

Abstract
The mode of action of reactive oxygen intermediates in cysosolic Ca2+ movements of cultured porcine aortic endothelial cells exposed to xanthine/xanthine oxidase (X/XO) was investigated.
Cytosolic Ca2+ movements provoked by X/XO consisted of an initial Ca2+ release from thapsigargin-sensitive intracellular Ca2+ stores and a sustained Ca2+ influx through cell-membrane Ca2+ channels. The Ca2+ movements from both sources were inhibited by catalase, cell-membrane permeable iron chelators (o-phenanthroline and deferoxamine), a •OH scavenger (5,5-dimethyl-1-pyrroline-N-oxide), or an anion channel blocker (disodium 4, 4′-diisothiocyano-2, 2′-stilbenedisulphonic acid), suggesting that •O2− influx through anion channels was responsible for the Ca2+ movements, in which •OH generation catalyzed by intracellular transition metals (i.e., Haber-Weiss cycle) was involved.
After an initial Ca2+ elevation provoked by X/XO, cytosolic Ca2+ concentration decreased to a level higher than basal levels. Removal of X/XO slightly enhanced the Ca2+ decrease. Extracellular addition of sulphydryl (SH)-reducing agents, dithiothreitol or glutathione, after the removal of X/XO accelerated the decrement. A Ca2+ channel blocker, Ni2+, abolished the sustained increase in Ca2+, suggesting that Ca2+ influx through cell-membrane Ca2+ channels was extracellularly regulated by the redox state of SH-groups.
The X/XO-provoked change in cellular respiration was inhibited by Ni2+ or dithiothreitol as well as inhibitors of Haber-Weiss cycle, suggesting that Ca2+ influx was responsible for •OH-mediated cytotoxicity. We concluded that intracellular •OH generation was involved in the Ca2+ movements in endothelial cells exposed to X/XO. Cytosolic Ca2+ elevation was partly responsible for the oxidants-mediated cytotoxicity.
Keywords: Endothelial cells, intracellular calcium concentration, calcium channel, anion channel, cytotoxicity, superoxide anion, hydrogen peroxide, hydroxyl radical, Haber-Weiss cycle, xanthine oxidase
Introduction
A growing body of evidence from cardiac myocytes (Josephson et al., 1991), vascular smooth muscle cells (Roveri et al., 1992; Krippeit-Drews et al., 1995), endothelial cells (Doan et al., 1994), and a variety of types of the other cells (Masumoto et al., 1990; Ikebuchi et al., 1991; Rojanasakul et al., 1993; Murata et al., 1994) indicates that the concentration of intracellular free calcium ([Ca2+]i) is increased by the exposure to reactive oxygen intermediates. It has also been reported that Ca2+ channel blockers inhibit the oxidant-provoked [Ca2+]i elevation of these cells, suggesting that functional regulation of Ca2+ channels occurs during the exposure to such oxidant species at concentrations lower than that provoke lethal cell membrane damage.
Several enzymic systems (e.g., NADPH oxidase in phagocytes or xanthine oxidase in endothelial cells) directly produce one- or two-electron reduced oxygen metabolites [i.e. superoxide anion (•O2−) and hydrogen peroxide (H2O2)] (Britigan et al., 1986; 1990a,1990b), contributing to acute inflammatory disorders such as ischaemia/reperfusion (Zweier et al., 1988; 1994a,1994b) or neutrophil-mediated lung injury (Fox, 1984; Kuroda et al., 1987). While the reactivity of these oxidant species with several biological structures is limited, mechanisms generating a further reduced oxygen metabolite, hydroxyl radical (•OH), are likely to enhance oxidative stress. We have previously described that the generation of •OH from •O2− and H2O2 catalyzed by transition metals around the target structures is involved in the endothelial cell injury provoked by the lethal oxidant exposure (Az-ma et al., 1996). However, the causal relationship of •OH generation in endothelial cells and the regulation of cytosolic Ca2+ movements by the exogenous oxidative stress has not yet been established.
The aim of this study was thus to elucidate the role of •OH generation in the cytosolic Ca2+ movements in endothelial cells exposed to reactive oxygen intermediates. The change in cellular respiration was also evaluated to index the involvement of [Ca2+]i in the oxidant-provoked cytotoxicity in endothelial cells.
