

Abstract
Experiments were designed to differentiate the mechanisms and subtype of kinin receptors mediating the changes in intracellular Ca2+ concentration ([Ca2+]i) induced by bradykinin (BK) in canine cultured tracheal epithelial cells (TECs).
BK and Lys-BK caused an initial transient peak of [Ca2+]i in a concentration-dependent manner, with half-maximal stimulation (pEC50) obtained at 7.70 and 7.23, respectively.
Kinin B2 antagonists Hoe 140 (10 nM) and [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK (1 μM) had high affinity in antagonizing BK-induced Ca2+ response with pKB values of 8.90 and 6.99, respectively.
Pretreatment of TECs with pertussis toxin (100 ng ml−1) or cholera toxin (10 μg ml−1) for 24 h did not affect the BK-induced IP accumulation and [Ca2+]i changes in TECs.
Removal of Ca2+ by the addition of EGTA or application of Ca2+-channel blockers, verapamil, diltiazem, and Ni2+, inhibited the BK-induced IP accumulation and Ca2+ mobilization, indicating that Ca2+ influx was required for the BK-induced responses.
Addition of thapsigargin (TG), which is known to deplete intracellular Ca2+ stores, transiently increased [Ca2+]i in Ca2+-free buffer and subsequently induced Ca2+ influx when Ca2+ was re-added to this buffer. Pretreatment of TECs with TG completely abolished BK-induced initial transient [Ca2+]i, but had slight effect on BK-induced Ca2+ influx.
Pretreatment of TECs with SKF96365 and U73122 inhibited the BK-induced Ca2+ influx and Ca2+ release, consistent with the inhibition of receptor-gated Ca2+ -channels and phospholipase C in TECs, respectively.
These results demonstrate that BK directly stimulates kinin B2 receptors and subsequently phospholipase C-mediated IP accumulation and Ca2+ mobilization via a pertussis toxin-insensitive G protein in canine TECs. These results also suggest that BK-induced Ca2+ influx into the cells is not due to depletion of these Ca2+ stores, as prior depletion of these pools by TG has no effect on the BK-induced Ca2+ influx that is dependent on extracellular Ca2+ in TECs.
Keywords: Bradykinin receptor, Ca2+, inositol phosphate, pertussis toxin, thapsigargin, tracheal epithelial cells
Introduction
Bradykinin (BK), one of the kinin family, is a classical mediator of inflammatory diseases of the airways and is implicated in allergic asthma (Christiansen et al., 1987; Farmer et al., 1991). In the airways, BK causes bronchoconstriction, pulmonary and bronchial vasodilatation, mucus secretion and microvascular leakage (Barnes, 1992). The physiological actions of BK are mediated through at least two distinct BK receptor subtypes, termed B1 and B2, which have been pharmacologically characterized using different kinin analogues with agonist and antagonist properties (Regoli & Barabe, 1980). Several lines of evidence demonstrate that activation of BK receptors leads to phospholipase C (PLC) activation resulting in phosphoinositide (PI) hydrolysis in the plasma membrane of different cell types (Balmforth et al., 1992; Marsh & Hill, 1992; Yang et al., 1994a; Smith et al., 1995). The resultant increase in inositol 1,4,5-trisphosphate (IP3) and diacylglycerol releases Ca2+ from internal stores and activates protein kinase C (PKC), respectively (Nishizuka, 1992; Horowitz et al., 1996).
BK has been shown to stimulate chloride secretion in native canine tracheal epithelium (Leikauf et al., 1985), but the mechanisms involved in BK-induced secretory response are not completely understood. One of possible mechanisms implicated in regulation of secretory function of trachea may be attributable to an increase in PI hydrolysis mediated by kinin B2 receptors (Marsh & Hill, 1992; Yang et al., 1994a; 1995; Luo et al., 1996) and a rise in intracellular Ca2+ ([Ca2+]i) (Marsh & Hill, 1993; Yang et al., 1994b; Smith et al., 1995; Mathis et al., 1996). The elevation of [Ca2+]i has been thoroughly characterized in these cells, which is typified by an immediate and transient peak followed by a sustained phase. Three mechanisms have been proposed to be involved in BK-induced elevation of [Ca2+]i in these cells. Mobilization of Ca2+ from internal stores is suggested by the observation that elevation of [Ca2+]i induced by BK is partially preserved despite reduction of external Ca2+ concentrations, and is associated with the generation of IP3 (Marsh & Hill, 1993; Buchan & Martin, 1991). Ca2+ influx is also likely to contribute to the BK response, because the rise in [Ca2+]i induced by BK is attenuated when external Ca2+ is removed (Marsh & Hill, 1993; Yang et al., 1994b). In many cases, agonist-induced Ca2+ influx is blocked by drugs that inhibit dihydropyridine-sensitive, voltage-gated Ca2+ channels (Nelson et al., 1988; Yang et al., 1994b). However, some workers have found no effect of such drugs on BK-induced [Ca2+]i response (Murray & Kotlikoff, 1991), and have proposed that Ca2+ influx involves dihydropyridine-insensitive, voltage-gated channels or nonspecific cation channels.
The biochemical mechanisms linking both the initial and sustained phases are not well defined in TECs. A capacitative entry model has been proposed by Putney (1993). In this model, Ca2+ influx can be activated by a process shunting second messengers such as IP metabolites, whereby the filling state of some Ca2+-stores alone is sufficient to induce Ca2+ entry. The evidence for supporting this hypothesis was obtained from studies using specific sarcoplasmic reticulum Ca2+-ATPase inhibitor, such as thapsigargin (TG), which has been proved to be useful in testing this hypothesis and emptying the intracellular Ca2+ pools without generating any known second messenger (Thastrup et al., 1990). The sustained increase in [Ca2+]i induced by TG is generally followed by an activation of Ca2+ influx, as shown in mast cells (Dar & Pecht, 1992), human platelets (Malcolm & Fitzpatrick, 1992), lacrimal acinar cells (Kwan et al., 1990) and neuronal cell lines (Takemura et al., 1991). These findings led us to suggest the existence, in canine TECs, of a similar signalling mechanism linking the filling state of intracellular Ca2+ pools and Ca2+ influx.
We have previously reported that BK can induce an increase in PI hydrolysis which appears to be mediated via the activation of kinin B2 receptors in canine cultured TECs (Luo et al., 1996). To further define the cellular mechanisms that modulate intracellular [Ca2+]i, therefore, we have undertaken these studies to clarify, in part, the nature of changes in [Ca2+]i during continued exposure to BK in TECs. These results suggest that BK induces Ca2+ release from internal stores and Ca2+ influx from the extracellular milieu by distinct mechanisms. Furthermore, these results demonstrate that the activation of a calcium-release from intracellular stores and the activation of a calcium-entry through channels are independent effects which occur following stimulation of kinin B2 receptor by BK in canine cultured TECs.
Methods
Materials
Dulbecco's modified Eagle's medium (DMEM)/Ham's nutrient mixture F-12 (F-12) medium and foetal bovine serum (FBS) were purchased from Gibco BRL (Gaithersburg, MD, U.S.A.). Myo-[3H]-inositol (18 Ci mmol−1) was from Amersham (Buckinghamshire, England). Fura-2/AM was from Molecular Probes (Eugene, OR, U.S.A.). 1-[β-[3-(4-methoxyphenyl)propoxy]-4- methoxyphenethyl]- 1H - imidazole (SKF96365), thapsigargin (TG), and 1-(6-((17β-3-methoxyestra -1,3,5(10)-trien- 17-yl)amino)hexyl) -1H -pyrrole-2,5-dione (U73122) were from Biomol (Plymouth Meeting, PA, U.S.A.). Enzymes and other chemicals were from Sigma (St. Louis, MO, U.S.A.).
