

Abstract
Activated microglial cells are believed to play an active role in most brain pathologies, during which they can contribute to host defence and repair but also to the establishment of tissue damage. These actions are largely mediated by microglial secretory products, among which are prostaglandins (PGs) and nitric oxide (NO).
The anti-inflammatory protein, lipocortin 1 (LC1) was reported to have neuroprotective action and to be induced by glucocorticoids in several brain structures, with a preferential expression in microglia. In this paper we tested whether the neuroprotective effect of LC1 could be explained by an inhibitory effect on microglial activation.
We have previously shown that bacterial endotoxin (LPS) strongly stimulates PGE2 and NO production in rat primary microglial cultures, by inducing the expression of the key enzymes cyclo-oxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS), respectively.
Dexamethasone (DEX, 1–100 nM) and LC1-derived N-terminus peptide (peptide Ac2-26, 1–100 μg ml−1) dose-dependently inhibited the production of both PGE2 and NO from LPS-stimulated microglia. The inhibitory effects of DEX on NO and of the peptide on NO and PGE2 synthesis were partially abrogated by a specific antiserum, raised against the N-terminus of human LC1. The peptide Ac2-26 did not affect arachidonic acid release from control and LPS-stimulated microglial cultures.
Western blot experiments showed that the LPS-induced expression of COX-2 and iNOS was effectively down-regulated by DEX (100 nM) and peptide Ac2-26 (100 μg ml−1).
In conclusion, our findings support the hypothesis that LC1 may foster neuroprotection by limiting microglial activation, through autocrine and paracrine mechanisms.
Keywords: Microglia, lipocortin 1, dexamethasone, prostaglandins, cyclo-oxygenase, nitric oxide synthase, nitric oxide, neuroprotection, inflammation, brain
Introduction
Lipocortin 1 (LC1) belongs to a family of calcium- and phospholipid-binding proteins, called either lipocortins or annexins, and it is thought to mediate some of the peripheral anti-inflammatory effects of glucocorticoids (Flower & Rothwell, 1994). Lipocortin 1 produces many steroid-like effects also at the level of the central nervous system: it has antipyretic activity (Davidson et al., 1991) and protects from ischaemic (Relton et al., 1991) as well as NMDA-induced brain damage (Black et al., 1992). LC1 is expressed at low levels throughout the brain of normal humans, but its expression increases in pathological conditions such as multiple sclerosis (MS) (Elderfield et al., 1992) or experimental allergic encephalomyelitis (EAE) (Elderfield et al., 1993). The precise biological role of LC1 and the molecular mechanisms of its neuroprotective effects, however, are still far from being elucidated.
Microglial cells, the brain resident macrophages, are constitutively rich in LC1 (McKanna, 1993) and administration of glucocorticoids to rats up-regulates LC1 expression in several brain structures, with a preferential effect on microglia (Go et al., 1994). Microglial cells play an active role in brain inflammatory, immune and degenerative processes. Due to their reactivity to a wide range of stimuli, they are believed to foster neuroprotective and repair processes, but also, depending on the type or intensity of the noxious stimulus and on the concurrence of other local factors, to contribute to the establishment or exacerbation of tissue damage (Kreutzberg, 1996).
Many of the effects of activated microglial cells are mediated by their numerous secretory products, among which are prostaglandins (PGs), a family of compounds derived from arachidonic acid (AA) through the cyclo-oxygenase (COX) pathway, and nitric oxide (NO), the formation of which from L-arginine is catalyzed by NO synthase (NOS). Both COX and NOS exist as constitutive isoforms (COX-1 and cNOS), and as inducible isoforms (COX-2 and iNOS). Cyclo-oxygenase-2 and iNOS are the major isoforms expressed in activated inflammatory cells, including microglia. Their expression enables these cells to release high levels of PGs and NO for sustained periods of time (Minghetti & Levi, 1995; Minghetti et al., 1997a).
Prostaglandins and NO are potent local mediators and, while low levels of these molecules may participate in protective responses leading to enhanced disease resistance, their excessive production may be involved in autotoxicity (Appleton et al., 1996). The temporal correlation between increased levels of prostanoids and various neuropathological processes has led to the hypothesis that prostanoids contribute to neurodegeneration (Shimizu & Wolfe, 1990). However, activation of the prostanoid cascade is accompanied by the generation of a broad range of other active molecules, such as cytokines, free radicals and AA itself, and a direct link between PGs and neurodegeneration remains controversial. Microglia-produced NO and reactive nitrogen oxide intermediates have been suggested to mediate neuronal degeneration occurring in ischaemic and neurodegenerative disorders (Dawson et al., 1991; Boje & Arora, 1992; Chao et al., 1992; Meda et al., 1995), as well as oligodendrocyte damage in MS (Merrill et al., 1993). Additional evidence for a role of NO in the pathogenesis of degenerative human diseases comes from the detection of iNOS protein in brain macrophage/microglia-like cells after viral infection and during EAE (Van Dam et al., 1995). Elevated iNOS mRNA and/or protein was also reported in demyelinating regions in MS (Bo et al., 1994; Bagasra et al., 1995; Degroot et al., 1997) and in the brain of AIDS patients with severe dementia (Adamson et al., 1996).
In the present study we evaluated the effects of a peptide derived from human LC1 N-terminus (peptide Ac2-26) (Perretti et al., 1993), on PGE2 and NO synthesis in cultured rat microglia activated by bacterial endotoxin (LPS). We found that peptide Ac2-26, which retains most of the functions of the parent protein, and the synthetic glucocorticoid dexamethasone (DEX) inhibit PGE2 and NO production mainly by down-regulating the expression of the inducible enzymes responsible for their synthesis.
Methods
Cell cultures
Microglial secondary cultures were prepared from mixed primary cultures obtained from cerebral cortex of 1-day-old rats as previously described (Levi et al., 1993). Briefly, microglial cells were detached by mild shaking from 10–12 day mixed primary cultures, plated at a density of 1.2×105 cells per cm2, allowed to adhere for 20 min, washed to remove non-adhering cells and cultured for 24 h. The cultures consisted of ⩾99% microglia/macrophages (positive for the macrophage marker ED1). Mixed primary cultures as well as microglial secondary cultures were maintained in basal Eagle's medium supplemented with 10% v v−1 foetal calf serum, 2 mM glutamine, 10 μg ml−1 gentamicin, in an atmosphere of 5% CO2–95% air, at 37°C. All media were virtually LPS free, as determined by Lymulus amebocytes lysate assay. One-day cultures were washed and then incubated for 24 h, with or without 10 ng ml−1 LPS or the indicated drugs. The dose of 10 ng ml−1 LPS and the incubation time of 24 h were chosen because they were previously shown to induce optimal de novo synthesis of COX-2 and iNOS. The production of PGE2 and NO was abrogated by the presence of specific inhibitors of COX and NOS activity, such as indomethacin (1 μM and L-NG-monomethyl-arginine (200 μM), respectively (Minghetti & Levi, 1995; Minghetti et al., 1996; 1997b).
