

Abstract
Spinal prostanoids are implicated in the development of thermal hyperalgesia after peripheral injury, but the specific prostanoid species that are involved are presently unknown. The current study used an in vitro spinal superfusion model to investigate the effect of substance P (SP), N-methyl-d-aspartate (NMDA), and capsaicin on multiple prostanoid release from dorsal spinal cord of naive rats as well as rats that underwent peripheral injury and inflammation (knee joint kaolin/carrageenan).
In naive rat spinal cords, PGE2 and 6-keto-PGF1α, but not TxB2, levels were increased after inclusion of SP, NMDA, or capsaicin in the perfusion medium.
Basal PGE2 levels from spinal cords of animals that underwent 5–72 h of peripheral inflammation were elevated relative to age-matched naive cohorts. The time course of this increase in basal PGE2 levels coincided with peripheral inflammation, as assessed by knee joint circumference. Basal 6-keto-PGF1α levels were not elevated after injury.
From this inflammation-evoked increase in basal PGE2 levels, SP and capsaicin significantly increased spinal PGE2 release in a dose-dependent fashion. Capsaicin-evoked increases were blocked dose-dependently by inclusion of S(+) ibuprofen in the capsaicin-containing perfusate.
These data suggest a role for spinal PGE2 and NK-1 receptor activation in the development of hyperalgesia after injury and demonstrate that this relationship is upregulated in response to peripheral tissue injury and inflammation.
Keywords: Spinal, prostaglandin, substance P, capsaicin, NMDA, kaolin/carrageenan, knee joint inflammation, prostanoid
Introduction
Peripheral inflammation and injury-induced hyperalgesia are associated with local prostaglandin synthesis by the enzyme, cyclo-oxygenase (COX), (Vane, 1971; Moncada et al., 1973). After peripheral injury, prostaglandins are synthesized at the site of injury (Ohuchi et al., 1976) and sensitize nociceptors through increases in sodium current-mediated depolarization and increases in the rate of sodium channel activation (Gold et al., 1996a,1996b). Administration of antibodies against PGE2 (Portanova et al., 1996) or deletion of the PGI2 receptor (IP) gene (Murata et al., 1997) prevents hyperalgesic behaviour after injury, suggesting a role for peripheral prostanoid synthesis in injury-mediated hyperalgesia. Demonstration that a structurally diverse class of agents called non-steroidal anti-inflammatory drugs (NSAIDs) inhibited COX-mediated synthesis of prostanoids and reduced the hyperalgesic state demonstrated the importance of this injury-induced cascade (Ferreira et al., 1971; Smith & Willis, 1971).
After peripheral injury, there is an increased sensitivity of nociceptors in the periphery (Kocher et al., 1987; Raja et al., 1988) and an increase in small calibre primary afferent activity (Puig & Sorkin, 1996). This increased peripheral activity leads to a spinally-mediated hypersensitivity (Woolf, 1983), such that innocuous tactile stimuli are interpreted as noxious (allodynia) and/or there is an increased responsiveness to noxious stimuli (hyperalgesia). In animal models of hyperalgesia, it has been clearly shown that such spinally-mediated states are dependent upon activation of spinal N-methyl-d-aspartate (NMDA) and neurokinin-1 (NK-1) receptors as well as spinal cyclo-oxygenase activity (for further review, see Yaksh et al., 1998).
Spinal COX inhibition blocks hyperalgesic responses to peripherally administered irritants such as zymosan (Yaksh, 1982), formalin (Malmberg & Yaksh, 1992a), or carrageenan (Dirig et al., 1998b). Moreover, intrathecal substance P (SP) evokes a thermal hyperalgesia (Yasphal et al., 1982; Dirig & Yaksh, 1996) that is blocked by spinal pretreatment with cyclo-oxygenase inhibitors (Malmberg & Yaksh, 1992b; Dirig et al., 1998a). Consistent with these observations, intrathecal prostanoids evoke a thermal hyperalgesia (Uda et al., 1990). Using chronic lumbar spinal microdialysis (Marsala et al., 1995) to elucidate spinal transmitter release, spinal PGE2 release was increased after peripheral injuries such as knee joint kaolin/carrageenan (Yang et al., 1996), formalin (Malmberg & Yaksh, 1995) or intrathecal substance P (Hua et al., 1998). While these observations support a possible role for spinal PGE2 in hyperalgesia, they do not exclude the likelihood that other prostanoids are synthesized and released from spinal cord (Smith & DeWitt, 1996; Willingale et al., 1997).
It is also unclear how spinal prostanoid synthesis is affected by peripheral injury that leads to spinal sensitization. The following points suggest that peripheral injury may increase the effects of afferent input on spinal prostanoid release: (i) Spinal cord SP immunoreactivity is increased after peripheral injury (Yaksh et al., 1980; Duggan et al., 1988), (ii) Intrathecal SP evokes a hyperalgesic state in rats (Yasphal et al., 1982), (iii) substance P increases PGE2 levels in an in vitro spinal superfusion assay (Dirig et al., 1996), and (iv) NK-1 receptor expression within the dorsal horn is upregulated after peripheral injury and inflammation (Schafer et al., 1993; Abbadie et al., 1996). Given these data, it is possible that SP-evoked spinal prostanoid levels may be increased in the presence of a protracted peripheral injury. The current study used an in vitro spinal tissue superfusion assay to address two hypotheses; (i) multiple prostanoids are released from dorsal spinal tissue in the presence of the hyperalgesic receptor ligands, SP, NMDA and capsaicin, and (ii) spinal prostanoid release is elevated after peripheral injury.
Methods
Animal preparation
Male Sprague Dawley rats (300–400 g; Harlan Industries; Indianopolis, IN, U.S.A.) were housed in pairs in cages and maintained on a 12 h light/dark cycle with free access to food and water. All studies were carried out using protocols approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. Animals used in the inflammation part of this study received an intra-articular injection of kaolin/carrageenan (4% w w−1) into the right hind knee under halothane anaesthesia (4%/oxygen). Kaolin and λ-carrageenan (0.4 g of each) were suspended in 10 ml of 0.9% w v−1 saline using sonication. This suspension (0.1 ml) was injected with a 22Ga needle into the knee joint capsule using the patellar tendon as a landmark. Animals undergoing inflammation were housed individually for up to 96 h after induction of inflammation. At the different time points after knee joint injection (5–96 h), the circumference of both hind knees of each rat was measured using a length of 3-O suture and the spinal cord was removed and perfused as described below. Spinal cords from inflamed animals and age-matched naive cohorts were harvested and perfused on the same day and prostanoid release from the spinal cords of these inflamed animals was compared to age-matched naive cohorts. Naive animals were used instead of intra-articular vehicle injected controls as it is well known that intra-articular injection of saline alone increases spinal transmitter release (Yang et al., 1996).