Animals
Mongrel dogs, 10–20 kg, both male and female, were purchased from a local supplier. Dogs were housed indoors in the animal facilities under automatically controlled temperature and light cycle conditions and fed standard laboratory chow and tap water ad libitum. Dogs were anaesthesized with ketamine (20 mg kg−1, intramuscularly) and pentobarbitone (30 mg kg−1, intravenously). The tracheae were surgically removed.
Isolation and culture of tracheal epithelial cells
Cells were isolated essentially as described by Wu et al. (1985). The trachea was cut longitudinally through the cartilage rings, and strip epithelium was pulled off the submucosa, rinsed with phosphate buffered saline (PBS) containing 5 mM dithiothreitol, and digested with 0.05% protease XIV in PBS at 4°C for 24 h; after vigorous shaking of the strips at room temperature, 5 ml of foetal bovine serum (FBS) was added to terminate the digestion. The released cells were collected and washed twice with 50% Dulbecco's modified Eagle's medium (DMEM) and 50% Ham's nutrient F-12 medium that contained 5% FBS, nonessential amino acids, penicillin (100 u ml−1), streptomycin (100 μg ml−1), gentamicin (50 μg ml−1), and fungizone (2.5 μg ml−1). Cell number was counted and diluted with DMEM/F-12 to 2×106 cells ml−1. The cells was plated onto (1 ml well−1) 12-well and 6-well (2 ml well−1) culture plates containing glass coverslips coated with collagen for IP accumulation and Ca2+ measurement, respectively. The culture medium was changed after 24 h and then changed every 2 days.
In order to characterize the isolated and cultured TECs, an indirect immunofluorescent staining was performed as described by O'Guin et al. (1985) using AE1 and AE3 mouse monoclonal antibodies and fluorescein isothiocyanate (FITC)-labelled goat anti-mouse IgG.
Accumulation of inositol phosphate
Effect of BK on the hydrolysis of PI was assayed by monitoring the accumulation of [3H]-IP as described by Yang et al. (1994a). Cultured TECs were incubated with 5 μCi ml−1 of myo-[2-3H]-inositol at 37°C for 24 h. TECs were washed two times with and incubated in Krebs-Henseleit buffer (KHS, pH 7.4) containing (in mM): NaCl 117, KCl 4.7, MgSO4 1.1, KH2PO4 1.2, NaHCO3 20, CaCl2 2.4, glucose 1, HEPES 20 and LiCl 10 at 37°C for 30 min. After BK added at the concentration indicated, incubation was continued for another 60 min in the presence of 2 μM indomethacin (to inhibit cyclo-oxygenase) and 10 μM phosphoramidon. Reactions were terminated by addition of 5% perchloric acid followed by sonication and centrifugation at 3000×g for 15 min.
The perchloric acid soluble supernatants were extracted four times with ether, neutralized with potassium hydroxide, and applied to a column of AG1-X8, formate form, 100–200 mesh (Bio-Rad). The resin was washed successively with 5 ml of water and 5 ml of 60 mM ammonium formate-5 mM sodium tetraborate to eliminate free [3H]-inositol and glycerophosphoinositol, respectively. Total IP was eluted with 5 ml of 1 M ammonium formate-0.1 M formic acid. The amount of [3H]-IP was determined in a radiospectrometer (Beckman LS5000TA, Fullerton, CA, U.S.A.).
Measurement of intracellular Ca2+ level
[Ca2+]i was measured in confluent monolayers with the calcium-sensitive dye fura-2/AM as described by Grynkiewicz et al. (1985). Upon confluence, the cells were cultured in DMEM/F-12 with 1% FBS one day before measurements were made. The monolayers were covered with 1 ml of DMEM/F-12 with 1% FBS containing 5 μM fura-2/AM and was incubated at 37°C for 45 min. At the end of the period, the coverslips were washed twice with the physiological buffer solution containing (mM): NaCl 125, KCl 5, CaCl2 1.8, MgCl2 2, NaH2PO4 0.5, NaHCO3 5, HEPES 10, and glucose 10, pH 7.4. The cells were incubated in PBS for further 30 min to complete dye de-esterification. The coverslip were inserted into a quartz cuvette at an angle of approximately 45° to the excitation beam and placed in the temperature controlled holder of a Hitachi F-4500 spectrofluorometer (Tokyo, Japan). Continuous stirring were achieved with a magnetic stirrer.
Fluorescence of Ca2+-bound and unbound fura-2 were measured by rapidly alternating the dual excitation wavelengths between 340 and 380 nm and electronically separating the resultant fluorescence signals at emission wavelength 510 nm. The autofluorescence of each monolayer was subtracted from the fluorescence data. The ratios (R) of the fluorescence at the two wavelengths are computed and used to calculate changes in [Ca2+]i. The ratios of maximum (Rmax) and minimum (Rmin) fluorescence of fura-2 were determined by adding ionomycin (10 μM) in the presence of PBS containing 5 mM Ca2+ and by adding (EGTA 5 mM) at pH 8 in a Ca2+-free PBS, respectively. The Kd of fura-2 for Ca2+ was assumed to be 224 nM (Grynkiewicz et al., 1985).
Analysis of data
Concentration-effect curves were fitted by Graph Pad Program (GraphPad, San Diego, CA, U.S.A.). EC50 values were estimated by the same program and expressed as the mean −logEC50 (M, unless stated otherwise)±s.e.mean. Data were expressed as the mean±s.e.mean and analysed with a two-tailed Student's t-test at a P<0.01 level of significance.
Results
[Ca2+]i response of TECs to BK
The BK receptor agonists, BK, Lys-BK, and [desArg9]-BK, were found to cause a concentration-dependent elevation of [Ca2+]i levels, as measured directly in cultured TECs loaded with Fura-2 (Figure 1). The resting level of [Ca2+]i in TECs was ranged from 100–300 nM (averaged about 210 nM). In the presence of extracellular Ca2+ (1.8 mM), agonist addition to the Fura-2-loaded cells resulted in a rapid increase in [Ca2+]i and declined to the resting level within 1 min. BK, Lys-BK, and [desArg9]-BK, each at a concentration of 10 μM, caused a maximal increase in the [Ca2+]i above the resting level of 250±21, 235±29 and 50±5 nM, n=8, respectively (Figure 1). The sustained phase of [Ca2+]i was not apparent in canine TECs. The pEC50 values of BK and Lys-BK in inducing the initial peak of [Ca2+]i were 7.23 and 7.70, respectively. The order of potency of these agonists in inducing the initial rise in [Ca2+]i was BK=Lys-BK> [desArg9]-BK.
Figure 1.
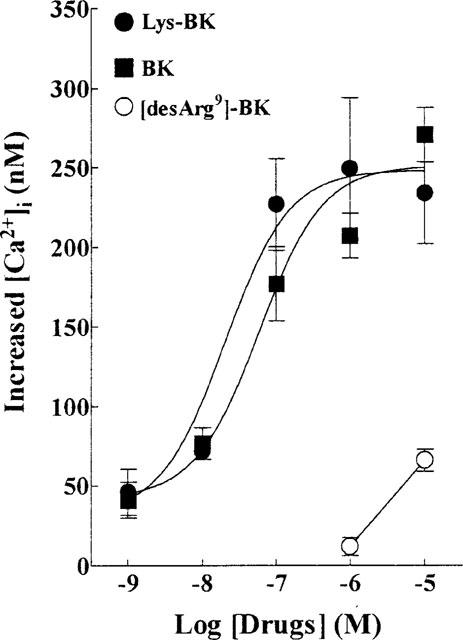
Dependence of the rise in [Ca2+]i on BK, Lys-BK, and [desArg9]-BK concentrations. Confluent cultures of TECs on glass coverslips were loaded with 5 μM Fura-2/AM and fluorescent measurement of [Ca2+]i was carried out in a dual excitation wavelength spectrophotometer, with excitation at 340 and 380 nm. The log concentration-effect curves of the BK agonists, BK-, Lys-BK-, and [desArg9]-BK-induced rising in [Ca2+]i were derived from six separate experiments. Results represent the mean±s.e.mean as the increase above the resting level (210±15 nM).