Drug treatments
The synthetic glucocorticoid DEX (1–100 nM) was resuspended in dimethylsulphoxide (DMSO) and diluted in serum-containing medium before addition to microglial cultures. Control samples were prepared with equal dilution of the vehicle. Maximal DMSO concentration was 0.001% v v−1 and did not affect cell viability.
Peptide Ac2-26 derived from the N-terminus of LC1 (aminoacids 2-26, Perretti et al., 1993), was dissolved in sterile PBS and added to the cultures to obtain final concentrations ranging from 1–100 μg ml−1.
The polyclonal sheep antiserum raised against the biologically active N-terminus of LC1 (LCPS1) (Perretti et al., 1996), was shown to neutralize the effects of both DEX and lipocortin peptide in rats (Ferreira et al., 1997) and mice (Getting et al., 1997). To analyse the effect of the antiserum on DEX and peptide Ac2-26, the cultures were pre-incubated with LCPS1 (1 : 100) and DEX (100 nM) for 2 h, or with LCPS1 and peptide Ac2-26 (100 μg ml−1) for 30 min before the addition of LPS. Control cultures were prepared by using a normal pre-immune sheep serum. All the treatments with the above drugs were performed in the presence or in the absence of LPS.
Analysis of PGE2 and NO production
Culture media were collected, centrifuged to remove residual cells and stored at −20°C until tested. Prostaglandin E2 released into culture media was measured using a specific radioimmunoassay (Minghetti & Levi, 1995). The detection limit was 25 pg ml−1. The level of PGE2 present in 10% v v−1 FCS-containing medium was measured (less than 50 pg ml−1) and subtracted from the value obtained for each sample. The production of NO was determined in cell supernatants by measuring the content of nitrite, one of the end-products of NO oxidation, by a procedure based on the diazotidation of nitrite by sulphanilic acid (Griess reaction), as previously described (Minghetti et al., 1996). The detection limit was 0.3 μM.
Western blot analysis
Microglial proteins were prepared and analysed as previously described (Minghetti et al., 1996). Briefly, microglial cells were lysed in a buffer solution containing 1% Nonidet P-40 and the protease inhibitors leupeptin (10 μg ml−1), aprotinin (30 μg ml−1) and phenylmethyl-sulphonyl fluoride (100 μg ml−1). Cell lysates were analysed for protein content and equal amounts of protein from each sample (10 or 25 μg) were subjected to SDS–PAGE and transferred on nitrocellulose membranes. After blocking with 5% w v−1 milk proteins, membranes were incubated with primary antibodies for 1 h at 25°C (anti-COX-2, 1 : 500 and anti-iNOS 1 : 2500), and, after several washes, with secondary antibodies (anti-IgG conjugated to horseradish peroxidase). The Amersham enhanced chemiluminescence (ECL) system was used to detect the antibodies. Purified COX-2 (from sheep placenta) and iNOS (from mouse macrophages) were used as standard controls (0.5 μg lane−1). The optical density of the bands (integrated area, arbitrary units) was measured by a GS-700 Imaging Densitometer (Bio-Rad) and referred to the corresponding control samples (taken as 100%), which were run in the same gel. It has to be noted that such analysis is semiquantitative and is intended only to give a numerical indication of the ratios of band intensities and their variability. Therefore, statistical analysis of the data would be inappropriate and has not been performed.
Analysis of LC1 expression
Lipocortin 1 expression was evaluated by Western blot analysis, using a rabbit polyclonal antibody raised against human LC1 (Ab 842, 1 : 2000) (Duncan et al., 1993). Total (intracellular plus membrane bound) LC1 expression was analysed using total microglial proteins, prepared and processed as described in the previous section, after culturing the cells for 24 h in a medium containing either 10% v v−1 serum or 10% v v−1 dextran/charcoal-stripped serum (steroid-depleted serum). Membrane-bound and intracellular LC1 pools were prepared from microglial cultures maintained in 10% v v−1 serum-containing medium. Briefly, membrane-bound LC1 fraction was prepared by washing microglial monolayers with phosphate buffered saline (PBS) and, subsequently, with PBS containing 10 mM ethylenediaminetetracetic acid (EDTA), which, by chelating Ca2+, release LC1 bound to the cell surfaces into the medium (Philip et al., 1997). After 5 min incubation at room temperature under mild agitation, EDTA/PBS washes were collected and centrifuged to remove all cellular components potentially present. Proteins were then precipitated by adding an equal volume of 50% v v−1 trichloroacetic acid (TCA, 25% v v−1 final concentration). After 30 min at 4°C, proteins were collected by centrifugation (12,000×g, 15 min), re-suspended in 10% v v−1 TCA and, after a second centrifugation, neutralized in 1 M Tris buffer. Proteins were then measured and diluted in SDS–PAGE sample buffer. The intracellular LC1 fraction was prepared from EDTA/PBS-washed monolayers, as described for total microglial proteins in the previous section.
[3H]-arachidonic acid release from activated microglial cells
In order to estimate AA release, microglial cells were incubated for 20–22 h in complete medium supplemented with 0.5 μCi ml−1 [3H]-AA, washed (three times) with the same medium, and incubated at 37°C for 2 h, in the presence of 10 ng ml−1 LPS and the indicated concentration of peptide Ac2-26, alone or in combination. The supernatants were then collected and counted for radioactivity. To evaluate the incorporation pattern of [3H]-AA, cellular phospholipids were extracted and separated by high performance thin layer chromatography as previously described (Minghetti & Levi, 1995). Typically, [3H]-AA was incorporated into membrane phospholipids as follows: phosphatidylcholine, 40–42%; phosphatidylethanolamine, 33–35%, phosphatidylinositol, 10–12%; phosphatidylserine, 5–7%.
Cell viability assays
After incubation with the tested substances, cells were stained with propidium iodide (50 μg ml−1; 20 min, 37°C) which labels the nuclei of dead cells. Measurement of reduction of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) by active mitochondria of living cells was carried out essentially according to Mossmann (1993). MTT (0.25 mg ml−1) was added to the cultures during the final 4 h of incubation. The medium was then removed and the dark blue crystals dissolved in 100 μl DMSO. Optical density was read using a test wavelength of 570 nm and a reference wavelength of 630 nm.
Materials
All cell culture reagents were from Gibco (Grand Island, NY, U.S.A.). Purified iNOS (from mouse macrophages), and COX-2 (sheep placenta) and specific antibodies for COX-2 and iNOS were obtained from Cayman Chemical Company (Ann Arbor, MI, U.S.A.). Specific antibodies for PGE2 were obtained from Biomakor (Rehovot, Israel) and [3H]-PGE2 (specific activity 171 Ci mmol−1) was from New England Nuclear-Du Pont (Boston, MA, U.S.A.). Peptide Ac2-26 (acetyl-AMVSEFLKQAWFIENEEQEYVQTVK) was prepared by The Advanced Biotechnology Centre (The Charing Cross and Westminster Medical School, London, U.K.) by use of solid phase step-wise synthesis. Purity was more than 90% as assessed by high performance liquid chromatography and capillary electrophoresis (data provided by the manufacturer). Western blot ECL detection system was from Amersham International (U.K.). All other chemicals, including LPS (from Escherichia Coli, serotype 026:B6) and DEX, were from Sigma Chemicals (St. Louis, MO, U.S.A.).