In vitro dorsal spinal cord superfusion
The spinal cord superfusion methodology has been previously published (Dirig et al., 1997). Briefly, rats were decapitated after terminal halothane anaesthesia (4%/oxygen) and spinal cords were hydraulically extruded (Sousa & Horrocks, 1979). Spinal cords were placed in ice-cold artificial cerebrospinal fluid (ACSF) and then dissected on a filter paper-covered glass plate placed on crushed ice. A 2 cm segment of the lumbar enlargement was isolated and hemisected longitudinally into lateral halves. These halves were hemisected again, and the dorsal quadrants were retained. These dorsal segments were chopped cross-sectionally into 2 mm prisms. Prisms were dispersed on Millipore filters (13 mm diam, 5 μm pore size) which were placed inside perfusion chambers (modified Millipore filter units, Bedford, MA, U.S.A.). The prisms of one lumbar enlargement were used in each perfusion study and dispersed at random to three or four perfusion chambers (five prisms per chamber). Perfusion chambers were perfused with ACSF at a rate of 200 μl min−1 using a peristaltic pump. ACSF consisted of (in mM): NaHCO3, 21.0; Na2HPO4, 2.5; NaCl, 125.0; KCl, 2.6; MgCl2, 0.9; CaCl2, 1.3; d-Glucose, 3.9. The ACSF reservoir for each study was placed in the same water bath as the perfusion chambers and pH was adjusted to 7.4 by bubbling with 5% CO2/95% O2 for 30 min prior to and throughout the study. After an initial washout period of 45 min, two perfusate samples were collected; a 10 min baseline and a 10 min stimulation sample. The stimulation samples contained ACSF including substance P (100 nM, 1 μM), NMDA (100 μM), or capsaicin (100 nM, 1 μM, 10 μM). Chamber temperature was monitored continuously and maintained at 37°C using a thermocouple (36Ga, Type T, Omega Instruments) permanently implanted in one perfusion chamber.
Prostanoid radioimmunoassay
In vitro samples were collected on ice, frozen at −70°C, lyophilized, and stored at −70°C until reconstitution in 0.1 ml of assay buffer (0.01 M phosphate, 0.1% w v−1 bovine gamma globulin, and 0.1% w v−1 sodium azide, pH 7.0) for competitive radioimmunoassay (RIA) using a polyclonal rabbit anti-prostanoid antibody in conjunction with a magnetic particle-coupled, goat anti-rabbit antibody (PerSeptive Biosystems, Framington, MA, U.S.A.). Separate RIA kits were used to assess sample content of PGE2, 6-keto-PGF1α (primary metabolite of PGI2), and TxB2 (primary metabolite of TxA2). There is <1.8% cross-reactivity between these different RIA kits and details of this methodology have been previously published (Jobke et al., 1973; Granstrom & Kindahl, 1978). Briefly, lyophilized perfusate samples or prostanoid (41–104 pg ml−1 as standard) were reconstituted and split three ways in assay buffer and incubated with 0.1 ml of Perseptives stock rabbit anti-prostanoid antibody solution for 2 h at 4°C. After precipitation of the bound complex using goat anti-rabbit serum and centrifugation (15 min, 4°C, 1000×g), the pellet was counted in a γ-counter (Packard Cobra). Assays were carried out with non-specific binding and blanks, and minimum assay sensitivity was 4 pg per assay tube for each kit. Individual experimental samples were run singly, and all standard curves were run in duplicate.
Drug delivery and statistics
Substance P and NMDA were dissolved in ACSF. Capsaicin was dissolved in 25% w v−1 beta-hydroxyl cyclodextrin to a concentration of 1 mg ml−1, and S(+) ibuprofen was dissolved to 5 mg ml−1 in 100% w v−1 dimethyl sulphoxide. These stock solutions were then diluted to final concentrations in ACSF. All drugs were reconstituted on the morning of testing and used only on that day. Inclusion of the solvent (i.e. either cyclodextrin or dimethyl sulphoxide) did not increase PGE2 levels relative to basal levels.
Basal and evoked levels for each prostanoid were compared in the presence of substance P, NMDA or capsaicin using a paired Student's t-test. To analyse the effect of peripheral inflammation on spinal PGE2 release, basal PGE2 release was compared across treatment groups (i.e. time after knee joint injury) using a one way ANOVA with Dunnet's post-hoc comparison to control (naive basal release). For the construction of capsaicin, SP, and S(+) ibuprofen dose response curves, each animal provided enough tissue to run four chambers in parallel; this allowed tissue from each animal to be exposed to two (substance P) or three different doses of the drug (capsaicin and capsaicin plus ibuprofen), as well as a negative control chamber containing drug solvent, but no drug. Thus, each animal provided data for all points of the dose response curves. Basal PGE2 release was compared in naive animals and was not different across drug groups (i.e. different drug concentrations). After 24 h of knee joint inflammation, basal PGE2 levels were significantly elevated but were not different across drug groups. Thus, in each case (naive and 24 h after knee joint inflammation), there was a stable basal PGE2 release that could be pooled and then compared to drug-evoked release using a one-way ANOVA design with Dunnet's post-hoc comparison to control (basal levels). In all tests, the minimum criterion for statistical significance was P<0.05.
Results
Multiple prostanoid release
Spinal PGE2 levels were increased from basline after inclusion of capsaicin (10 μM), SP (1 μM), or NMDA (100 μM) in the second 10 min stimulation sample (P<0.05 in each case). Similar results were observed for 6-keto PGF1α upon stimulation with capsaicin and SP (P<0.05), but this numerical trend did not reach significance for NMDA stimulation (P=0.08). No changes in T×B2 levels were observed after any stimulation condition (Figure 1).
Figure 1.