Effect of kinin B2 antagonists on BK-induced [Ca2+]i
To further elucidate the type of BK receptor that is involved in the increase of [Ca2+]i, selective kinin B2 antagonists [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK and Hoe 140 were used to antagonize the BK-induced [Ca2+]i response. As shown in Figure 2, preincubation of TECs with these antagonists inhibited the BK-induced increase in [Ca2+]i. The log concentration-effect curves of BK were shifted to the right in a nearly parallel fashion, without changing the maximal response, upon addition of 1 μM [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK and 10 nM Hoe 140. The pEC50 values of BK were decreased from 7.06 to 6.03 and 6.11, in the presence of [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK and Hoe 140. The dissociation constants (pKB) were calculated from the dose-ratios and were 6.99 and 8.90 for [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK and Hoe 140, respectively. In contrast, the kinin B1 receptor antagonist, [desArg9, Leu8]-BK (10 μM), did not change the BK-induced [Ca2+]i response (data not shown). These results indicate that the receptors mediating BK-induced increase in [Ca2+]i had similar pharmacological properties to those of kinin B2 receptors (Regoli & Barabe, 1980; Marsh & Hill, 1993; Yang et al., 1994b).
Figure 2.
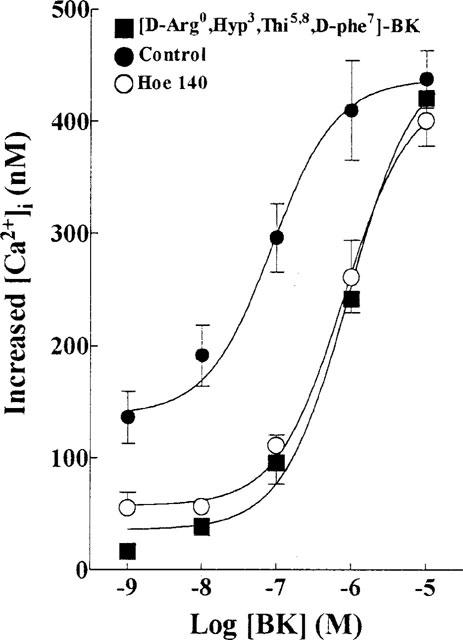
Antagonism of BK-induced changes in [Ca2+]i in TECs by Hoe 140 and [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK. The log concentration-effect curves of BK for the increase in [Ca2+]i were constructed in the absence (control) and presence of Hoe 140 (10 nM) or [D-Arg0, Hyp3, Thi5,8, D-Phe7]-BK (1 μM). TECs were incubated with each of these antagonists for 30 min before the addition of increasing concentrations of BK. Values represent mean±s.e.mean derived from six separate experiments as the increase above the resting level (221±17 nM).
Effect of pertussis toxin and cholera toxin on BK-induced IP accumulation and Ca2+ mobilization
To characterize the identity of the coupling G protein in the BK-induced signal transduction mechanism, cultured TECs were preincubated with either PTX (100 ng ml−1) or CTX (10 μg ml−1) for 24 h, then IP accumulation and [Ca2+]i changes were measured. As shown in Figure 3, both IP accumulation and [Ca2+]i changes evoked by BK were not significantly attenuated by pretreating TECs with either PTX or CTX. These toxins alone did not change the basal levels of IP and [Ca2+]i in TECs.
Figure 3.
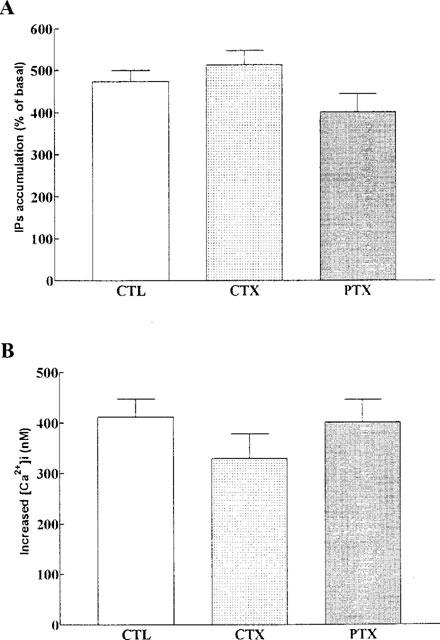
Effects of pertussis toxin and cholera toxin on BK-induced IPs accumulation and [Ca2+]i changes in TECs. Cells were preincubated with either pertussis toxin (PTX, 100 ng ml−1) or cholera toxin (CTX, 10 μg ml−1) for 24 h and then exposed to BK (10 μM). (A) [3H]-IPs and (B) [Ca2+]i were determined as described under Methods. Values represent mean±s.e.mean of six separate experiments. The basal level of IPs accumulation was 15,300±2300 d.p.m. well−1.
Effects of extracellular Ca2+ on BK-induced IP accumulation
To determine whether Ca2+ influx is required for the activation of PLC, BK-induced IP accumulation was conducted under Ca2+-free KHS buffer. Results in Figure 4 show the dependence of BK-induced IP accumulation on extracellular Ca2+. TECs preincubated in Ca2+-free KHS or in Ca2+-free KHS plus EGTA (0.5 mM) for 30 min, caused a significant reduction of the BK-induced IP accumulation (P<0.001, n=3, as compared with control). Furthermore, BK-induced IP accumulation in TECs was significantly inhibited by pretreatment with Ca2+-channel blockers verapamil (10 μM), nifedipine (10 μM), diltiazem (10 μM), and Ni2+ (5 mM) (Figure 4, P<0.001, n=3, as compared with control).
Figure 4.
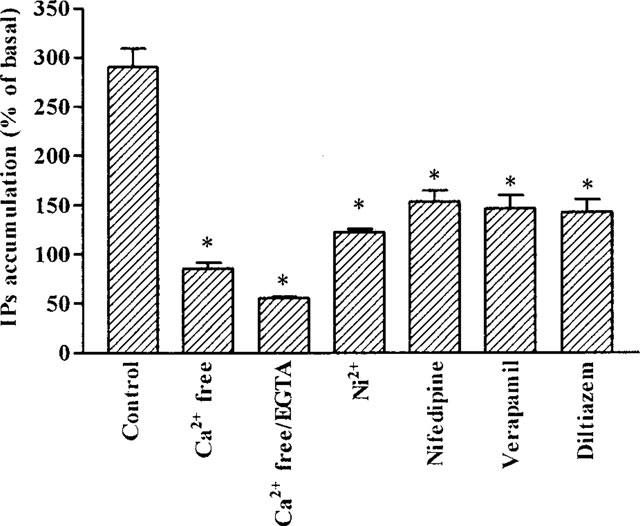
Calcium dependence of BK-stimulated IPs accumulation in TECs. Cells were incubated either in KHS, Ca2+-free KHS, Ca2+-free KHS plus 0.5 mM EGTA, Ni2+ (5 mM), nifedipine (10 μM), verapamil (10 μM) or diltiazem (10 μM) with 10 mM LiCl for 30 min. Then BK (10 μM) was added and continuously incubated at 37°C for 60 min. [3H]-IPs were determined as described under Methods. Values represent mean±s.e.mean of triplicate determinations in three separate experiments. *P<0.001, as compared with that of cells treated with BK alone in KHS buffer. The basal level of IPs accumulation was 17350±1670 d.p.m. well.−1
Effects of extracellular Ca2+ on BK-induced Ca2+ mobilization
To further define the mechanisms underlying the [Ca2+]i changes induced by BK, experiments were performed to examine the changes in [Ca2+]i induced by BK with or without Ca2+. As shown in Figure 5, trace A indicates the increase in [Ca2+]i in the presence of extracellular Ca2+ (1.8 mM). Upon addition of BK, an immediate increase in [Ca2+]i was seen and reached a peak (780±60 nM) within 15 s. The peak was then declined close to the resting level within 30 s. In the absence of extracellular Ca2+, BK induced an immediate, transient, increase in [Ca2+]i similar to that observed in the presence of extracellular Ca2+, but of smaller magnitude (490±57 nM). The peak then declined close to the resting level within 30 s (Figure 5B). When Ca2+ (1.8 mM) was added after the addition of BK to the solution, a small and sustained increase in [Ca2+]i was seen (Figure 5B). Reversal of the addition order (i.e. Ca2+ followed by BK) revealed that re-addition of Ca2+ to Ca2+-free buffer did not significantly change in [Ca2+]i, but subsequent addition of BK yielded a profile that was similar to control (data not shown). These results suggest that the increase in [Ca2+]i required two sources of Ca2+: (a) stored intracellular Ca2+ that is immediately but transiently mobilized upon receptor occupation; and (b) an influx of external Ca2+ into the cells that accounts for the sustained high level of [Ca2+]i. It was worth noting that there was no apparently sustained phase of [Ca2+]i induced by BK in the presence of Ca2+ in the medium. Therefore, the agonist-mediated Ca2+ influx was observed by re-addition of Ca2+ (1.8 mM) to the Ca2+-free buffer in the following studies.