Statistical analysis
Assay data are expressed as mean±the standard error of the mean (s.e.mean), with the number of observations in each group indicated in parenthesis i.e. (n=). Comparison data from two treatment groups was made by Student's two-tailed t-test. When comparison data from more than two treatment groups was required, two factor analysis of variance (two-way ANOVA) was used. A two-tailed probability of less than 5% (i.e. P<0.05) was taken as statistically significant.
Results
Effect of dexamethasone and peptide Ac2-26 on microglial PGE2 and NO synthesis
The presence of the synthetic glucocorticoid DEX (1–100 nM) strongly inhibited LPS-induced production of NO and PGE2 (Figure 1), in a dose-dependent manner. The inhibition was more pronounced in the case of PGE2, the production of which was almost totally abrogated with a concentration of DEX as low as 1 nM. The effect of DEX on the very low basal production of both NO and PGE2 was not appreciable. Like DEX, the bioactive peptide Ac2-26 (1–100 μg ml−1) inhibited both LPS-induced PGE2 (Figure 2, upper panel) and NO (Figure 2, lower panel) production in a dose-dependent way, without affecting in a statistically significant way the basal production of the two metabolites. In the concentration range tested (1–100 μg ml−1), peptide Ac2-26 did not affect cell viability, as judged by propidium iodide staining (dead cells were less than 5% in all samples) or cell metabolism, as measured by reduction of MTT (not shown).
Figure 1.
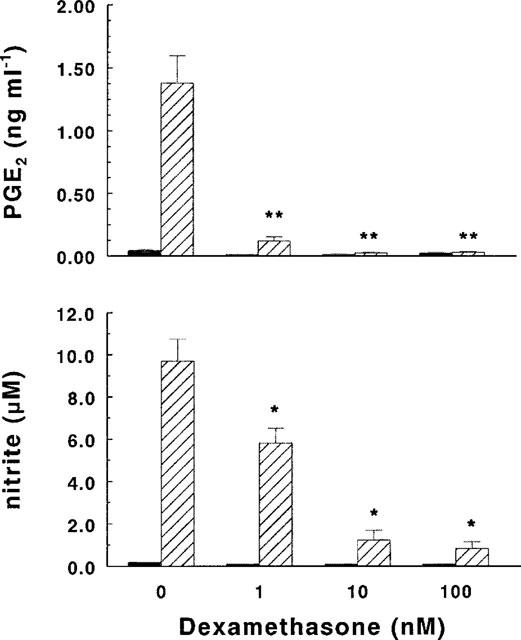
Effect of dexamethasone (DEX) on PGE2 and NO synthesis. Microglial cells (2.5×105 cells ml−1) were subcultured for 24 h in 10% FCS-containing medium, which was replaced with fresh medium before stimulation. Cultures were then incubated for 24 h in the presence of increasing concentrations of DEX with (hatched columns) or without (solid columns) 10 ng ml−1 of LPS. Supernatants were collected and analysed for PGE2 (upper panel) and nitrite (lower panel) accumulation. Data are means±s.e.mean of three independent experiments, run in duplicate. (*P<0.025, **P<0.01 vs LPS, two-way ANOVA).
Figure 2.
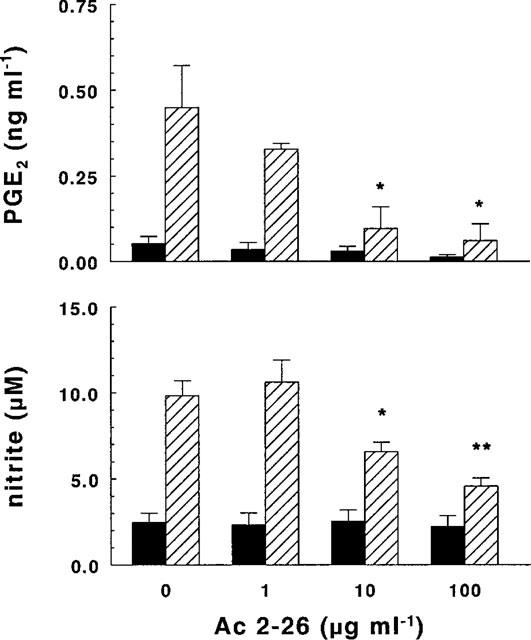
Effect of peptide Ac2-26 on PGE2 and NO synthesis. Microglial cells (2.5×105 cells ml−1) were subcultured for 24 h in 10% FCS-containing medium, which was replaced with fresh medium before stimulation. Cultures were then incubated for 24 h in the presence of increasing concentrations of peptide Ac2-26 with (hatched columns) or without (solid columns) 10 ng ml−1 of LPS. Supernatants were collected and analysed for PGE2 (upper panel) and nitrite (lower panel) accumulation. Data are means±s.e.mean of four (PGE2) and six (nitrite) independent experiments, run in duplicate. (*P<0.05, **P<0.025 vs LPS, two-way ANOVA).
When microglial cultures were exposed to 100 nM DEX and LPS in the presence of a specific antiserum (LCPS1, 1 : 100) raised against the N-terminus region of human LC1, the inhibitory effect of DEX on NO production was attenuated, but the effect on PGE2 formation remained unchanged (Figure 3), even when DEX was used at the lowest concentration (1 nM, not shown). At a higher concentration LCPS1 (1 : 50) caused an increase in NO and PGE2 accumulation on its own as well as in the presence of LPS. It is difficult to determine whether this was due to a neutralizing effect of the antiserum on endogenous LC1 or to a non specific interaction of the antiserum with microglial cells, since in separate experiments a pre-immune serum, at the same concentration, had a similar effect. The same LCPS1 antiserum (1 : 100) effectively counteracted the inhibitory action of peptide Ac2-26 (100 μg ml−1) on both NO and PGE2 synthesis (Table 1).
Figure 3.