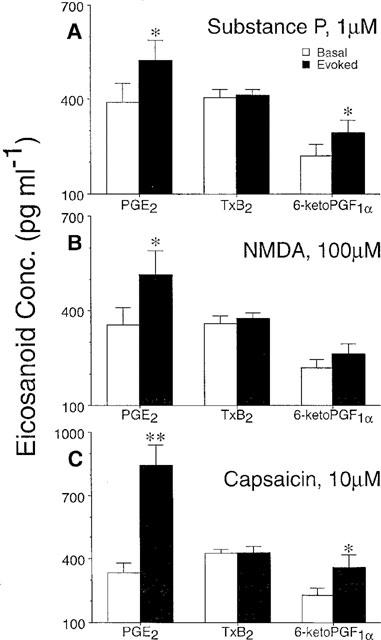
Spinal prostanoid release. (A) Substance P- and (C) capsaicin-evoked increases in perfusate prostanoid content were observed for PGE2 and 6-keto-PGF1α, but not TxB2. (B) NMDA increased PGE2 levels significantly, however, the numerical increase in 6-keto-PGF1α levels did not reach significance. Each data point represents the mean±s.e.mean of 4–8 animals. Significant increases (Student's Paired t-test) from basal eicosanoid levels indicated by *P<0.05 or **P<0.01.
Basal prostanoid release after knee joint injury
Spinal PGE2 basal levels were increased as early as 5 h after induction of knee joint inflammation and this elevation persisted for 72 h after injury (P<0.01, Figure 2A). Basal PGE2 levels were not significantly elevated by 96 h after injury. Similar to the time course of basal spinal PGE2 levels at 5–72 h post-injury, knee joint oedema (circumference) was also significantly increased from 5–72 h after injury for the injected knee (P<0.01) and returned to baseline by 96 h after induction of knee joint inflammation. The contralateral knee joint circumference was not different from baseline values at any time point (Figure 2B).
Figure 2.
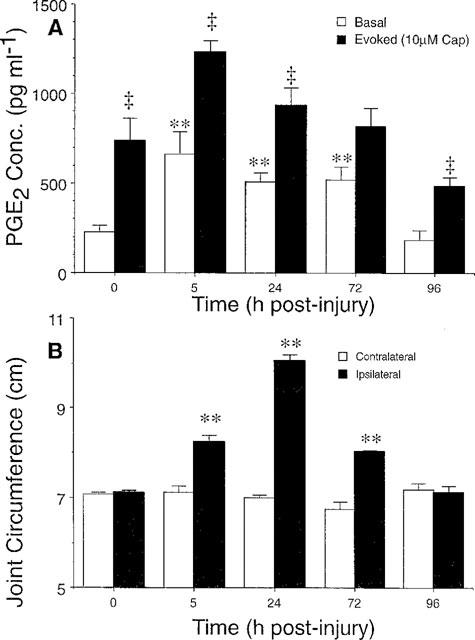
Basal and capsaicin-evoked spinal PGE2 levels after 5–72 h of knee joint inflammation. (A) PGE2 levels from spinal cords of naive cohorts are presented at T=0. Note that basal PGE2 levels increase from 5–72 h after injury and return to naive PGE2 levels by 96 h post-injury. (B) Knee joint circumference after injection of kaolin/carrageenan to the knee joint capsule increases (ipsilateral) as compared to the un-injected knee (contralateral) with a similar time course to increases in spinal PGE2 level. Each data point represents the mean±s.e.mean of 4–8 animals. Significant increases in basal PGE2 release (A) and knee joint circumference (B) after inflammation are indicated by **P<0.01 as compared to naive cohorts. Significant capsaicin-evoked increases in PGE2 at each time point after inflammation are indicated by ‡P<0.005.
SP-evoked prostanoid release after injury
After 24 h of knee joint inflammation, basal and SP-evoked PGE2 levels were significantly elevated relative to naive cohorts (P<0.05, Figure 3A), but basal 6-keto-PGF1α levels were not elevated 24 h after knee joint injury. In contrast to the PGE2 results, basal and SP-evoked 6-keto-PGF1α release did not differ when comparing spinal release from naive and injured animals (Figure 3B, P>0.05). Given the marked increase in basal and evoked spinal PGE2 release, the remainder of the study focused on PGE2 release from spinal cords of rats that had undergone 24 h of knee joint inflammation. Substance P evoked a significant increase in PGE2 release from spinal cords of naive animals (P<0.05, 1 μM), and this release was significantly elevated after 24 h of knee joint inflammation (P<0.001). Importantly, 24 h of knee joint inflammation significantly decreased the SP dose necessary to evoke a significant increase in PGE2 levels (100 nM for the inflamed animal versus 1 μM for naive animals, see Figure 3A).
Figure 3.
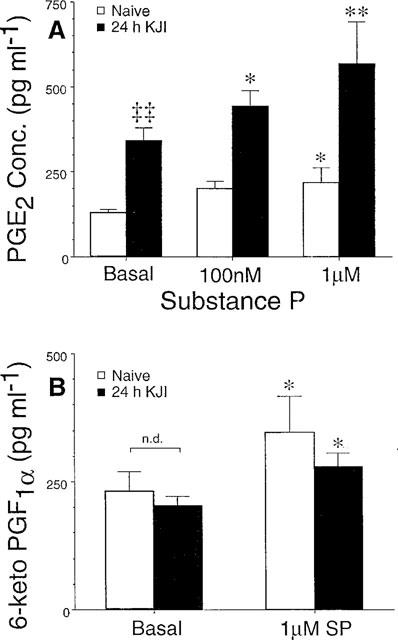
Substance P-evoked increases in eicosanoid levels after injury. Although basal PGE2 levels were elevated after knee joint inflammation (relative to naive cohorts), basal levels were not different within each treatment group (i.e. injured and naive). Given the stable, condition-dependent basal PGE2 release, basal levels were pooled within each treatment group and compared to substance P-evoked release. After 24 h of knee joint inflammation (black bars), basal and evoked PGE2 levels were elevated relative to naive cohorts (white bars), and lower doses of SP were required to produce significant increases in PGE2. (B) SP evoked a slight, but significant, increase in 6-keto-PGF1α from spinal cords of naive animals (white bars), but there was no increase in this eicosanoid after 24 h of inflammation (black bars). Each data point represents the mean±s.e.mean of 4–8 animals, and significant increases from basal levels are indicated by *P<0.05 and **P<0.01. Comparison of basal release across naive and injured animals indicated by ‡‡P<0.0001.
Capsaicin-evoked prostanoid release after injury
In addition to the increase in basal PGE2 levels 5–72 h after knee joint injury, stimulation with capsaicin (10 μM) in the second 10 min sample evoked significant increases in PGE2 release. This capsaicin-evoked release was evident in naive animals as well as after 5, 24, and 96 h of knee joint inflammation (P<0.005, Figure 2A). This trend was also observed at 72 h post-injury, but the capsaicin-evoked PGE2 release did not reach statistical significance (P=0.06, Figure 2A). Capsaicin-evoked PGE2 release demonstrated a dose dependence as shown in Figure 4A, and the amount of PGE2 release evoked by capsaicin was greater than that of exogenous SP described in Figure 3A. Since the greatest PGE2 release was evoked by capsaicin, the ability of the non-selective COX inhibitor S(+) ibuprofen to block this release was explored. S(+) ibuprofen was included with capsaicin in the stimulation sample (i.e. 10 μM capsaicin+0.1, 1.0 or 10.0 μM ibuprofen in the second 10 min sample). As shown in Figure 4B, capsaicin-evoked PGE2 release was significantly decreased by S(+) ibuprofen in a dose-dependent manner.