Figure 5.
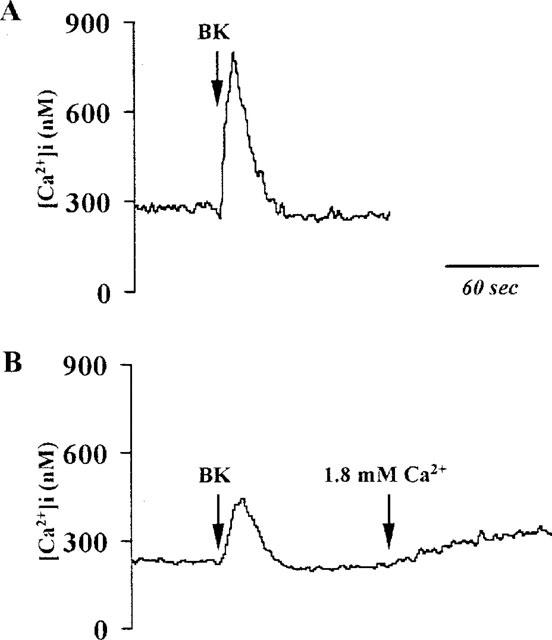
Effect of extracellular Ca2+ on BK-stimulated changes in [Ca2+]i Trace (A): Cells were stimulated by BK (10 μM) to the buffer containing Ca2+ (1.8 mM). An immediate increase in [Ca2+]i was seen. Trace (B): Cells were incubated in the absence of extracellular Ca2+ and stimulated by BK (10 μM). The response showed a transient increase of [Ca2+]i similar to trace (A) but to a substantially smaller degree. When Ca2+ (1.8 mM) was added, a sustained increase of [Ca2+]i occurred. The traces shown are typical of six separate experiments.
Effect of TG on [Ca2+]i
In a number of cells, emptying of intracellular Ca2+ stores by TG stimulates Ca2+ influx into the cytosol (Putney, 1993). Thus, the characteristics of Ca2+ influx in TECs were investigated using a Ca2+ removal-Ca2+ restoration protocol. As shown in Figure 6, cells were loaded with fura-2/AM in the presence of external Ca2+ and then incubated in Ca2+-free buffer to reach a steady baseline (100±29 nM, n=5). The subsequent addition of TG resulted in an increase in [Ca2+]i (362±25 nM, n=5) and then decayed to the resting levels of [Ca2+]i within 1 min. The re-addition of 1.8 mM Ca2+ to the Ca2+-free buffer resulted in an increase in [Ca2+]i to 325±37 nM (n=5). This magnitude of increase with re-addition of Ca2+ was not seen in TECs that were not exposed to TG (data not shown). These results indicate that depletion of intracellular Ca2+ stores by TG induces Ca2+ influx into the cytosol in TECs.
Figure 6.
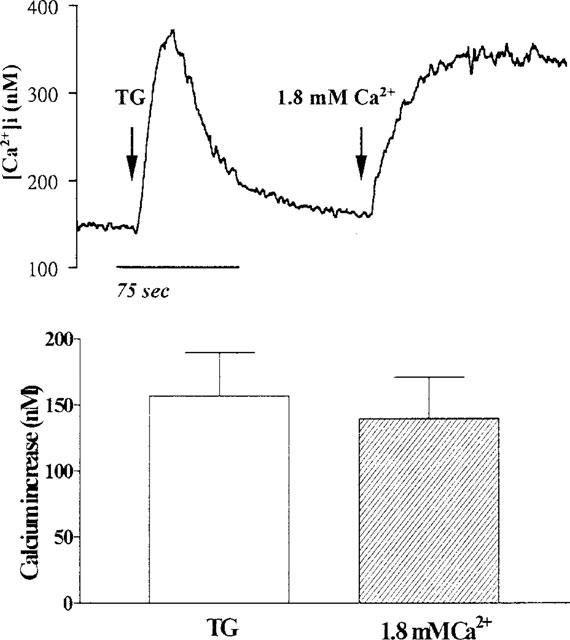
Effect of TG on [Ca2+]i in TECs. The cells were incubated in Ca2+-free buffer, exposed to TG (10 μM), and then Ca2+ (1.8 mM) was re-added to the buffer, as indicated by arrows. The data expressed as mean±s.e.mean of five separate experiments are shown in bar graph.
Effect of TG on BK-induced increase in [Ca2+]i
To further investigate the effect of depleting intracellular Ca2+ stores on BK-induced Ca2+ influx in TECs, the cells were pretreated with TG for 30 min and then exposed to BK in Ca2+-free buffer. As shown in Figure 7, pretreatment of TECs with TG completely abolished the BK-induced increase in [Ca2+]i which was seen in non-TG treated cells. However, re-addition of Ca2+ (1.8 mM) to the cells in Ca2+-free buffer caused a sustained increase in [Ca2+]i (186±21 nM, n=5) similar to that of non-TG treated cells (Figure 5). These results suggest that TG and BK release Ca2+ from the same intracellular stores in TECs.
Figure 7.
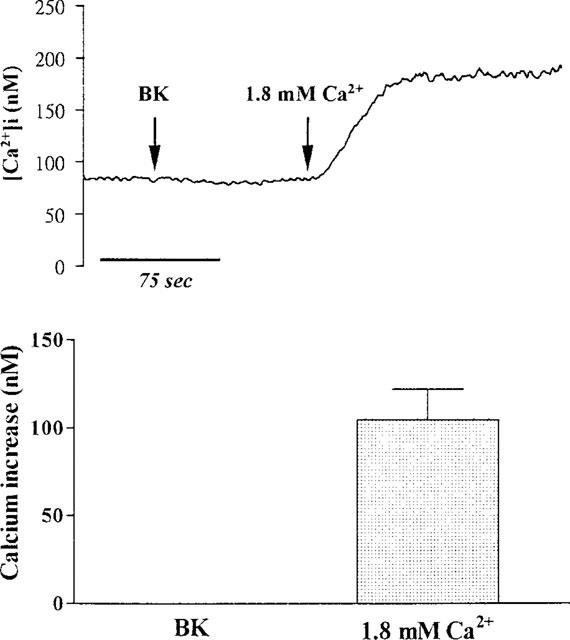
Effect of TG on BK-induced increase in [Ca2+]i. TECs were pretreated with 10 μM TG for 30 min and then incubated in Ca2+-free buffer. After reaching the steady baseline, the cells were exposed to BK (10 μM), and then Ca2+ (1.8 mM) was re-added to the buffer, as indicated by arrows. The data expressed as mean±s.e.mean of five separate experiments are shown in bar graph.