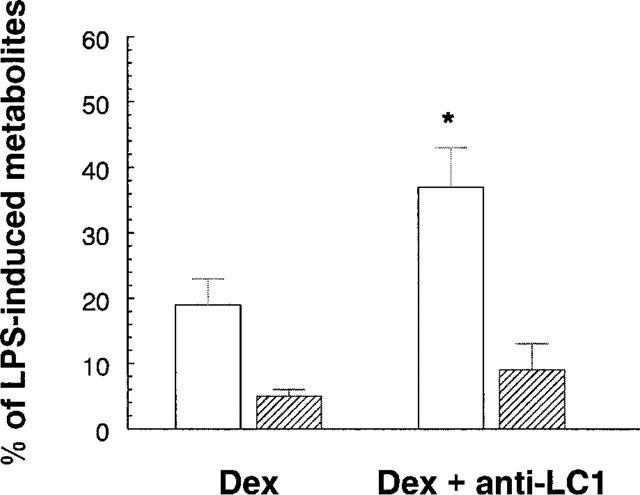
Effect of a specific anti-LC1 antiserum on dexamethasone (DEX) inhibitory activity. Microglial cells (2.5×105 cells ml−1) were subcultured for 24 h in 10% FCS-containing medium, which was replaced with fresh medium before stimulation. Dexamethasone (100 nM) and the anti-LC1 antiserum (1 : 100) were added to the microglial cultures for 2 h before the addition of 10 ng ml−1 of LPS. After 24 h supernatants were collected and analysed for PGE2 (hatched columns) and nitrite (open columns) accumulation. Data, expressed as percentage of LPS-induced PGE2 (0.68±0.15 ng ml−1) or nitrite (9.48±1.81 μM) accumulation, are means±s.e.mean of six independent experiments, run in duplicate. (*P<0.025 vs LPS+DEX, Student's paired t-test).
Table 1.
Effect of specific anti-LC1 antiserum on peptide Ac2-26 activity

Effect of dexamethasone and peptide Ac2-26 on microglial COX-2 and iNOS expression
To test whether peptide Ac2-26 or DEX influenced the expression of the key enzymes in PGs and NO synthesis, we performed Western blot analysis of microglial proteins after incubating the cells for 24 h in the presence of 100 μg ml−1 peptide or 100 nM DEX, with or without 10 ng ml−1 LPS. While DEX totally abolished COX-2 and iNOS expression, peptide Ac2-26 completely abrogated COX-2 and only partially down-regulated iNOS expression. The basal expression of the two enzymes was barely detectable and was not influenced by either DEX or peptide Ac2-26 (Figure 4).
Figure 4.
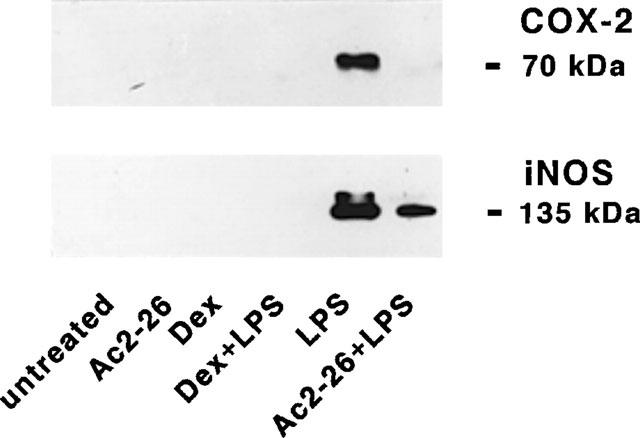
Effects of peptide Ac2-26 and dexamethasone (DEX) on LPS-induced COX-2 and iNOS expression. Microglial cells were subcultured for 24 h in 10% FCS-containing medium, which was replaced with fresh medium before stimulation in the absence or in the presence of 10 ng ml−1 LPS with or without 100 μg ml−1 peptide Ac2-26 and 100 nM DEX. After 24 h, cell lysates were prepared and equal amounts of proteins were analysed by Western blot using anti-COX-2 or anti-iNOS antibodies. Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies and ECL. One experiment representative of four is shown.
Effect of peptide Ac2-26 on arachidonic acid release
An inhibitory effect of peptide Ac2-26 on AA release from membrane phospholipids could, in principle, contribute to the observed inhibition of LPS-induced PGE2 synthesis. In order to test this possibility, microglial cells were pre-incubated with [3H]-AA, washed to remove unincorporated [3H]-AA, and then treated with 10 or 100 μg ml−1 of peptide Ac2-26, alone or in combination with LPS. After 2 h of incubation, the radioactivity released into cell supernatants, corresponding mainly to [3H]-AA (Minghetti & Levi, 1995), was measured. Consistent with our previous data (Minghetti & Levi, 1995), LPS induced an increased release of [3H]-AA (132±10% of the control in three independent experiments, run in triplicate). The peptide did not significantly affect either basal or LPS-stimulated [3H]-AA release (Table 2).
Table 2.
Effect of peptide Ac2-26 on [3H]-arachidonic acid release

Microglial expression of LC1
Since microglial cells have been shown to express LC1 both in vitro (Gebicke-Haerter et al., 1991) and in vivo (Go et al., 1994), we assessed the level of expression of the protein in our culture conditions by semi-quantitative Western blot analysis. Microglial cells grown in a standard medium containing 10% v v−1 serum strongly expressed a LC1 immunoreactive band with a molecular weight of 37 kDa. This constitutive expression remained unchanged when cells were cultured in the presence steroid-depleted serum (Figure 5 upper panel).
Figure 5.
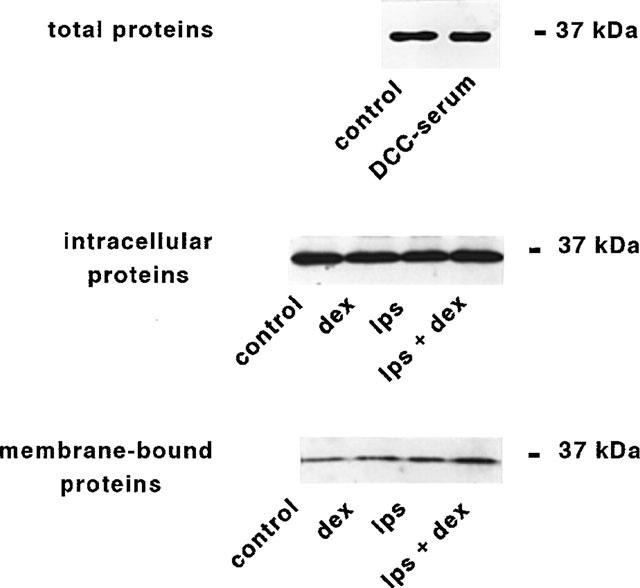
Expression of lipocortin 1 in microglial cultures. Expression and externalization of LC1 immunoreactivity from microglial cells. (Upper panel) Microglial cells were grown for 24 h in culture medium containing either 10% normal serum (control), or 10% steroid-depleted serum (DCC-serum). Cell lysates were prepared and equal amounts of proteins were analysed by Western blot using a rabbit polyclonal antibody raised against human LC1 (Ab 842, 1 : 2000). Protein bands were visualized using horseradish peroxidase-conjugated secondary antibodies and ECL. One experiment representative of three is shown. Microglial cells were cultured in 10% serum containing medium in the absence (control) or in the presence of DEX and LPS, alone or in combination. (Central panel) Protein from EDTA-washed microglial cells were prepared and analysed by Western blot as described above. One experiment representative of six is shown. (Lower panel) Protein from EDTA-washes were precipitated by TCA (see Methods) and analysed by Western blot as above. One experiment representative of four (DEX or LPS) or three (LPS+DEX) is shown.