Figure 4.
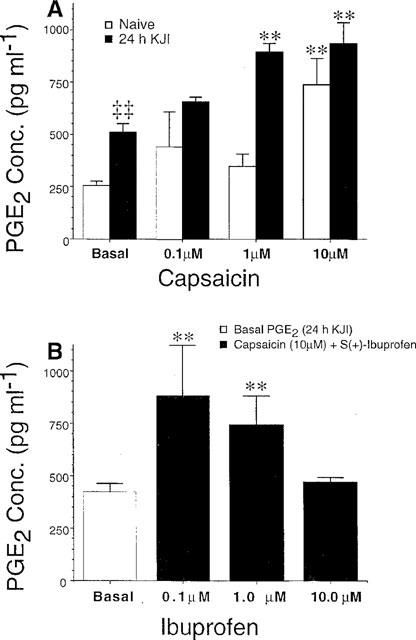
As in Figure 3, basal PGE2 levels within both treatment groups (naive and injured) did not differ and were pooled for comparison to capsaicin-evoked release. (A) Capsaicin evoked increases in PGE2 release in a dose dependent fashion, and this release was greater (both in basal and evoked samples) after 24 h of knee joint inflammation (black bars) relative to naive animals (white bars). (B) Capsaicin-evoked PGE2 release after 24 h of knee joint inflammation was blocked by increasing doses of S(+) ibuprofen. Each data point represents the mean±s.e.mean of 4–8 animals, and significant increases from basal levels are indicated by *P<0.05 and **P<0.01. Comparison of basal PGE2 levels across naive and injured animals indicated by ‡‡P<0.0001.
Discussion
Spinal prostanoid release
In the present study, PGE2, 6-keto PGF1α, and TXB2-like immunoreactivity were observed in the in vitro superfusates of rat dorsal spinal cords. These results are in accord with previous in vitro and in vivo work in which spinal synthesis of multiple prostanoids has been reported (Dirig et al., 1996; Willingale et al., 1997). Delivery of capsaicin and SP to the spinal parenchyma increased PGE2 and 6-keto PGF1α, but not TXB2, concentrations in spinal perfusates. Of the three eicosanoids evaluated, the most abundant eicosanoid observed under basal and evoked conditions was PGE2. This agrees with previous in vitro studies on capsaicin-evoked spinal PGE2 release (Malmberg & Yaksh, 1994) as well as in vivo studies describing increased spinal microdialysate PGE2 after activation of spinal NK1 receptors (Hua et al., 1998), hind paw formalin injection (Malmberg & Yaksh, 1995) and knee joint injection of kaolin/carrageenan (Yang et al., 1996) in the rat. These data support the hypothesis that multiple eicosanoids are released from spinal cord and this increase is enhanced after direct activation of spinal terminal systems associated with hyperalgesia.
It should be noted that the effects of capsaicin (1 and 10 μM) were greater than that of substance P (100 nM and 1 μM) whether compared under naive or inflamed conditions. This may be interpreted as the difference between a specific post-synaptic NK-1 receptor effect versus primary afferent depolarization by capsaicin. Depolarization of primary afferent terminals would be expected to involve not only SP release, but also excitatory amino acids and other neuropeptides. As shown in Figure 1B, an NMDA-receptor mediated component to this system is likely (Malmberg & Yaksh, 1992b).
Origin of spinal prostanoid release
There are at least two isoforms of cyclo-oxygenase (COX-1 and COX-2), the enzyme responsible for synthesis of PGH2 from arachidonic acid. In spinal cord, COX-2 is the predominant COX isozyme present at the mRNA and protein level, but COX-1 has also been localized to the dorsal root ganglion (DRG) and spinal cord (Beiche et al., 1996; Willingale et al., 1997). These results are consistent with our own work in which it was shown that intrathecal COX-2, but not COX-1 inhibitors will block intrathecal substance P-evoked hyperalgesia and PGE2 release (Dirig et al., 1998a). It is not certain whether these prostanoids uniformly originate from neuronal, glial or vascular endothelial sources. Although it has not been clearly delineated, neuronal prostanoid sources are likely, based on the localization of the COX isozymes to neurons within the DRG and spinal cord. Marriott and colleagues (Marriott et al., 1991a,1991b; Marriott & Wilkin, 1993) have reported that SP can evoke release of PGE2 from cultured spinal astrocytes, but in vivo studies report NK-1 receptors on neurons (Brown et al., 1995; Mantyh et al., 1995), but not astrocytes. Astrocytes are only reported to express NK-1 receptors after a central nervous system injury, such as ischaemia, anoxia, or mechanical trauma (see Palma et al., 1997 for details). Thus, while SP-evoked PGE2 from astrocytes may play a role in central nervous system responses to neuronal injury and ischaemia, such sources are not likely in an acute spinal cord preparation as described herein.
Upregulation of PGE2 release after injury
Injection of carrageenan and kaolin into the knee joint resulted in a marked inflammatory reaction at 5–72 h as indicated by increased joint circumference. This enlarged knee joint has been shown to be extremely sensitive to mechanical compression with an inflated cuff. Graded compression of the inflamed knee yields blood pressure responses consistent with an autonomic response to a painful stimulus (Nagasaka et al., 1996). As with other models of peripheral injury and inflammation, knee joint inflammation has also been associated with thermal hyperalgesia that has a distinct spinal pharmacology (Sluka & Westlund, 1993a,1993b).