Effects of Ca2+-channel blockers on BK-induced increase in [Ca2+]i
In order to determine whether voltage-gated Ca2+ channels could contribute to the Ca2+ entry induced by the emptying of intracellular Ca2+ stores, we tested the effects of verapamil and diltiazem, inhibitors of voltage-gated Ca2+ channels, on BK-induced Ca2+ influx in TECs. As shown in Figure 8, in Ca2+-free buffer, addition of BK produced an increase in [Ca2+]i (212±19 nM, n=5), indicative of Ca2+ release from intracellular Ca2+ stores. [Ca2+]i reached a peak within 15 s and subsequently returned to the resting level within 30 s after addition of BK. Re-addition of Ca2+ (1.8 mM) to Ca2+-free buffer after BK led to an increase in [Ca2+]i (225±23 nM, n=5), indicative of Ca2+ influx through an agonist-activated pathway. Pretreatment of TECs with verapamil and diltiazem did not significantly change the BK-induced increase in [Ca2+]i compared with control, whereas the rise in [Ca2+]i seen after re-addition of Ca2+ (1.8 mM) to the cells in Ca2+-free buffer was significantly attenuated by these two Ca2+-channel blockers. Ni2+ (5 mM) also displayed a similar effect to those of verapamil and diltiazem on BK-induced Ca2+ response in TECs, when added prior to BK. These results suggest that BK-induced Ca2+ influx is partially mediated through the voltage-gated Ca2+ channels in TECs.
Figure 8.
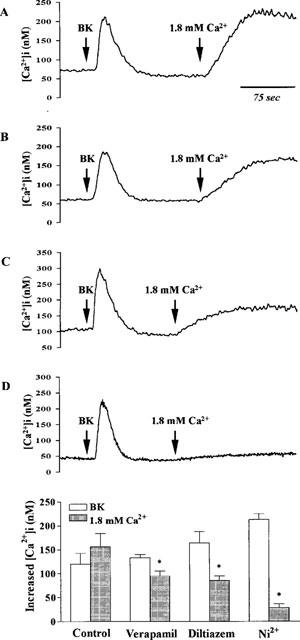
Effects of Ca2+-channel blockers on BK-induced increase in [Ca2+]i. TECs were preincubated with (A) vehicle, (B) verapamil (10 μM), (C) diltiazem (10 μM), and (D) Ni2+ (5 mM) for 30 min and then incubated in Ca2+-free buffer. After reaching the steady baseline, the cells were exposed to BK (10 μM), and then Ca2+ (1.8 mM) was re-added to the buffer, as indicated by arrows. The data expressed as mean±s.e.mean of five separate experiments are shown in bar graph. *P<0.01, as compared with Ca2+ influx induced by BK in control cells.
Effect of SKF96365 on BK-induced increase in [Ca2+]i
As a further comparison to reports in other cell types of another Ca2+ influx pathway dependent on the emptying of intracellular Ca2+ stores, TECs were incubated with SKF96365, an agent that has been found to selectively inhibit receptor-gated Ca2+ influx (Merrit et al., 1990). As shown in Figure 9, pretreatment of TECs with SKF96365 (10 μM) did not significantly change the BK-induced release of sequestered Ca2+ from intracellular Ca2+ stores. However, BK-induced Ca2+ influx into TECs was significantly inhibited by pretreatment with SKF96365, when Ca2+ (1.8 mM) was re-added to the cells in Ca2+-free buffer, as compared with non-treated cells as shown in Figure 8. These results demonstrate that SKF96365 blocks BK-induced Ca2+ influx but has no significant effect on BK-induced Ca2+ release from intracellular stores.
Figure 9.
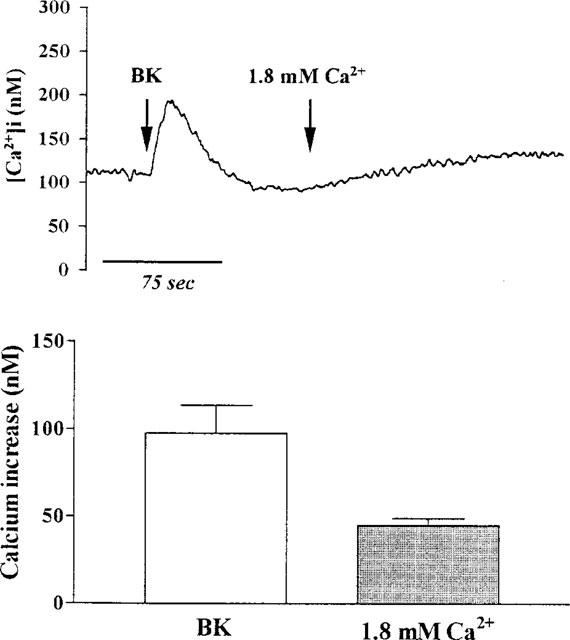
Effect of SKF96365 on BK-induced increase in [Ca2+]i. TECs were pretreated with SKF96365 (10 μM) for 30 min and then incubated in Ca2+-free buffer. After reaching the steady baseline, the cells were exposed to BK (10 μM), and then Ca2+ (1.8 mM) was re-added to the buffer, as indicated by arrows. The data expressed as mean±s.e.mean of five separate experiments are shown in bar graph. *P<0.01, as compared with Ca2+ influx induced by BK in control cells.
Effect of PLC inhibitor U73122 on BK-induced increase in [Ca2+]i
BK is known to cause release of intracellular Ca2+ by activation of PLC and generation of IP3. We therefore examined whether PLC was involved in the influx of Ca2+ induced by BK. Data in Figure 10 show that addition of U73122 (a specific PLC inhibitor) alone had no effect of [Ca2+]i in TECs incubated in Ca2+-free buffer. In contrast, pretreatment of TECs with U73122 (10 μM) almost completely abolished the BK-induced rise in [Ca2+]i and did not prevent the increase in [Ca2+]i following addition of Ca2+ (1.8 mM) to the cells in Ca2+-free buffer, as compared with non-treated cells as shown in Figure 8. These results suggest that the effect of U73122 appeared to involve a direct action on Ca2+ release from intracellular stores since BK-induced rise in [Ca2+]i was inhibited.
Figure 10.
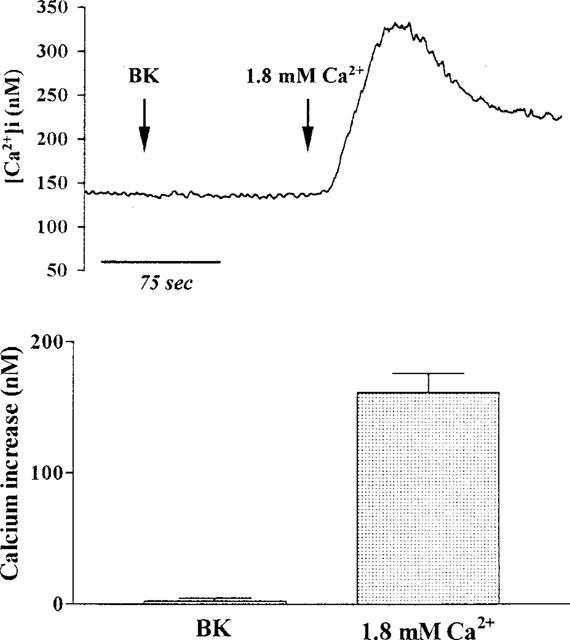
Effect of U73122 on BK-induced increase in [Ca2+]i. TECs were pretreated with U73122 (10 μM) for 30 min and then incubated in Ca2+-free buffer. After reaching the steady baseline, the cells were exposed to BK (10 μM), and then Ca2+ (1.8 mM) was re-added to the buffer, as indicated by arrows. The data expressed as mean±s.e.mean of five separate experiments are shown in bar graph. *P<0.01, as compared with BK-induced increase in [Ca2+]i in control cells.