The effect of DEX on LC1 expression in unstimulated and LPS-activated microglial cultures was evaluated using both intracellular (Figure 5 central panel) and membrane-bound (Figure 5 lower panel) protein fractions. In six independent experiments, addition of DEX or LPS alone did not alter the intensity of the 37 kDa band corresponding to LC1 in the intracellular fractions. In contrast, when the two agents were added together a moderate increase in the intracellular expression of LC1 could be observed ranging from 1.1–2.4 folds of control cultures (n=6). On the other hand, the amount of LC1 associated to the membranes and harvested in the EDTA washes was increased after treatment with DEX (from 1.2–8 folds, n=4) or LPS (from 1.4–5.7 folds, n=4) alone. The highest mobilization of LC1 from internal pool to the membranes was observed when microglial cultures were treated with 100 nM DEX plus 10 ng ml−1 LPS, up to a maximum of 10 fold increase in LC1 recovered from EDTA washed membranes (n=3 independent experiments). The presence of serum in the culture media did not influence the expression and distribuiton of LC1, as indicated by experiments performed in serum-free media (n=3, not shown).
Discussion
The identification of LC1 as a potential mediator of some anti-inflammatory effects of glucocorticoids (Flower & Rothwell, 1994) has stimulated the search for short peptides retaining the activities of the parent protein. The N-terminus region of lipocortins is poorly conserved among family members, and has been proposed to account for the selectivity of action of the different lipocortins (Barton et al., 1991). In this light, a peptide corresponding to the N-terminal portion of LC1 (amino acids 2-26) has been shown to retain most of the anti-inflammatory actions of the whole protein (for a review, see Perretti, 1994).
In the present study the peptide Ac2-26 has been used to test the hypothesis that the described neuroprotective effect of LC1 (Flower & Rothwell, 1994) could be related to a down-regulation of the production of inflammatory substances by activated microglial cells. We have previously shown that microglial cells stimulated by endotoxin release substantial amounts of prostanoids and NO, following the up-regulation of the inducible enzymes COX-2 and iNOS, respectively (Minghetti & Levi, 1995; Minghetti et al., 1997a). The addition of peptide Ac2-26 inhibited in a dose-dependent manner the release of both PGE2 and NO, whilst the specific antibody LCPS1 abrogated this effect. Western blotting studies indicated that the inhibitory effects of peptide Ac2-26 were largely due to the down-regulation of COX-2 and iNOS. Interestingly, COX-2 induction was more sensitive to inhibition by LC1 fragment than induction of iNOS. We suggest that this different sensitivity may be due to different mechanisms involved in the regulation of COX-2 and iNOS gene expression or, alternatively, to the level of expression of the two genes. Indeed, iNOS showed a stronger expression in response to LPS when compared to COX-2, which may be more difficult to inhibit. Further experiments are needed to clarify this point.
At variance with our results, a long LC1 fragment spanning 1-188 aminoacids depressed LPS-induced iNOS expression but not COX-2 expression in the macrophage cell line J774.2 (Wu et al., 1995). At present, it is difficult to say whether this apparent discrepancy is due to the different cell type, to the different LC1 fragment used or to other unknown factors.
The peptide mimicked the action of the potent anti-inflammatory steroid DEX, which also inhibited the release of PGE2 and NO and the expression of the two inducible enzymes. In the case of DEX, however, the specific antibody LCPS1 only partially prevented the inhibition of NO synthesis and did not affect that of PGE2 suggesting that the effect of this drug can be only partly ascribed to an enhanced production of LC1. Our observations were in agreement with Newman et al. (1994), who, in a different experimental model (the epithelioma A459), showed that the inhibitory effect of DEX on COX-2 was not mediated by LC1, but rather by a direct effect of DEX at the transcriptional level. However, in the same study, the authors reported that a neutralizing anti-LC1 antibody did revert the inhibitory effect of DEX on PGE2 synthesis, suggesting that LC1 might mediate the DEX suppression of PGE2 synthesis by inhibiting AA release. Indeed, peptides from the LC1 N-terminal domains inhibit AA release in A459 cells stimulated by epidermal growth factor (Croxtall et al., 1998 and references therein). In our experimental conditions, peptide Ac2-26 did not affect the release of AA from unstimulated and LPS-stimulated cultures, although the same peptide effectively inhibited AA release from human neutrophils, when used in a comparable concentration range (Perretti et al., 1995). Again, this discrepancy could stem from the different cell type used or from the type of cellular phospholipase A2 involved in AA release.
Our observations suggest that LC1, like glucocorticoids, may exert a protective action through its ability to concomitantly block both COX-2 and iNOS expression in activated microglial cells. Indeed, several in vitro findings suggest that NO participates in the establishment of neurone and oligodendrocyte damage in ischaemic and neurodegenerative disorders (Dawson et al., 1991; Boje & Arora, 1992; Chao et al., 1992; Meda et al., 1995; Merrill et al., 1993). Nitric oxide can induce call damage by several mechanisms, including DNA strand breaks, inactivation of iron-containing enzymes, depletion of ATP and lipid peroxidation (Stamler, 1994). Merrill and co-workers have shown that NO, generated by the synthetic donor S-nitroso N-acetyl-DL-penicillamine, is able to induce cell damage and necrotic cell death in oligodendrocyte cultures, whereas microglia and, to a lesser extent, astrocytes are particularly resistant to NO damage (Mitrovic et al., 1995). The detection of iNOS mRNA and protein in brain macrophage/microglia-like cells after viral infection and in demyelinating regions of MS patients provides further evidence for a role for NO in the pathogenesis of neurological human diseases (Bo et al., 1994; Bagasra et al., 1995; Van Dam et al., 1995; Degroot et al., 1997). The part played by COX metabolites in neurodegeneration is more controversial. Indeed, exogenous PGE2 and PGI2 have been shown to protect neuronal cultures from hypoxia, glutamate-induced excitotoxicity and microglial products (Cazeivieille et al., 1993; 1994; Akaike et al., 1994; Thery et al., 1994). The administration of COX inhibitors has been shown to either worsen or ameliorate ischaemic brain damage and it has been postulated that the neuroprotective efficacy of prostanoid inhibition may depend on the severity of the ischaemic episode (Patel et al., 1993). An indirect toxic effect association with COX activity could also be due to the generation of oxygen free radicals during the synthesis of prostaglandins as suggested by the remarkable efficacy of antioxidant agents in reducing ischaemic brain damage (Chan, 1996).