Methods
Preparation of endothelial cells
Isolation and primary culture of porcine aortic endothelial (PAE) cells were performed as previously described (Az-ma et al., 1995a; 1996). The culture medium used was RD medium [1 : 1 (v v−1) RPMI 1640 medium/Dulbecco's modified Eagle's medium (DMEM)] supplemented with bicarbonate, 2 mg ml−1, HEPES, 15 mM, ampicillin, 90 μg ml−1, kanamycin, 90 μg ml−1 and 10% (v v−1) foetal bovine serum (FBS) equilibrated with 5% CO2 in air under a humidified atmosphere at 37°C (pH 7.4). PAE cells were subcultured at a 1 : 3 split ratio in collagen-coated 25 cm2 plastic flasks. The resulting subconfluent monolayers of PAE cells (passage 2) were harvested with trypsin [0.125% (w v−1)/EDTA [0.02% (w v−1) in Ca2+- and Mg2+-free Dulbecco's phosphate buffered saline (PBS), and cryopreserved in FBS containing 10% (v v−1) dimethyl sulphoxide (DMSO) at a density of 1–2×106 cells ml−1 under liquid nitrogen. PAE cells were re-suspended in the culture medium (1–2×105 cells ml−1) 3–4 days before the experiments, and seeded on fibronectin-coated glass coverslips (45×45 mm) attached to silicon rubber septa separated into four 15 mmΦ chambers (300 per well) for the measurement of [Ca2+]i. The cells were also cultured in 48-well cluster dishes for the spin trapping study and the cellular respiration assay. The endothelial cell identity was confirmed post-cryopreservation by the uptake of diiodoacetyl-low-density lipoprotein using fluorescence microscopy (>99% of the cells) (Doan et al., 1994), and by a bradykinin-dependent increase in [Ca2+]i and nitric oxide (•NO) production as previously described (Az-ma et al., 1995a, 1995b). Experiments were performed within 24 h after the cells reached to confluent monolayers.
Exogenous oxidant generating system
Production of •O2− and H2O2 was achieved by adding various amounts of xanthine oxidase to modified Hanks Balanced Salt Solution (HBSS) in the presence of xanthine at a final concentration of 100 μM. The composition of HBSS was (in mM): NaCl, 138; KCl, 4.7; CaCl2, 1.3; MgSO4, 0.8; KH2PO4, 0.4; K2HPO4, 0.3; D-glucose, 5.6; HEPES, 4.2; and diethylenetriaminepentaacetic acid (DTPA), 0.02 (pH 7.4). HBSS contained the iron chelator (DTPA) at a minimum but significant concentration to eliminate an exogenous transition metal-dependent production of •OH from commercially available xanthine oxidase (Britigan et al., 1990b; Az-ma et al., 1996). The amount of xanthine oxidase in HBSS was adjusted using a spectrophotometer (DU 640, Beckman, Fullerton, CA, U.S.A.) by the rate of uric acid generation for the initial 1 min at 25°C (λmax=295 nm, ε=11 mM−1 cm−1) before each batch of experiments. Production of •O2− was also measured as superoxide dismutase (SOD)-inhibitable reduction of ferricytochrome c and monitored spectrophotometrically at 550 nm (Pou et al., 1989). The rate of •O2− production in the reaction mixture of xanthine/xanthine oxidase containing 20 μM ferricytochrome c was calculated using an extinction coefficient of 21 mM−1 cm−1 (Table 1). The generation of oxygen free radicals (i.e., •O2− and •OH) in the incubation buffer of PAE cells was also qualitatively determined by a spin trapping study using electron paramagnetic resonance spectrometry as previously described (Az-ma et al., 1996). The exogenous generation of •OH from xanthine/xanthine oxidase was not observed except when Fe3+ was added to HBSS.
Table 1.