Discussion
It has been established that kinin B2 receptors are coupled to PLC and IP3-dependent mobilization of Ca2+ (Marsh & Hill, 1993; Yang et al., 1994b), but, in addition, the influx of extracellular Ca2+ also plays an important role in the regulation of [Ca2+]i (Marsh & Hill, 1993; Yang et al., 1994b). The present study demonstrates that, in canine TECs, BK, Lys-BK, and [desArg9]-BK produced a concentration-dependent rapid increase in [Ca2+]i and declined to the resting level. The present differences in the negative log concentration-response curves for the effects of BK, Lys-BK, and [desArg9]-BK on the [Ca2+]i changes in canine TECs are similar to those reported by others using different tissues and cell preparations (Regoli & Barabe, 1980; Marsh & Hill, 1993; Yang et al., 1994b). The results obtained from the Ca2+ mobilization induced by these agonists show that the order of potency was BK=Lys-BK>[desArg9]-BK (Figure 1). BK has been suggested to induce its effects through at least two types of receptors, which have been characterized as kinin B1 and B2 receptors (Regoli & Barabe, 1980). BK and Lys-BK have high affinity for B2 receptors and low affinity for B1 receptors. In contrast, [desArg9]-BK has high affinity for B1 receptors, but low affinity for the B2 receptors. Therefore, our findings reflect the presence of kinin B2 receptors in canine TECs. Furthermore, our results demonstrate that the BK receptors coupled to Ca2+ signal have high affinity for Hoe 140 and [D-Arg0, Hyp3, Thi5.8, D-Phe7]-BK with pKB values of 8.9 and 6.99 (Figure 2), respectively, which are of the same order of magnitude as those obtained in functional studies using the kinin B2 receptor enriched systems (Regoli & Barabe, 1980; Marsh & Hill, 1993; Lembeck et al., 1991; Yang et al., 1994a,1994b). Therefore, these receptors can be classified as kinin B2 receptors in canine cultured TECs.
It has been well known that G proteins are involved in receptor coupling to PLC activity for many agonists (Sternweis & Smrcka, 1992). Our results demonstrate that BK-stimulated IP accumulation and Ca2+ mobilization is not sensitive to inhibition by PTX and CTX (Figure 3). These results suggests that a coupling process occurs in these cells through a mechanism which is mediated through a PTX- or CTX-insensitive G protein and consistent with the results reported for several cell types by others (Etscheid & Villereal, 1989; Gutowski et al., 1991; Yang et al., 1994a).
BK-stimulated IP accumulation and [Ca2+]i change is partially dependent on the presence of extracellular Ca2+ (Figure 4). This calcium dependence is comparable to the IP response to agonists in several types of cells (Fisher et al., 1987; Eberhard & Holz, 1991; Yang et al., 1994a,1994b). In the present study, TECs exposed to BK, caused an accumulation of IP in the presence of external Ca2+. However, Ca2+-free buffer or inclusion of EGTA (0.5 mM) almost completely abolished the BK-induced increase in IP accumulation. In addition, the BK-induced IP accumulation and [Ca2+]i change was significantly inhibited by the Ca2+-channel blockers verapamil and diltiazem (Figures 4 and 8), suggesting that Ca2+ influx may be required for the maximal activation of PLC. Furthermore, both influx of Ca2+ and mobilization of sequestered intracellular Ca2+ have been implicated in BK-induced elevation of [Ca2+]i, but the relative importance of these two pathways may be different in various cell types (Nelson et al., 1988; Somlyo & Somlyo, 1994). In our study, depletion of external Ca2+ attenuated the initial peak [Ca2+]i response to BK and the sustained [Ca2+]i was not seen (Figure 5), suggesting that influx of extracellular Ca2+ may contribute to both phases of [Ca2+]i in response to BK. The initial elevation of [Ca2+]i still occurs in the absence of external Ca2+ but to a substantially less extent than that measured in the presence of the external Ca2+ (Figure 5), indicating that BK-induced release of Ca2+ from the intracellular stores is due to the generation of IP3. This hypothesis was further conformed by the results that pretreatment of TECs with U73122 completely abolished the BK-induced increase in [Ca2+]i (Figure 10). The other important observation was that addition of Ca2+ (1.8 mM) to the cells in Ca2+-free buffer rapidly brought [Ca2+]i to the sustained plateau level of [Ca2+]i (Figure 5). Our data provide the evidence that the two phases of BK-stimulated increase in [Ca2+]i are mediated by two different mechanisms. An increase in intracellular [Ca2+]i is generally caused by its release from the intracellular stores and by entry through the cell membrane from extracellular milieu (van Breemen & Saida, 1989; Horowitz et al., 1996).
However, the nature of the Ca2+ pools involved in the regulation of Ca2+ entry has not been identified in TECs. Several lines of evidence display the ability of TG to deplete the IP3-sensitive Ca2+ pools. TG-induced stimulation of Ca2+ entry appears to be a consequence of the depletion of IP3-sensitive Ca2+ stores in rat parotid acinar cells (Takemura et al., 1991), macrophages (Randriamampita & Trautman, 1990), endothelial cells (Schilling et al., 1992), and vascular smooth muscle cells (Xuan et al., 1992). The results in this study show that TG depletes intracellular Ca2+ stores and activates an influx pathway for Ca2+ in TECs (Figure 6). The transient rise in [Ca2+]i induced by BK was abolished by pretreatment with TG (Figure 7), suggesting that BK and TG release sequestered Ca2+, at least in part, from a common intracellular Ca2+ stores. Moreover, pretreatment of TECs with TG did not affect the BK-induced Ca2+ influx when Ca2+ was re-added to the cells in Ca2+-free buffer (Figure 7). We therefore hypothesize that mobilization of Ca2+ from TG-sensitive intracellular Ca2+ pools is a key signal for initiating the occurrence of the sustained phase. This was conformed by our results that BK stimulated Ca2+ release from intracellular stores in canine TECs in the absence of extracellular Ca2+ and subsequent addition of Ca2+ to the cells in Ca2+-free buffer induced a rapid transient increase in [Ca2+]i (Figure 8), suggesting that the depletion of intracellular Ca2+ pools is a signal sufficient for the activation of Ca2+ influx. This is consistent with previous studies in rat parotid acinar cells (Takemura et al., 1989), vascular smooth muscle cells (Xuan et al., 1992), and human tracheal smooth muscle cells (Amrani et al., 1995), indicating a putative role of TG-sensitive Ca2+ stores in the regulation of Ca2+ influx. Nevertheless, the fact that the TG per se activates the Ca2+ entry pathway addresses the question whether the effect of TG on Ca2+ influx could superimpose the effect of BK on this response.
The characteristics of the Ca2+ entry mechanism are not completely understood in TECs. In several cell types, it is conceivable that both voltage-gated and receptor-gated Ca2+-channels contribute to elevate the intracellular [Ca2+]i (Horowitz et al., 1996). In the current study, voltage-gated Ca2+-channels blockers, verapamil and diltiazem, partially inhibited BK-induced initial transient [Ca2+]i in the absence of extracellular Ca2+ and also the sustained [Ca2+]i when Ca2+ was re-added to the cells in Ca2+-free buffer (Figure 8). These results suggest that Ca2+ influx induced by BK is mediated at least in part by voltage-gated Ca2+-channels in TECs. On the other hand, the existence of Ca2+-channels distinct from voltage-gated Ca2+-channels referred to as receptor-gated Ca2+-channels, is likely in TECs. This hypothesis was conformed by the results that SKF96365 inhibited the sustained [Ca2+]i response to BK again without affecting its ability to release Ca2+ from the intracellular stores (Figure 9). SKF96365 has been shown to inhibit the agonist-induced Ca2+ entry in several cell types (Merritt et al., 1990). The mechanism by which a decrease in Ca2+ content in the endoplasmic reticulum induces the opening of the plasma membrane Ca2+ channels is still unknown in TECs. In other cell types, it has been suggested that the depletion of intracellular Ca2+ pools induces the release of a soluble mediator called ‘Calcium Influx Factor' (Parekh et al., 1994; Randriamampita & Tsien, 1993). In human neutrophils and rat hepatocytes, it has been shown that TG is able to activate receptor-operated Ca2+ channels (Foder et al., 1989; Kass et al., 1990).
