCHEN F.M., DOYLE P.M., GOUGH S.L., HARRIS C.J., MARSHALL I. Structure-activity relationship of analogues of the antagonist hα CGRP8–37 in rat prostatic vas deferens. Br. J. Pharmacol. 1994;112:239P. doi: 10.1038/sj.bjp.0702432. [DOI] [PMC free article] [PubMed] [Google Scholar]