Production of superoxide anion from xanthine/xanthine oxidase

Measurement of [Ca2+]i
The [Ca2+]i of PAE cells was measured using a fluorescent Ca2+ indicator dye, fura-2, as previously described with modification (Az-ma et al., 1995a). PAE cell monolayers attached on a coverslip were loaded with 5 μM acetoxymethyl ester form of fura-2 in HBSS for 1 h at 25°C. The cells were then washed with HBSS to remove the dye from the extracellular space. The coverslip was placed on the stage of a fluorescence inverted microscope, combined with a computer Ca2+ analysing system (ARGUS-50/CA2, Hamamatsu Photonics, Hamamatsu, Japan). The cells were continuously perfused with HBSS containing xanthine/xanthine oxidase in the absence or the presence of various agents (1 ml min−1, 37°C). The fluorescence intensity ratio with excitation at 340/380 nm and emission at 510 nm was converted to [Ca2+]i by using an in-vitro calibration curve obtained from standard Ca2+/EGTA solutions containing 5 μM fura-2 free acid. The mean value of [Ca2+]i obtained from randomly selected 21 cells in a microscope field was considered as the [Ca2+]i of each experiment. In the preliminary experiments, we confirmed that disodium 4, 4′-diisothiocyano-2, 2′-stilbenedisulphonic acid (DIDS) possesses an autofluorescence, which influenced the change in fluorescence intensities of fura-2. Thus, PAE cells were vigorously washed with HBSS before the commencement of [Ca2+]i measurement when the cells were preincubated with DIDS.
Measurement of cellular respiration
The cellular respiration of PAE cells was assessed by the conversion of a tetrazolium dye, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT; Dojindo, Kumamoto, Japan), to its formazan by the intact electron transport systems of mitochondria in the cells, according to the assay protocol provided by Dojindo. PAE cell monolayers in 48-well cluster dishes (0.8–1.2×105 cells cm2) were washed and pre-equilibrated with HBSS containing xanthine in the absence or the presence of various drugs for 10–15 min prior to the addition of xanthine oxidase. Following 30 min exposure to xanthine oxidase at 37°C, PAE cells were gently washed with HBSS to remove xanthine/xanthine oxidase and further incubated with 0.5 mg ml−1 MTT in HBSS for 4 h. The cells were then rinsed with HBSS for the removal of MTT in the extracellular space, and the intracellularly yielded formazan was extracted by isopropanol containing 40 μM HCl. The extracts were transferred to a 96-F microtiter plate, and the absorbance related to formazan (λmax=565 nm, ε=20 mM−1 cm−1) was measured at 550 nm using a spectroscopic microplate reader (MTP-120, Corona Electric, Ibaragi, Japan). Cellular respiration was determined according to the following equation:
where the control OD represented the optical density of the extract from control culture wells without exposure to xanthine/xanthine oxidase, while the sample OD represented that from culture wells exposed to the oxidant-generating system in the presence or the absence of various agents. The blank OD was obtained from the extract of culture wells without incubation with MTT. Each sample was measured in duplicate.
Statistical analysis
Data were expressed as mean±s.e.mean. One- or two-factor(s) multiple comparisons were performed using analysis of variance followed by the t-test with Bonferroni's correction.
Reagents
Dulbecco's modified Eagle's medium (DMEM) and RPMI 1640 were purchased from Gibco BRL (Grand Island, NY, U.S.A.). N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulphonic acid] (HEPES), ampicillin, kanamycin, diethylenetriaminepentaacetic acid (DTPA), trypsin, xanthine, xanthine oxidase, allopurinol, superoxide dismutase (SOD), catalase, deferoxamine mesylate, and reduced form of glutathione (GSH) were obtained from Sigma (St. Louis, MO, U.S.A.). Foetal bovine serum (FBS) was from JRH Biosciences (Lenaxa, KS, U.S.A.). Di-iodoacetyl-low-density lipoprotein was from Funakoshi (Tokyo, Japan). Ferricytochrome c was from Boehringer Mannheim (Tokyo, Japan). Acetoxymethyl ester form of fura-2 (fura-2/AM), 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), disodium 4, 4′-diisothiocyano-2, 2′-stilbenedisulphonic acid (DIDS), and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT) were from Dojindo Laboratories (Kumamoto, Japan). Dimethyl sulphoxide (DMSO), disodium ethylenediaminetetraacetate (EDTA), glycoletherdiaminetetraacetic acid (EGTA), 1,10-phenanthroline hydrochloride (o-phenanthroline), and dithiothreitol (DTT) were from Katayama (Osaka, Japan). All other chemicals were of analytical quality.