It has been reported that the LC1 fragment 1–188 has protective effects in ischaemic brain damage (Relton et al., 1991) as well as in excitotoxic neuronal damage (Black et al., 1992). The mechanism of this protection has been attributed to inhibition of phospholipase A2 activity (Rothwell & Relton, 1993) but other actions of LC1 might also contribute. Our finding that in microglial cultures peptide Ac2-26 failed to inhibit AA release induced by LPS, but effectively reduced COX-2 and iNOS expression, is consistent with the observation that the inhibition by peptide Ac2-26 of IL-1-induced hyperalgesic response in rats is mediated by down-regulation of COX-2 expression, rather than by decreased activity of PLA2 (Ferreira et al., 1997).
Dexamethasone was reported to inhibit COX-2 and iNOS expression in vivo (Masferrer et al., 1992; Mitchell et al., 1994; Salvemini et al., 1995) and in several types of cultured cells (Fu et al., 1990; O'Banion et al., 1992; Di Rosa et al., 1990; Bauer et al., 1997). It is not yet known how DEX acts to inhibit COX-2 gene expression, and the presence of glucocorticoid response elements in the promoter regions of COX-2 gene has not yet been unambiguously identified (Smith et al., 1996; Goppelt-Struebe, 1997). Recent experiments have demonstrated that glucocorticoids can interfere with essential transcription factors (Beauparlant & Hiscott, 1996). In particular, they have been shown to inhibit iNOS expression by down-regulating the cytokine-induced activity of nuclear factor-κB (NF-κB) (Kleinert et al., 1996). It will be important to assess whether LC1 and related peptides interfere with the activity of NF-κB, that appears to be involved in microglial COX-2 and iNOS gene expression (Bauer et al., 1997; Colasanti et al., 1995).
Finally, we found that microglial cells express LC1 in absence of stimulation. Such basal expression of LC1 could not be ascribed to the activity of steroids present in serum, and may at least in part represent a true constitutive LC1 expression by resting microglia. This would be consistent with other observations made in in vivo experimental models (McKanna, 1993; Go et al., 1994). On the other hand, DEX treatment increased the amount of LC1 associated to the outer surface of cells, both in untreated and LPS-stimulated microglial cultures, suggesting that DEX promotes the translocation of LC1 from intracellular to pericellular sites both in resting and activated microglia, as observed in other cells (Solito et al., 1994). Interestingly, we also found that LPS-treatment increases the LC1 membrane-bound fraction. This observation is consistent with the reported increased expression of LC1 during brain pathologies, in which microglia activation takes place (Elderfield et al., 1992; 1993). It is tempting to suggest that this could be part of a negative feed-back mechanism to limit microglial activation.
In conclusion, our data support the hypothesis that LC1 contributes to the down-regulation of microglia by limiting the expression of the two inducible enzymes COX-2 and iNOS. The presence of LC1 in microglia may represent an endogenous mechanism of neuroprotection that limits microglial activation in pathological conditions and may also participate in keeping microglia in a resting state in healthy brain. As a consequence, the pharmacological manipulation of LC1 expression may potentially be exploited to control the extent of microglial activation in inflammatory and neurodegenerative diseases.
Acknowledgments
This work was supported by the following grants: Project on AIDS of the Italian Ministry of Health, grant no. 10/A/H and Project on Multiple Sclerosis of the Istituto Superiore di Sanità. LP is supported by grants from the Italian National Research Council (C.N.R.) N. 95.02382.CT04 and 96.03339.CT04. MP is a Post-doctoral Fellow of the Arthritis & Rheumatism Council.
Abbreviations
- COX
cyclo-oxygenase
- DEX
dexamethasone
- DMSO
dimethylsulphoxide
- EAE
experimental allergic encephalomyelitis
- iNOS
inducible nitric oxide synthase
- LC1
lipocortin 1
- LPS
bacterial endotoxin
- MS
multiple sclerosis
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NO
nitric oxide
- NOS
nitric oxide synthase
- PGs
prostaglandins
References
- ADAMSON D.C., WILDEMANN B., SASAKI M., GLASS J.D., MCARTHUR J.C., CHRISTOV V.I., DAWSON T.M., DAWSON V.L. Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-a gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- AKAIKE A., KANEKO S., TAMURA Y., NAKATA N., SHIOMI H., USHIKUBI F., NARUMIYA S. Prostaglandin E2 protects cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;663:237–243. doi: 10.1016/0006-8993(94)91268-8. [DOI] [PubMed] [Google Scholar]
- APPLETON I., TOMLINSON A., WILLOUGHBY D.A. Induction of cyclo-oxygenase and nitric oxide synthase in inflammation. Adv. Pharmacol. 1996;35:27–78. doi: 10.1016/s1054-3589(08)60274-4. [DOI] [PubMed] [Google Scholar]
- BAGASRA O., MICHAELS F.H., ZHENG Y.M., BOBROSKI L.E., SPITSIN S.V., FU Z.F., TAWADROS R., KOPROWSKI H. Activation of the inducible form of nitric oxide synthase in the brains of patients with multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 1995;92:12041–12045. doi: 10.1073/pnas.92.26.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARTON G.J., NEWMAN R.H., FREEMONT P.S., CRUMPTON M.J. Amino acid sequence of the annexin super-gene family of proteins. Eur. J. Biochem. 1991;198:749–760. doi: 10.1111/j.1432-1033.1991.tb16076.x. [DOI] [PubMed] [Google Scholar]
- BAUER M.K.A., LIEB K., SCHULZE-OSTHOFF K., BERGER M., GEBICKE-HAERTER P.J., BAUER J., FIEBICH B.L. Expression and regulation of cyclooxygenase-2 in rat microglia. Eur. J. Biochem. 1997;243:726–731. doi: 10.1111/j.1432-1033.1997.00726.x. [DOI] [PubMed] [Google Scholar]
- BEAUPARLANT P., HISCOTT J. Biological and biochemical inhibitors of the NF-κB/Rel proteins and cytokine synthesis. Cyto. Growth Factor Rev. 1996;7:175–190. doi: 10.1016/1359-6101(96)00020-2. [DOI] [PubMed] [Google Scholar]
- BLACK M.D., CAREY F., CROSSMAN A.R., RELTON J.K., ROTHWELL N.J. Lipocortin-1 inhibits NMDA receptor-mediated neuronal damage in the striatum of the rat. Brain Res. 1992;585:135–140. doi: 10.1016/0006-8993(92)91198-n. [DOI] [PubMed] [Google Scholar]