Basal PGE2 release from the dorsal lumbar spinal cord was significantly elevated by 24 h after initiation of a peripheral inflammatory state. This may be considered as the in vitro equivalent of observations reported by Yang et al. (1996), who reported increased spinal lumbar intrathecal microdialysate PGE2 content 24 h after knee joint injury in rats. Interestingly, the substance P and capsaicin doses necessary to evoke significant increases in spinal PGE2 release were reduced 10 fold after 24 h of knee joint inflammation (Figures 3A and 4A). It is well accepted that spinal neuronal activity is dependent on the effective concentration of excitatory amino acids and substance P (Dougherty & Willis, 1991), and that this release is dependent upon depolarization and calcium-dependent exocytosis of synaptic vesicles (Miller, 1987; Smith & Augustine, 1988) from the central terminal of the primary afferent (De Biasi & Rustioni, 1988). The decreased substance P and capsaicin dosage requirement reported herein suggests that after peripheral injury and inflammation, a sub-threshold noxious stimulus, which is associated with lower frequency C-fibre activity (Handwerker et al., 1991) and thereby less substance P release, could still evoke significant spinal PGE2 release. Given the effectiveness of intrathecal COX inhibitors (Neugebauer et al., 1995; Dirig et al., 1998b) and PGE2 receptor antagonists (Malmberg et al., 1994) in reducing injury-induced hyperalgesia, these data suggest that increased spinal PGE2 release is necessary for the development of hyperalgesia after peripheral injury and inflammation.
Given the effectiveness of capsaicin-evoked PGE2 release and the implication that spinal PGE2 synthesis is necessary for the development of hyperalgesia in vivo, we explored the sensitivity of this PGE2 release to COX inhibition. In agreement with the effectiveness of intrathecal COX inhibitors to block hyperalgesic behaviour in vivo (Yaksh, 1982; Malmberg & Yaksh, 1992b; Dirig et al., 1998b) and the effect of S(+)ibuprofen in vitro (Malmberg & Yaksh, 1994), S(+) ibuprofen reduced capsaicin-evoked PGE2 release in a dose-dependent manner. While this result implicates spinal COX activity in PGE2 release, the specific COX isoforms cannot be concluded from this work. Other studies (Dirig et al., 1998a,1998b) suggest that this spinal PGE2 release may be due to activity of COX-2, but not COX-1. Regardless of the COX isoform implicated, the elevated spinal PGE2 release after peripheral injury suggest that spinally mediated hyperalgesia after injury may be due to one of the following mechanisms of substance P mediated spinal PGE2 synthesis.
First, elevated basal PGE2 levels after injury may indicate an increased tonic substance P release and ongoing activation of PGE2 synthesis after injury (i.e. due to an increased tonic NK-1 receptor activation). This hypothesis is consistent with reports of Yaksh and colleagues (Yaksh et al., 1980; Go & Yaksh, 1987) and Duggan and colleagues (Duggan & Johnston, 1970; Hope et al., 1990) that spinal substance P levels are elevated after peripheral injury. Consistent with this hypothesis, Hargreaves and colleagues have reported that capsaicin-evoked SP release from a similar in vitro spinal slice apparatus is elevated after a peripheral injury (Garry & Hargreaves, 1992). Second, following peripheral soft tissue or nerve injury, dorsal horn NK-1 receptor protein increases (Schafer et al., 1993; Abbadie et al., 1996). While NK-1 antagonists were not employed in the present study, substance P has been shown to bind preferentially to the NK-1 receptor (Henry, 1993) and the behavioural effects of intrathecal substance P are fully reversed by intrathecal NK-1 antagonists (Malmberg & Yaksh, 1992b; Dirig et al., 1998a). Thus, one possible explanation for the increased tonic PGE2 release after injury as well as the increased sensitivity of the spinal cord to substance P after peripheral injury may be an increased NK-1 receptor expression within the dorsal spinal cord. Third, increased basal or evoked PGE2 release may arise from an increase in COX-2 enzyme expression within the dorsal horn. As early as 6 h after peripheral injury and inflammation, COX-2 peptide within the spinal cord is increased (Goppelt-Struebe & Beiche, 1997). Thus, the observed increase in basal and evoked spinal PGE2 release after injury may be due to an increase in spinal SP synthesis and release from primary afferent terminals, an increase in post-synaptic NK-1 receptor expression, or increasese in the post-synaptic PGE2 synthetic machinery (i.e. increases in COX-2 enzyme expression).
Spinal prostanoid-mediated feedback loops
Substance P evoked increases in spinal PGE2 and PGI2 release are consistent with a role for substance P and multiple eicosanoids in the development of hyperalgesic states. The substance P precursor, preprotachykinin, is present in DRG cells (Oida et al., 1995), and substance P is present in primary afferent spinal terminals (De Biasi & Rustioni, 1988). Substance P binds preferentially to the NK-1 receptor (Henry, 1993) which is present on neurons within the superficial dorsal horn (Lamina I) as well as in the dendrites of deep dorsal horn neurons which project to the superficial laminae (Brown et al., 1995; Mantyh et al., 1995). Suggestive of prostanoid action on the primary afferent terminal, mRNA for both PGE2 and PGI2 receptors (EP and IP, respectively) co-localize with preprotachykinin mRNA in rat DRG cells. PGE2 binds to several subtypes of PGE2 (EP) receptors (Coleman et al., 1994) and binds with high affinity to the superficial laminae (Matsumura et al., 1992) of the rat dorsal horn. Further supporting a role for PGE2 receptors in sensory processing, Beiche et al. (1998) recently reported EP receptor immunoreactivity on primary afferent terminals within the superficial dorsal horn in rats. For dorsal horn prostanoid synthesis to be implicated in sensory processing, the synthetic enzyme, COX, must also be present. In the past year, several studies have reported immunoreactivity for COX-2 within superficial dorsal horn neurons (Willingale et al., 1997; Beiche et al., 1998). These localization studies suggest a substance P-mediated, COX-2 dependent synthesis of prostanoids in the development of hyperalgesia. This possible relationship is supported by experiments demonstrating the effectiveness of spinal inhibitors of COX-2, but not COX-1, in blocking substance P-induced thermal hyperalgesia (Dirig et al., 1998a).
Beyond characterization studies, cultured DRG neurons have also been used to study the relationship between spinal prostanoids and neuropeptide release. Prostanoid administration to cultured rat DRG cells does not evoke release or change basal neuropeptide release, but addition of prostanoids to the cell culture medium increases capsaicin-evoked release of substance P from cultured DRG cells or a spinal slice preparation similar to that described herein (Vasko et al., 1993; Hingtgen & Vasko, 1994; Vasko, 1995). This substance P release was augmented by pre-conditioning with PGE2 and PGI2, the same two eicosanoids that were increased after exposure of spinal tissue to substance P in the current work. As shown in Figure 1, exogenous substance P or NMDA evokes synthesis and release of PGE2 and PGI2 (Dirig et al., 1996) from the dorsal horn of the spinal cord, arguably from neurons within the superficial laminae. This synthesis and release of prostanoids may then feedback on primary afferent terminals (at pre-synaptic prostanoid receptors) to augment evoked transmitter release of any subsequent action potentials invading the primary afferent terminals. This positive feedback loop may partially explain the relationship of substance P and eicosanoids in the development of spinally-mediated hyperalgesic states after peripheral injury and inflammation.