Results
Reciprocal action of •O2− and H2O2 in the cytosolic Ca2+ movements provoked by xanthine/xanthine oxidase
The [Ca2+]i, of PAE cells was not influenced by a single application of 100 μM xanthine, while it was promptly increased by the simultaneous exposure to xanthine oxidase at concentrations equal or higher than 0.01 u ml−1 (Figure 1a). However, PAE cells were lysed or detached from monolayers by exposure to 0.04 u ml−1 xanthine oxidase within 15 min. Therefore, effects of various drug interventions on the cytosolic Ca2+ movements were evaluated by using 0.02 u ml−1 xanthine oxidase. During the exposure of PAE cells to xanthine/xanthine oxidase, [Ca2+]i was elevated to a maximum level, then gradually declined to levels higher than that observed before the exposure. Allopurinol (400 μM), an inhibitor of xanthine oxidase, abolished the rise in [Ca2+]i (Figure 1b and Table 2), confirming that the observed [Ca2+]i elevation was due to the exposure to xanthine oxidase. Non-selective Ca2+ channel blockers, Ni2+ (2.0 mM; Figure 2a) or Co2+ (2.0 mM, not shown), decreased [Ca2+]i to basal levels after an initial transient increase in [Ca2+]i. The peak [Ca2+]i of the transient was significantly smaller than that observed in the absence of Ni2+ (Table 2). The removal of this Ca2+ channel blocker caused a sustained increase in [Ca2+]i (Figure 2a). To further evaluate these modulatory effects of Ni2+ on xanthine/xanthine oxidase-provoked cytosolic Ca2+ movements, thapsigargin which is known to deplete intracellular Ca2+ stores (Doan et al., 1994) was added to PAE cells in the presence of Ni2+. Preloading of PAE cells with 0.2 μM thapsigargin induced a transient increase in [Ca2+]i, while potently suppressed the [Ca2+]i elevation provoked by the following addition of xanthine oxidase (Figure 2b). These results indicated that the [Ca2+]i elevation provoked by xanthine/xanthine oxidase at concentrations used in the present experiment was dependent on a sustained Ca2+ influx from the extracellular space through Ca2+ channels during and following the initial Ca2+ release from thapsigargin-sensitive intracellular Ca2+ sources.
Figure 1.
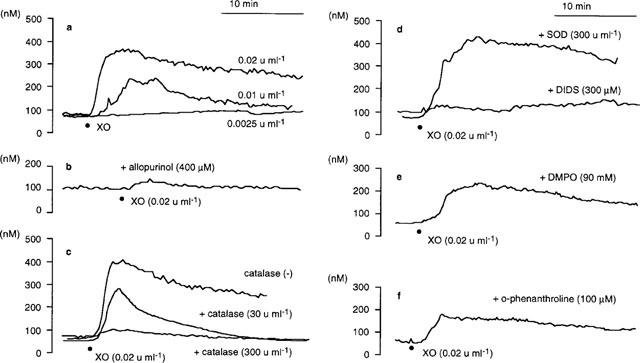
Representative recordings of the change in [Ca2+]i of cultured porcine aortic endothelial (PAE) cells exposed to xanthine/xanthine oxidase in the absence or the presence of various oxidant inhibitors at 37°C. [Ca2+]i was measured by using fura-2 fluorometry: (a) PAE cells were perfused with 100 μM xanthine-containing HBSS (pH 7.4, 1 ml min−1). (•) Addition of xanthine oxidase (XO) at indicated concentrations; (b) same condition as in (a) except that HBSS contained allopurinol (400 μM); (c) same condition as in (a) except that HBSS contained catalase (30, 300 u ml−1); (d) PAE cells were perfused with xanthine-containing HBSS in the presence of superoxide dismutase (SOD, 300 u ml−1) or 4, 4′-diisothiocyano-2, 2′-stilbenedisulphonic acid (DIDS, 300 μM). DIDS was removed with xanthine-containing HBSS following a 15 min pre-incubation before the exposure of the cells to xanthine oxidase; (e) same condition as in (a) except that HBSS contained 5,5-dimethyl-1-pyrroline-N-oxide (DMPO, 90 mM); (f) same condition as in (a) except that HBSS contained o-phenanthroline (100 μM). The shown traces were obtained from a batch of experiments using PAE cell monolayers split from the same origin. Similar results were obtained from at least three separate cell preparations cultured from different donors.