- BÖ L., DAWSON T.M., WESSELINGH S., MÖRK S., CHOI S., KONG P.A., HANLEY D., TRAPP B.D. Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann. Neurol. 1994;36:778–786. doi: 10.1002/ana.410360515. [DOI] [PubMed] [Google Scholar]
- BOJE K.M., ARORA P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- CAZEVIEILLE C., MULLER A., BONNE C. Prostacyclin (PGI2) protects rat cortical neurons in culture against hypoxia/reoxygenation and glutamate-induced injury. Neurosci. Lett. 1993;160:106–108. doi: 10.1016/0304-3940(93)90924-a. [DOI] [PubMed] [Google Scholar]
- CAZEVIEILLE C., MULLER A., MEYNIER F., DUTRAIT N., BONNE C. Protection by prostaglandins from glutamate toxicity in cortical neurons. Neurochem. Int. 1994;24:395–398. doi: 10.1016/0197-0186(94)90118-x. [DOI] [PubMed] [Google Scholar]
- CHAN P.H. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- CHAO C.C., HU S., MOLITOR T.W., SHASKAN E.G., PETERSON P.K. Activated microglia mediate neuronal cell death injury via a nitric oxide mechanism. J. Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- COLASANTI M., PERSICHINI T., MENEGAZZI M., MARIOTTO S., GIORDANO E., CALDARERA C.M., SOGOS V., LAURO G.M., SUZUKI H. Induction of nitric oxide synthase mRNA expression. J. Biol. Chem. 1995;270:26731–26733. doi: 10.1074/jbc.270.45.26731. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., FLOWER R.J. Inhibitory effect of peptides derived from the N-terminus of lipocortin 1 on arachidonic acid release and proliferazione in the A549 cell line: identification of E-Q-E-Y-T as a crucial component. Br. J. Pharmacol. 1998;123:975–983. doi: 10.1038/sj.bjp.0701679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIDSON J., FLOWER R.J., MILTON A.S., PEERS S.H., ROTONDO D. Antipyretic actions of human recombinant lipocortin-1. Br. J. Pharmacol. 1991;102:7–9. doi: 10.1111/j.1476-5381.1991.tb12122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAWSON V.L., DAWSON T.M., LONDON E.D., BREDT D.S., SNYDER S.H. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEGROOT C.J.A., RUULS S.R., THEUWES J.W.M., DIJKSTRA C.D., VANDERVALK P. Immunocytochemical characterization of the expression of inducible and constitutive isoforms of nitric oxide synthase in demyelinating multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 1997;56:10–20. doi: 10.1097/00005072-199701000-00002. [DOI] [PubMed] [Google Scholar]
- DI ROSA M., RADOMSKI M., CARNUCCIO R., MONCADA S. Glucocorticoids inhibit the induction of nitric oxide synthase in macrophages. Biochem. Biophys. Res. Commun. 1990;172:1246–1252. doi: 10.1016/0006-291x(90)91583-e. [DOI] [PubMed] [Google Scholar]
- DUNCAN G.S., PEERS S.H., CAREY F., FORDER R., FLOWER R.J. The local anti-inflammatory action of dexamethasone in the rat carrageenin oedema model is reversed by an antiserum to lipocortin 1. Br. J. Pharmacol. 1993;108:62–65. doi: 10.1111/j.1476-5381.1993.tb13440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELDERFIELD A.J., BOLTON C., FLOWER R.J. Lipocortin 1 (annexin 1) immunoreactivity in the cervical spinal cord of Lewis rats with acute experimental allergic encephalomyelitis. J. Neurolog. Sci. 1993;119:146–153. doi: 10.1016/0022-510x(93)90127-k. [DOI] [PubMed] [Google Scholar]
- ELDERFIELD A.J., NEWCOMBE J., BOLTON C., FLOWER R.J. Lipocortins (annexins) 1, 2, 4 and 5 are increased in the central nervous system in multiple sclerosis. J. Neuroimmunol. 1992;39:91–100. doi: 10.1016/0165-5728(92)90178-n. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., CUNHA F.Q., LORENZETTI B.B., MICHELIN M.A., PERRETTI M., FLOWER R.J., POOLE S. Role of lipocortin-1 in the anti-hyperalgesic actions of dexamethasone. Br. J. Pharmacol. 1997;121:883–888. doi: 10.1038/sj.bjp.0701211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLOWER R.J., ROTHWELL N.J. Lipocortin-1: cellular mechanisms and clinical relevance. Trends Pharmacol. Sci. 1994;15:71–76. doi: 10.1016/0165-6147(94)90281-x. [DOI] [PubMed] [Google Scholar]
- FU J.Y., MASFERRER J.L., SEIBERT K., RAZ A., NEEDLEMAN P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J. Biol. Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- GEBICKE-HAERTER P.J., SHOBERT A., DIETER P., HONEGGER P., HERTTING P. Regulation and glucocorticoid-independent induction of lipocortin I in cultured astrocytes. J. Neurochem. 1991;57:175–183. doi: 10.1111/j.1471-4159.1991.tb02113.x. [DOI] [PubMed] [Google Scholar]
- GETTING S.J., FLOWER R.J., PERRETTI M. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br. J. Pharmacol. 1997;120:1075–1082. doi: 10.1038/sj.bjp.0701029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GO K.G., TER HAAR J.G., DE LEY L., ZUIDERVEEN F., PARENTE L., SOLITO E., MOLENAAR W.M. The effect of steroid treatment on lipocortin immunoreactivity of rat brain. Med. Inflamm. 1994;3:177–180. doi: 10.1155/S0962935194000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPPELT-STRUEBE M. Molecular mechanisms involved in the regulation of prostaglandin biosynthesis by glucocorticoids. Biochem. Pharmacol. 1997;53:1389–1395. doi: 10.1016/s0006-2952(97)00018-x. [DOI] [PubMed] [Google Scholar]
- KLEINERT H., EUCHENHOFER C., IHRIG-BIEDERT I., FORSTERMANN U. Glucocorticoids inhibit the induction of nitric oxide synthase II by down-regulating cytokine-induced activity of transcription factor nuclear factor-kB. Mol. Pharmacol. 1996;49:15–21. [PubMed] [Google Scholar]
- KREUTZBERG G.W. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- LEVI G., PATRIZIO M., BERNARDO A., PETRUCCI T.C., AGRESTI C. Human immunodeficiency virus coat protein gp 120 inhibits the β-adrenergic regulation of astroglial and microglial functions. Proc. Natl. Acad. Sci. U.S.A. 1993;90:1541–1545. doi: 10.1073/pnas.90.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MASFERRER J.L., SEIBERT K., ZWEIFEL B.S., NEEDLEMAN P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3917–3921. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKANNA J.A. Lipocortin 1 immunoreactivity identifies microglia in adult rat brain. J. Neurosci. Res. 1993;36:491–500. doi: 10.1002/jnr.490360415. [DOI] [PubMed] [Google Scholar]