In conclusion, this study demonstrates that multiple eicosanoids are released from rat dorsal spinal cord after challenge with receptor ligands associated with hyperalgesia in vivo (i.e. substance P, NMDA, and capsaicin). PGE2 was the predominant species released, and this eicosanoid was further elevated (both basal and evoked levels) from 5–72 h after a peripheral injury. These data suggest a role for substance P-evoked PGE2 synthesis in the development of spinally-mediated hyperalgesia after peripheral injury. Upregulation of spinal PGE2 release in the presence of the persistent afferent input generated by a peripheral injury provides additional support for a spinal site of action for the potent anti-hyperalgesic effects of both intrathecally and systemically administered COX inhibitors.
Acknowledgments
The authors would like to express their thanks to Dr C.M. Conway for his critical review and suggestions during manuscript preparation. Research supported in part by NIH grant NIDA02110 (TLY), and Pre-Doctoral National Research Service Award NIDA05726 (DMD).
Abbreviations
- ACSF
artificial cerebrospinal fluid
- COX
cyclo-oxygenase
- DRG
dorsal root ganglion
- EP
receptor for PGE2
- IP
receptor for PGI2
- KJI
knee joint inflammation
- NK
neurokinin
- NMDA
N-methyl-d-aspartate
- NSAID
non-steroidal anti-inflammatory drug
- PG
prostaglandin
- SP
substance P
- Tx
thromboxane
References
- ABBADIE C., BROWN J.L., MANTYH P.W., BASBAUM A.I. Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience. 1996;70:201–209. doi: 10.1016/0306-4522(95)00343-h. [DOI] [PubMed] [Google Scholar]
- BEICHE F., KLEIN T., NUSING R., NEUHUBER W., GOPPELT-STRUEBE M. Localization of cyclooxygenase-2 and prostaglandin E2 receptor EP3 in the rat lumbar spinal cord. J. Neuroimmunol. 1998;89:26–34. doi: 10.1016/s0165-5728(98)00061-7. [DOI] [PubMed] [Google Scholar]
- BEICHE F., SCHEUERER S., BRUNE K., GEISSLINGER G., GOPPELT-STRUEBE M. Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS Lett. 1996;390:165–169. doi: 10.1016/0014-5793(96)00604-7. [DOI] [PubMed] [Google Scholar]
- BROWN J.L., LIU H., MAGGIO J.E., VIGNA S.R., MANTYH P.W., BASBAUM A.I. Morphological characterization of substance P receptor-immunoreactive neurons in the rat spinal cord and trigeminal nucleus caudalis. J. Comp. Neurol. 1995;356:327–344. doi: 10.1002/cne.903560302. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A., SMITH W.L., NARUMIYA S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- DE BIASI S., RUSTIONI A. Glutamate and substance coexist in primary afferent terminals in the superficial laminae of spinal cord. Proc. Natl. Acad. Sci. U.S.A. 1988;85:7820–7824. doi: 10.1073/pnas.85.20.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIRIG D.M., CONWAY C.M., ISAKSON P.C., YAKSH T.L. Spinal cyclooxygenase-2 (COX-2) inhibition blocks intrathecal Substance P-evoked thermal hyperalgesia in rats. Soc. for Neurosci. (Abstract) 1998a. p. 155.12.
- DIRIG D.M., DUFFIN K., ROSSI S., ISAKSON P., YAKSH T.L. Evoked release of eicosanoids from rat spinal cord in vitro with correlative quantitation by radioimmunoassay (RIA) and negative-ion electrospray mass spectrometry (ESI-MS) 8th World Congress on Pain (Abstract) 1996. p. 135.
- DIRIG D.M., HUA X.Y., YAKSH T.L. Temperature dependency of basal and evoked release of amino acids and calcium gene-related peptide from rat dorsal spinal cord. J. Neurosci. 1997;17:4406–4414. doi: 10.1523/JNEUROSCI.17-11-04406.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIRIG D.M., ISAKSON P.C., YAKSH T.L. Effect of COX-1 and COX-2 inhibition on induction and maintenance of carrageenan-evoked thermal hyperalgesia in rats. J. Pharmacol. Exp. Ther. 1998b;285:1031–1038. [PubMed] [Google Scholar]
- DIRIG D.M., YAKSH T.L. Thermal hyperalgesia in rat evoked by intrathecal substance P at multiple stimulus intensities reflects an increase in the gain of nociceptive processing. Neurosci. Lett. 1996;220:93–96. doi: 10.1016/s0304-3940(96)13230-4. [DOI] [PubMed] [Google Scholar]
- DOUGHERTY P.M., WILLIS W.D. Enhancement of spinothalamic neuron responses to chemical and mechanical stimuli following combined micro-iontophoretic application of N-methyl-D-aspartic acid and substance P. Pain. 1991;47:85–93. doi: 10.1016/0304-3959(91)90015-P. [DOI] [PubMed] [Google Scholar]
- DUGGAN A.W., HENDRY I.A., MORTON C.R., HUTCHISON W.D., ZHAO Z.Q. Cutaneous stimuli releasing immunoreactive substance P in the dorsal horn of the cat. Brain Res. 1988;451:261–273. doi: 10.1016/0006-8993(88)90771-8. [DOI] [PubMed] [Google Scholar]
- DUGGAN A.W., JOHNSTON G.A. Glutamate and related amino acids in cat spinal roots, dorsal root ganglia and peripheral nerves. J. Neurochem. 1970;17:1205–1208. doi: 10.1111/j.1471-4159.1970.tb03369.x. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., MONCADA S., VANE J.R. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nature New Biol. 1971;231:237–239. doi: 10.1038/newbio231237a0. [DOI] [PubMed] [Google Scholar]
- GARRY M.G., HARGREAVES K.M. Enhanced release of immunoreactive CGRP and substance P from spinal dorsal horn slices occurs during carrageenan inflammation. Brain Res. 1992;582:139–142. doi: 10.1016/0006-8993(92)90328-7. [DOI] [PubMed] [Google Scholar]