The putative PLC inhibitor U73122 was found to inhibit the initial transient [Ca2+]i induced by BK in TEC incubated in Ca2+-free buffer (Figure 10). However, there was no effect on BK-induced Ca2+ influx when Ca2+ was re-added to the cells in this buffer (Figure 10). This effect of U73122 is consistent with the proposed mechanism of action of the aminosteroid, acting as an inhibitor of PLC (Thompson et al., 1994). It is generally accepted that BK-induced release of intracellular Ca2+ in TECs is linked to activation of PLC and is probably mediated by IP3. A similar effect of U73122 inhibiting IP generation and Ca2+ mobilization has been reported in cultured neuroblastoma cells (Thompson et al., 1994).
In conclusion, our results demonstrate that, in canine TECs, BK stimulates PLC-mediated PI hydrolysis and Ca2+ mobilization via kinin B2 receptors and that this coupling process is not sensitive to PTX- or CTX-treatment. BK stimulates Ca2+ release from the intracellular stores and Ca2+ entry through calcium channels. These results suggest that the initial peak results from IP3-mediated Ca2+ release from intracellular stores and that sustained elevation of [Ca2+]i is due to an agonist-dependent influx from the extracellular milieu. Furthermore, pretreatment of TECs with TG or U73122 is able to prevent the BK-induced Ca2+ release from intracellular stores while it has no effect on the BK-induced Ca2+ entry, indicating that the BK-induced Ca2+ entry is indeed independent from the depletion of intracellular Ca2+. This is further supported by experiments performed with SKF96365, which did not affect the BK-induced Ca2+ release from intracellular stores while it strongly inhibited the BK-induced Ca2+ entry. Taken together, these results suggest that the activation of a Ca2+-release from intracellular stores and the activation of Ca2+-entry through channels are independent effects which might occur following stimulation of kinin B2 receptor by BK in cultured canine TECs.
Acknowledgments
This work was supported by grants CMRP-680 from Chang Gung Medical Research Foundation and NSC86-2314-B182-107 (CMY), and NSC86-2314-B182-024 (LSF) from National Science Council, Taiwan.
Abbreviations
- BK
bradykinin
- CTX
cholera toxin
- DMEM
Dulbecco's modified Eagle's medium
- F-12
Ham's nutrient F-12 medium
- IP
inositol phosphate
- IP3
inositol 1,4,5-trisphosphate
- KHS
Krebs-Henseleit buffer
- PBS
phosphate-buffered saline
- PI
phosphoinositide
- PKC
protein kinase C
- PLC
phospholipase C
- PTX
pertussis toxin
- SKF96365
1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole
- TECs
tracheal epithelial cells
- TG
thapsigargin
- U73122
1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione
References
- AMRANI Y., MAGNIER C., ENOUF J., WUYTACK F., BRONNER C. Ca2+ increase and Ca2+-influx in human tracheal smooth muscle cells: role of Ca2+ pools controlled by sarco-endoplasmic reticulum Ca2+-ATPase 2 isoform. Br. J. Pharmacol. 1995;115:1204–1210. doi: 10.1111/j.1476-5381.1995.tb15026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALMFORTH A.J., PARKINSON F.E., ALTIOK N., FREDHOLM B.B. Identification of a B2-bradykinin receptor linked to phospholipase C and inhibition of dopamine stimulated cyclic AMP accumulation in the human astrocytoma cell line D384. Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;346:303–310. doi: 10.1007/BF00173543. [DOI] [PubMed] [Google Scholar]
- BARNES P.J. Modulation of neurotransmission in airways. Physiol. Rev. 1992;72:699–729. doi: 10.1152/physrev.1992.72.3.699. [DOI] [PubMed] [Google Scholar]
- BUCHAN K.W., MARTIN W. Modulation of agonist-induced calcium mobilization in bovine aortic endothelial cells by phorbol myristate and cyclic AMP but not cyclic GMP. Br. J. Pharmacol. 1991;104:361–366. doi: 10.1111/j.1476-5381.1991.tb12436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHRISTIANSEN S.C., PROOD D., COCHRANE C.G. Detection of tissue kallikrein in the bronchoalveolar lavage fluid of asthmatic patient. J. Clin. Invest. 1987;79:188–197. doi: 10.1172/JCI112782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAR O., PECHT I. Fcε receptor mediated calcium influx into mast cells is modulated by the concentration of cytosolic free Ca2+ ions. FEBS Lett. 1992;222:123–128. doi: 10.1016/0014-5793(92)81311-9. [DOI] [PubMed] [Google Scholar]
- EBERHARD D.A., HOLZ R.W. Regulation of the formation of inositol phosphates by calcium, guanine nucleotides and ATP in digitonin-permeabilized bovine adrenal chromaffin cells. Biochem. J. 1991;279:447–453. doi: 10.1042/bj2790447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ETSCHEID B.G., VILLEREAL M.L. Coupling of bradykinin receptors to phospholipase C in cultured fibroblasts is mediated by a G-protein. J. Cell. Physiol. 1989;140:264–271. doi: 10.1002/jcp.1041400211. [DOI] [PubMed] [Google Scholar]
- FARMER S.G., BURCH R.M., KYLE D.J., MARTIN J.A., MEEKER S.A., TOGO J. D-Arg[Hyp3-Thi5-D-Tic7-Tic8]-bradykinin, a potent antagonist of smooth muscle BK2 receptors and BK3 receptors. Br. J. Pharmacol. 1991;102:785–787. doi: 10.1111/j.1476-5381.1991.tb12251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FISHER S.K., DOMASK L.M., ROLAND R.M. Muscarinic receptor regulation of cytoplasmic Ca2+ concentrations in human SK-N-SH neuroblastoma cells: Ca2+ requirements for phospholipase C activation. Mol. Pharmacol. 1989;35:195–204. [PubMed] [Google Scholar]
- FODER B., SCHARFF O., THASTRUP O. Ca2+ transients and Mn2+ entry in human neutrophils induced by thapsigargin. Cell Calcium. 1989;10:477–490. doi: 10.1016/0143-4160(89)90025-0. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- GUTOWSKI S., SMRCKA A., LOWAK L., WU D., SIMON M., STERNWEIS P.C. Antibodies to the αq subfamily of guanine nucleotide-binding regulatory protein α subunits attenuate activation of phosphatidylinositol 4,5-bisphosphate hydrolysis by hormones. J. Biol. Chem. 1991;266:20519–20524. [PubMed] [Google Scholar]
- HOROWITZ A., MENICE C.B., LAPORTE R., MORGAN K.G. Mechanisms of smooth muscle contraction. Physiol. Rev. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- KASS G.E.N., LLOPIS J., CHOW S.C., DUDDU S.K., ORRENIUS S. Receptor-operated calcium influx in rat hepatocytes. J. Biol. Chem. 1990;265:17486–17492. [PubMed] [Google Scholar]
- KWAN C.Y., TAKEMURA H., OBIE J.F., THASTRUP O., PUTNEY J.W., JR Effects of Mech, thapsigargin and La3+ on plasmalemmal and intracellular Ca2+ ion transport in lacrimal acinar cells. Am. J. Physiol. 1990;258:C1006–C1015. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- LEIKAUF G.D., UEKI I.F., NADEL J.A., WIDDICOMBE J.H. Bradykinin stimulates Cl secretion and prostaglandin E2 release by canine tracheal epithelium. Am. J. Physiol. 1985;248:F48–F55. doi: 10.1152/ajprenal.1985.248.1.F48. [DOI] [PubMed] [Google Scholar]