- MEDA L., CASSATELLA M.A., SZENDREI G.I., OTVOS L., Jr, BARON P., VILLALBA M., FERRARI D., ROSSI F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- MERRILL J.E., IGNARRO L.J., SHERMAN M.P., MELINEK J., LANE T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- MINGHETTI L., LEVI G. Injection of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J. Neurochem. 1995;65:2690–2698. doi: 10.1046/j.1471-4159.1995.65062690.x. [DOI] [PubMed] [Google Scholar]
- MINGHETTI L., NICOLINI A., POLAZZI E., CRÉMINON C., MACLOUF J., LEVI G. Inducible oxide synthase expression in activated rat microglial cultures is downregulated by exogenous prostaglandin E2 and by cyclooxygenase inhibitors. Glia. 1997a;19:152–160. [PubMed] [Google Scholar]
- MINGHETTI L., POLAZZI E., NICOLINI A., CRÉMINON C., LEVI G. Interferon-γ and nitric oxide down-regulate lipopolysaccharide-induced prostanoid production in cultured rat microglial cells by inhibiting cyclooxygenase-2 expression. J. Neurochem. 1996;66:1963–1970. doi: 10.1046/j.1471-4159.1996.66051963.x. [DOI] [PubMed] [Google Scholar]
- MINGHETTI L., POLAZZI E., NICOLINI A., CRÉMINON C., LEVI G. Upregulation of cyclooxygenase-2 expression in cultured microglia by prostaglandin E2, cyclic AMP and non steroidal anti-inflammatory drugs. Eur. J. Neurosci. 1997b;9:934–940. doi: 10.1111/j.1460-9568.1997.tb01444.x. [DOI] [PubMed] [Google Scholar]
- MITCHELL J.A., BELVISI M.G., AKARASEREENONT P., ROBBINS R.A., KWON O.-J., CROXTALL J., BARNES P.J., VANE J.R. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: Regulation by dexamethasone. Br. J. Pharmacol. 1994;113:1008–1014. doi: 10.1111/j.1476-5381.1994.tb17093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITROVIC B., IGNARRO L.J., VINTERS H.V., AKERS M.-A., SCHMID I., UITTENBOGAART C., MERRILL J.E. Nitric oxide induces necrotic but not apoptotic cell death in oligodendrocytes. Neuroscience. 1995;65:531–539. doi: 10.1016/0306-4522(94)00491-m. [DOI] [PubMed] [Google Scholar]
- MOSMANN T.R. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assay. J. Immunol. Methods. 1993;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- NEWMAN S.P., FLOWER R.J., CROXTALL J.D. Dexamethasone suppression of IL-1b-induced cyclooxygenase 2 expression is not mediated by lipocortin-1 in A549 cells. Biochem. Biophys. Res. Commun. 1994;202:931–939. doi: 10.1006/bbrc.1994.2019. [DOI] [PubMed] [Google Scholar]
- O'BANION M.K., WINN V.D., YOUNG D.A. cDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4888–4892. doi: 10.1073/pnas.89.11.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATEL P.M., DRUMMOND J.C., SANO T., COLE D.J., KALKMAN C.J., YAKSH T.L. Effect of ibuprofen on regional eicosanoid production and neuronal injury after forebrain ischemia in rats. Brain Res. 1993;614:315–324. doi: 10.1016/0006-8993(93)91050-3. [DOI] [PubMed] [Google Scholar]
- PERRETTI M. Lipocortin-derived peptides. Biochem. Pharmacol. 1994;47:931–938. doi: 10.1016/0006-2952(94)90402-2. [DOI] [PubMed] [Google Scholar]
- PERRETTI M., AHLUWALIA A., HARRIS J.G., GOULDING N.J., FLOWER R.J. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse. A qualitative comparison with an anti-CD11b monoclonal antibody. J. Immunol. 1993;151:4306–4314. [PubMed] [Google Scholar]
- PERRETTI M., AHLUWALIA A., HARRIS J.G., HARRIS H.J., WHELLER S.K., FLOWER R.J. Acute inflammatory response in the mouse: exacerbation by immunoneutralization of lipocortin 1. Br. J. Pharmacol. 1996;117:1145–1154. doi: 10.1111/j.1476-5381.1996.tb16709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERRETTI M., WHELLER S.K., CHOUDHURY Q., CROXTALL J.D., FLOWER R.J. Selective inhibition of neutrophil functions by a peptide derived from lipocortin 1 terminus. Biochem. Pharmacol. 1995;50:1037–1042. doi: 10.1016/0006-2952(95)00238-u. [DOI] [PubMed] [Google Scholar]
- PHILIP G.G., FLOWER R.J., BUCKINGHAM J.C. Glucocorticoids modulate the cellular disposition of lipocortin 1 in the rat brain in vivo and in vitro. Neuroreport. 1997;8:1871–1876. doi: 10.1097/00001756-199705260-00016. [DOI] [PubMed] [Google Scholar]
- RELTON J.K., STRIJBOS P.J.L.M., O'SHAUGHNESSY C.T., CAREY F., FORDER R.A., TILDERS F.J.H., ROTHWELL N.J. Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. J. Exp. Med. 1991;174:305–310. doi: 10.1084/jem.174.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROTHWELL N.J., RELTON J.K. Involvement of interleukin-1 and lipocortin-1 in ischemic brain damage. Cerebrovasc. Brain Metab. Rev. 1993;5:178–198. [PubMed] [Google Scholar]
- SALVEMINI D., MANNING P.T., ZWEIFEL B.S., SIEBERT K., CONNOR J., CURRIE M.G., NEEDLEMAN P., MASFERRER J.L. Dual inhibition of nitric oxide and prostaglandin production contributed to the antiinflammatory properties of nitric oxide synthase inhibitors. J. Clin. Invest. 1995;96:301–308. doi: 10.1172/JCI118035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU T., WOLFE L.S. Arachidonic acid cascade and signal transduction. J. Neurochem. 1990;55:1–15. doi: 10.1111/j.1471-4159.1990.tb08813.x. [DOI] [PubMed] [Google Scholar]
- SMITH W.L.R., GARAVITO M., DEWITT D.L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- SOLITO E., NUTI S., PARENTE L. Dexamethasone induces the translocation of lipocortin (annexin) 1 to the cell membrane of U-937 cells. Br. J. Pharmacol. 1994;112:347–348. doi: 10.1111/j.1476-5381.1994.tb13075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAMLER J.S. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- THERY C., DOBBERTIN A., HALLAT H. Downregulation of in vitro neurotoxicity of brain macrophages by prostaglandin E2 and β-adrenergic agonist. GLIA. 1994;11:383–386. doi: 10.1002/glia.440110411. [DOI] [PubMed] [Google Scholar]
- VAN DAM A.-M., BAUER J., MAN-A-HING W.K.H., MARQUETTE C., TILDERS F.J.H., BERKENBOSCH F. Appearance of inducible nitric oxide synthase in the rat central nervous system after rabies virus infection and during experimental allergic encephalomyelitis but not after peripheral administration of endotoxin. J. Neurosci. Res. 1995;40:251–260. doi: 10.1002/jnr.490400214. [DOI] [PubMed] [Google Scholar]
- WU C.-C., CROXTALL J., PERRETTI M., BRYANT C.E., THIEMERMANN C., FLOWER R.J., VANE J.R. Lipocortin 1 mediates the inhibition by dexamethasone of the induction by endotoxin of nitric oxide synthase in the rat. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3473–3477. doi: 10.1073/pnas.92.8.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]