- GO V.L., YAKSH T.L. Release of substance P from the cat spinal cord. J. Physiol. 1987;391:141–167. doi: 10.1113/jphysiol.1987.sp016731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLD M.S., DASTMALCHI S., LEVINE J.D. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience. 1996a;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- GOLD M.S., REICHLING D.B., SHUSTER M.J., LEVINE J.D. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc. Natl. Acad. Sci. U.S.A. 1996b;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPPELT-STRUEBE M., BEICHE F. Cyclooxygenase-2 in the spinal cord: localization and regulation after a peripheral inflammatory stimulus. Adv. Exp. Med. Biol. 1997;433:213–216. doi: 10.1007/978-1-4899-1810-9_45. [DOI] [PubMed] [Google Scholar]
- GRANSTROM E., KINDAHL H. Radioimmunoassay of prostaglandins and thromboxanes. Adv. Prostaglandin Thromboxane Res. 1978;5:119–210. [PubMed] [Google Scholar]
- HANDWERKER H.O., KILO S., REEH P.W. Unresponsive afferent nerve fibres in the sural nerve of the rat. J. Physiol. 1991;435:229–242. doi: 10.1113/jphysiol.1991.sp018507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HENRY J.L. Participation of substance P in spinal physiological responses to peripheral aversive stimulation. Regul. Pept. 1993;46:138–143. doi: 10.1016/0167-0115(93)90024-3. [DOI] [PubMed] [Google Scholar]
- HINGTGEN C.M., VASKO M.R. Prostacyclin enhances the evoked-release of substance P and calcitonin gene-related peptide from rat sensory neurons. Brain Res. 1994;655:51–60. doi: 10.1016/0006-8993(94)91596-2. [DOI] [PubMed] [Google Scholar]
- HOPE P.J., JARROTT B., SCHAIBLE H.G., CLARKE R.W., DUGGAN A.W. Release and spread of immunoreactive neurokinin A in the cat spinal cord in a model of acute arthritis. Brain Res. 1990;533:292–299. doi: 10.1016/0006-8993(90)91352-h. [DOI] [PubMed] [Google Scholar]
- HUA X.-Y., CHEN P., MARSALA M., YAKSH T.L. Intrathecal substance P-induced thermal hyperalgesia and spinal release of prostaglandin E2 and amino acids. Neuroscience. 1998;89:525–534. doi: 10.1016/s0306-4522(98)00488-6. [DOI] [PubMed] [Google Scholar]
- JOBKE A, , PESKAR B.A., PESKAR B.M. On the specificity of antisera against prostaglandins A2 and E2. FEBS Lett. 1973;37:192–196. doi: 10.1016/0014-5793(73)80456-9. [DOI] [PubMed] [Google Scholar]
- KOCHER L., ANTON F., REEH P.W., HANDWERKER H.O. The effect of carrageenan-induced inflammation on the sensitivity of unmyelinated skin nociceptors in the rat. Pain. 1987;29:363–373. doi: 10.1016/0304-3959(87)90051-0. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., RAFFERTY M.F., YAKSH T.L. Antinociceptive effect of spinally delivered prostaglandin E receptor antagonists in the formalin test on the rat. Neurosci. Lett. 1994;173:193–196. doi: 10.1016/0304-3940(94)90181-3. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Antinociceptive actions of spinal nonsteroidal anti-inflammatory agents on the formalin test in the rat. J. Pharmacol. Exp. Ther. 1992a;263:136–146. [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992b;257:1276–1279. doi: 10.1126/science.1381521. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Capsaicin-evoked prostaglandin E2 release in spinal cord slices: relative effect of cyclooxygenase inhibitors. Eur. J. Pharmacol. 1994;271:293–299. doi: 10.1016/0014-2999(94)90786-2. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Cyclooxygenase inhibition and the spinal release of prostaglandin E2 and amino acids evoked by paw formalin injection: a microdialysis study in unanesthetized rats. J. Neurosci. 1995;15:2768–2776. doi: 10.1523/JNEUROSCI.15-04-02768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANTYH P.W., DEMASTER E., MLHOTRA A., GHILARDI J.R., ROGERS S.D., MANTYH C.R., LIU H., BASBAUM A.I., VIGNA S.R., MAGGIO J.E., SIMONE D.A. Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- MARRIOTT D.R., WILKIN G.P. Substance P receptors on O-2A progenitor cells and type-2 astrocytes in vitro. J. Neurochem. 1993;61:826–834. doi: 10.1111/j.1471-4159.1993.tb03593.x. [DOI] [PubMed] [Google Scholar]
- MARRIOTT D., WILKIN G.P., COOTE P.R., WOOD J.N. Eicosanoid synthesis by spinal cord astrocytes is evoked by substance P; possible implications for nociception and pain. Adv. Prostaglandin Thromboxane Leukot. Res. 1991a;21B:739–741. [PubMed] [Google Scholar]
- MARRIOTT D.R., WILKIN G.P., WOOD J.N. Substance P-induced release of prostaglandins from astrocytes: regional specialisation and correlation with phosphoinositol metabolism. J. Neurochem. 1991b;56:259–265. doi: 10.1111/j.1471-4159.1991.tb02590.x. [DOI] [PubMed] [Google Scholar]
- MARSALA M., MALMBERG A.B., YAKSH T.L. A chronic spinal dialysis catheter for use in the unanesthetized rat: Methodology and application. J. Neurosci. Meth. 1995;62:43–53. doi: 10.1016/0165-0270(95)00053-4. [DOI] [PubMed] [Google Scholar]
- MATSUMURA K., WATANABE Y., IMAI-MATSUMURA K., CONNOLLY M., KOYAMA Y., ONOE H., WATANABE Y. Mapping of prostaglandin E2 binding sites in rat brain using quantitative autoradiography. Brain Res. 1992;581:292–298. doi: 10.1016/0006-8993(92)90720-t. [DOI] [PubMed] [Google Scholar]
- MILLER R.J. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- MONCADA S., FERREIRA S.H., VANE J.R. Prostaglandins, aspirin-like drugs and the oedema of inflammation. Nature. 1973;246:217–219. doi: 10.1038/246217a0. [DOI] [PubMed] [Google Scholar]
- MURATA T., USHIKUBI F., MATSUOKA T., HIRATA M., YAMASAKI A., SUGIMOTO Y., ICHIKAWA A., AZE Y., TANAKA T., YOSHIDA N., UENO A., OH-ISHI S., NARUMIYA S. Altered pain perception and inflammatory responses in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- NAGASAKA H., AWAD H., YAKSH T.L. Peripheral and spinal actions of opioids in the blockade of the autonomic response evoked by compression of the inflamed knee joint. Anesthesiology. 1996;85:808–816. doi: 10.1097/00000542-199610000-00016. [DOI] [PubMed] [Google Scholar]