- LEMBECK F., GRIESBACHER T., ECKHARDT M., HENKE S., BRIEPOHL G. New, long-acting, potent bradykinin antagonist. Br. J. Pharmacol. 1991;102:297–304. doi: 10.1111/j.1476-5381.1991.tb12169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUO S.-F., TSAI C.-T., WU W.-B., PAN S.-L., TSAI Y.-J., YANG C.M. Pharmacological and functional characterization of bradykinin receptors in canine cultured tracheal smooth muscle cells. Br. J. Pharmacol. 1996;119:439–445. doi: 10.1111/j.1476-5381.1996.tb16005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALCOLM K.A., FITZPATRICK F.P. Epoxieicosatrienoic acids inhibit Ca2+ entry into platelets stimulated by thapsigargin and thrombin. J. Biol. Chem. 1992;267:19854–19858. [PubMed] [Google Scholar]
- MARSH K.A., HILL S.J. Bradykinin B2 receptor-mediated phosphoinositide hydrolysis in bovine cultured tracheal smooth muscle cells. Br. J. Pharmacol. 1992;107:443–447. doi: 10.1111/j.1476-5381.1992.tb12765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARSH K.A., HILL S.J. Characteristics of the bradykinin-induced changes in intracellular calcium ion concentration of single bovine tracheal smooth muscle cells. Br. J. Pharmacol. 1993;110:29–35. doi: 10.1111/j.1476-5381.1993.tb13767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHIS S.A., CRISCIMAGNA N.L., LEEB-LUNDBERG L.M.F. B1 and B2 kinin receptors mediate distinct patterns of intracellular Ca2+ signaling in single cultured vascular smooth muscle cells. Mol. Pharmacol. 1996;50:128–139. [PubMed] [Google Scholar]
- MERRIT J.E., ARMSTRONG W.P., BENHAM C.D., HALLAM T.J., JACOB R., JAXA-CHAMIEC A., LEIGH B.K., MCCARTHY S.A., MOORES K.E., RINK T.J. SK&F96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 1990;271:515–522. doi: 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRAY R.K., KOTLIKOFF M.I. Receptor-activated calcium influx in human airway smooth muscle cells. J. Physiol. 1991;435:123–144. doi: 10.1113/jphysiol.1991.sp018501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NELSON M.T., STANDEN N.B., BRAYDEN J.E., WORLY J.F. Noradrenaline contracts arteries by activating voltage-dependent calcium channels. Nature. 1988;336:382–385. doi: 10.1038/336382a0. [DOI] [PubMed] [Google Scholar]
- NISHIZUKA Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- O'GUIN W.M., SCHERMER A., SUN T.-T. Immunofluorescence staining of keratin filaments in cultured epithelial cells. J. Tissue Culture Methods. 1985;9:123–128. [Google Scholar]
- PAREKH A.B., TERLAU H., STUHMER W. Depletion of InsP3 stores activates a Ca2+ and K+ current by means of a phosphatase and diffusable messenger. Nature. 1994;364:814–818. doi: 10.1038/364814a0. [DOI] [PubMed] [Google Scholar]
- PUTNEY J.W. Excitement about calcium signaling in inexcitable cells. Science. 1993;262:676–678. doi: 10.1126/science.8235587. [DOI] [PubMed] [Google Scholar]
- RANDRIAMAMPITA C., TRAUTMANN A. Arachidonic acid activates Ca2+ extrusion in macrophages. J. Biol. Chem. 1990;265:18059–18062. [PubMed] [Google Scholar]
- REGOLI D., BARABE J. Pharmacology of bradykinin and related kinins. Pharmacol. Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- SCHILLING W.P., CABELLO O.A., RAJAN L. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ stores in vascular endothelial cells activates the agonist-sensitive Ca2+-influx pathway. Biochem. J. 1992;284:521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH J.A.M., WEBB C., HOLFORD J., BURGESS G.M. Signal transduction pathways for B1 and B2 bradykinin receptors in bovine pulmonary endothelial cells. Mol. Pharmacol. 1995;47:525–534. [PubMed] [Google Scholar]
- SOMLYO A.P., SOMLYO A.V. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- STERWIES P.C., SMRCKA A.V. Regulation of phospholipase C by G proteins. Trends Biochem. Sci. 1992;17:502–506. doi: 10.1016/0968-0004(92)90340-f. [DOI] [PubMed] [Google Scholar]
- TAKEMURA H., HUGHES G.H., THASTRUP O., PUTNEY J.W., JR Activation of calcium entry by tumor promoter thapsigargin in parotid acinar cells. J. Biol. Chem. 1989;264:12266–12271. [PubMed] [Google Scholar]
- TAKEMURA H., OHSHIDA H., YOKASAWA N., OGUMA K., THASTRUP O. The thapsigargin-sensitive intracellular Ca2+ pool is more important in plasma membrane Ca2+ entry than the IP3-sensitive intracellular Ca2+ pool in neuronal cell lines. Biochem. Biophys. Res. Commun. 1991;180:1518–1526. doi: 10.1016/s0006-291x(05)81368-3. [DOI] [PubMed] [Google Scholar]
- THASTRUP O., CULLEN P.J., DROBAK B.K., HANLEY M.R., DAAWSON A.P. Thapsigargin, a tumor promotor discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON A.K., MOSTAFAPOUR S.P., DENLINGER L.C., BLEASDALE J.E., FISHER S.K. The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. A role for Gp in receptor compartmentation. J. Biol. Chem. 1994;266:23856–23862. [PubMed] [Google Scholar]
- VAN BREEMAN C., SAIDA K. Cellular mechanism in regulating [Ca2+]i in smooth muscle. Annu. Rev. Physiol. 1989;51:315–329. doi: 10.1146/annurev.ph.51.030189.001531. [DOI] [PubMed] [Google Scholar]
- WU R., YANKASKAS J., CHENG E., KNOWLES M.R., BOUCHER J.R. Growth and differentiation of human nasal epithelial cells in culture: serum-free, hormone-supplemented medium and proteoglycan synthesis. Am. J. Physiol. 1985;261:C1123–C1129. doi: 10.1164/arrd.1985.132.2.311. [DOI] [PubMed] [Google Scholar]
- XUAN Y.-T., WANG O.-L., WHORTON R. Thapsigargin stimulates Ca2+ entry in vascular smooth muscle cells: nicardipine-sensitive and -insensitive pathways. Am. J. Physiol. 1992;262:C1258–C1265. doi: 10.1152/ajpcell.1992.262.5.C1258. [DOI] [PubMed] [Google Scholar]
- YANG C.M., HSIA H.-C., CHOU S.-P., ONG R., HSIEH J.-T., LUO S.-F. Bradykinin-stimulated phosphoinositide metabolism in cultured canine tracheal smooth muscle cells. Br. J. Pharmacol. 1994a;111:21–28. doi: 10.1111/j.1476-5381.1994.tb14018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG C.M., HSIA H.-C., HSIEH J.-T., ONG R., LUO S.-F. Bradykinin-stimulated calcium mobilization in cultured canine tracheal smooth muscle cells. Cell Calcium. 1994b;16:59–70. doi: 10.1016/0143-4160(94)90001-9. [DOI] [PubMed] [Google Scholar]
- YANG C.M., LUO S.-F., HSIA H.C. Pharmacological characterization of bradykinin receptors in canine cultured tracheal smooth muscle cells. Br. J. Pharmacol. 1995;114:67–72. doi: 10.1111/j.1476-5381.1995.tb14906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]