- NEUGEBAUER V., GEISSLINGER G., RUMENAPP P., WEIRETTER F., SZELENYI I., BRUNE K., SCHAIBLE H.G. Antinociceptive effects of R(−)- and S(+)-flurbiprofen on rat spinal dorsal horn neurones rendered hyperexcitable by an acute knee joint inflammation. J. Pharmacol. Exp. Ther. 1995;275:618–628. [PubMed] [Google Scholar]
- OHUCHI K., SATO H., TSURUFUJI S. The content of prostaglandin E and prostaglandin F2alpha in the exudate of carrageenin granuloma of rats. Biochim. Biophys. Acta. 1976;424:439–448. doi: 10.1016/0005-2760(76)90033-3. [DOI] [PubMed] [Google Scholar]
- OIDA H., NAMBA T., SUGIMOTO Y., USHIKUBI F., OHISHI H., ICHIKAWA A., NARUMIYA S. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. Br. J. Pharmacol. 1995;116:2828–2837. doi: 10.1111/j.1476-5381.1995.tb15933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PALMA C., MINGHETTI L., ASTOLFI M., AMBROSINI E., SILBERSTEIN F.C., MANZINI S., LEVI G., ALOISI F. Functional characterization of substance P receptors on cultured human spinal cord astrocytes: synergism of substance P with cytokines in inducing interleukin-6 and prostaglandin E2 production. Glia. 1997;21:183–193. doi: 10.1002/(sici)1098-1136(199710)21:2<183::aid-glia2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- PORTANOVA J.P., ZHANG Y., ANDERSON G.D., HAUSER S.D., MASFERRER J.L., SEIBERT K., GREGORY S.A., ISAKSON P.C. Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J. Exp. Med. 1996;184:883–891. doi: 10.1084/jem.184.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUIG S., SORKIN L.S. Formalin-evoked activity in identified primary afferent fibers: Systematic lidocaine suppresses phase 2 activity. Pain. 1996;64:345–355. doi: 10.1016/0304-3959(95)00121-2. [DOI] [PubMed] [Google Scholar]
- RAJA S.N., MEYER R.A., CAMPBELL J.N. Peripheral mechanisms of somatic pain. Anesthesiology. 1988;168:571–590. doi: 10.1097/00000542-198804000-00016. [DOI] [PubMed] [Google Scholar]
- SCHAFER M.K., NOHR D., KRAUSE J.E., WEIHE E. Inflammation-induced upregulation of NK1 receptor mRNA in dorsal horn neurones. Neuroreport. 1993;4:1007–1010. doi: 10.1097/00001756-199308000-00003. [DOI] [PubMed] [Google Scholar]
- SLUKA K.A., WESTLUND K.N. Behavioral and immunohistochemical changes in an experimental arthritis model in rats. Pain. 1993a;55:367–377. doi: 10.1016/0304-3959(93)90013-F. [DOI] [PubMed] [Google Scholar]
- SLUKA K.A., WESTLUND K.N. Centrally administered non-NMDA but not NMDA receptor antagonists block peripheral knee joint inflammation. Pain. 1993b;55:217–225. doi: 10.1016/0304-3959(93)90150-N. [DOI] [PubMed] [Google Scholar]
- SMITH J.B., WILLIS A.L. Aspirin selectively inhibits prostaglandin production in human platelets. Nature New Biol. 1971;231:235–237. doi: 10.1038/newbio231235a0. [DOI] [PubMed] [Google Scholar]
- SMITH S.J., AUGUSTINE G.J. Calcium ions, active zones and synaptic transmitter release. Trends Neurosci. 1988;11:458–464. doi: 10.1016/0166-2236(88)90199-3. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., DEWITT D.L. Prostaglandin Endoperoxide H Synthases-1 and -2. Adv. Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- SOUSA B.N., HORROCKS L.A. Development of rat spinal cord: I. Weight and length with a method for rapid removal. Dev. Neurosci. 1979;2:115–121. [Google Scholar]
- UDA R., HORIGUCHI S., ITO S., HYODO M., HAYAISHI O. Nociceptive effects induced by intrathecal administration of prostaglandin D2, E2, or F2 alpha to conscious mice. Brain Res. 1990;510:26–32. doi: 10.1016/0006-8993(90)90723-o. [DOI] [PubMed] [Google Scholar]
- VANE J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- VASKO M.R. Prostaglandin-induced neuropeptide release from spinal cord. Prog. Brain Res. 1995;104:367–380. doi: 10.1016/s0079-6123(08)61801-4. [DOI] [PubMed] [Google Scholar]
- VASKO M.R., ZIRKELBACH S.L., WAITE K.J.Prostaglandins stimulate the release of substance P from rat spinal cord slices Progress in Pharmacology and Clinical Pharmacology 1993Stuttgart, New York: Gustav Fischers Verlag; 69–89.eds. Jurna, I. & Yaksh, T.L. [Google Scholar]
- WILLINGALE H.L., GARDINER N.J., MCLYMONT N., GIBLETT S., GRUBB B.D. Prostanoids synthesized by cyclo-oxygenase isoforms in rat spinal cord and their contribution to the development of neuronal hyperexcitability. Br. J. Pharmacol. 1997;122:1593–1604. doi: 10.1038/sj.bjp.0701548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOLF C.J. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686–688. doi: 10.1038/306686a0. [DOI] [PubMed] [Google Scholar]
- YAKSH T.L.Central and peripheral mechanism for the antialgesic action of acetylsalicylic acid Aectylsalicylic Acid: New Uses for an Old Drug 1982New York: Raven Press; 137–152.eds. Barnet, J.M., Hirsh, J. & Mustard, J.F. [Google Scholar]
- YAKSH T.L., DIRIG D.M., MALMBERG A.B. Mechanism of action of nonsteroidal anti-inflammatory drugs. Canc. Invest. 1998;16:509–527. doi: 10.3109/07357909809011705. [DOI] [PubMed] [Google Scholar]
- YAKSH T.L., JESSELL T.M., GAMSE R., MUDGE A.W., LEEMAN S.E. Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature. 1980;286:155–157. doi: 10.1038/286155a0. [DOI] [PubMed] [Google Scholar]
- YANG L., MARSALA M., YAKSH T.L. Characterization of time course of spinal amino acids, citrulline, and PGE2 release after carrageenan/kaolin-induced knee joint inflammation: a chronic microdialysis study. Pain. 1996;67:345–354. doi: 10.1016/0304-3959(96)03106-5. [DOI] [PubMed] [Google Scholar]
- YASPHAL K., WRIGHT D.M., HENRY J.L. Substance P reduces tail-flick latency: implications for chronic pain syndromes. Pain. 1982;14:155–167. doi: 10.1016/0304-3959(82)90096-3. [DOI] [PubMed] [Google Scholar]