Table 2.
Maximum [Ca2+]i of cultured porcine aortic endothelial cells exposed to xanthine/xanthine oxidase
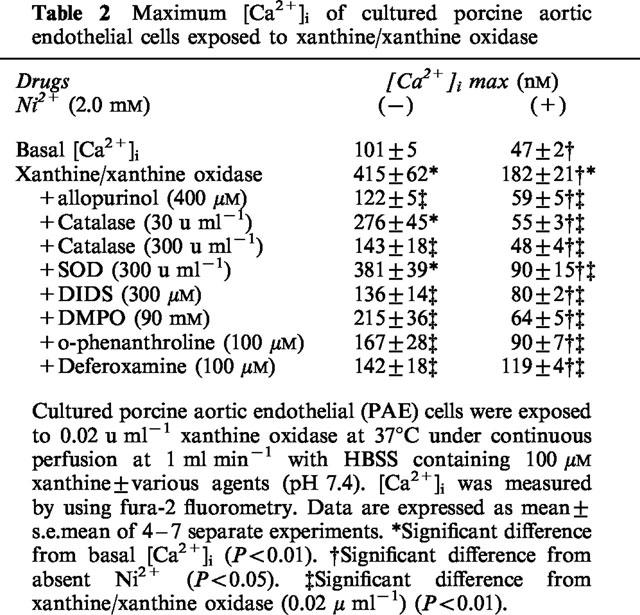
Figure 2.
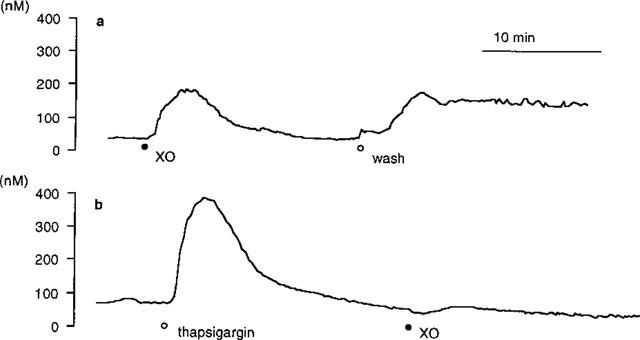
Representative recordings of the change in [Ca2+]i of cultured porcine aortic endothelial (PAE) cells exposed to xanthine/xanthine oxidase at 37°C in the presence of Ni2+. [Ca2+]i was measured by using fura-2 fluorometry: (a) PAE cells were perfused with HBSS containing 100 μM xanthine and 2.0 mM Ni2+ (pH 7.4, 1 ml min−1). (•) Addition of 0.02 u ml−1 xanthine oxidase (XO). (○) PAE cells were washed with HBSS to remove Ni2+ and xanthine/xanthine oxidase; (b) PAE cells were preloaded with 0.2 μM thapsigargin (○) before the addition of 0.2 u ml−1 xanthine oxidase (•). The traces shown were obtained from a batch of experiments using PAE cell monolayers split from the same origin. Similar results were obtained from at least three separate cell preparations cultured from different donors.
A 5 min pre- and co-incubation of PAE cells with a H2O2 scavenger, catalase, at 30 u ml−1 did not significantly decrease the maximum [Ca2+]i provoked by xanthine/xanthine oxidase, while it abolished the sustained increase in [Ca2+]i. Addition of 300 u ml−1 catalase potently inhibited the initial [Ca2+]i elevation as well as the sustained increase in [Ca2+]i (Figure 1c). In contrast, superoxide dismutase (SOD, 300 u ml−1) failed to influence the cytosolic Ca2+ movements (Figure 1d), although SOD potently and concentration-dependently decreased •O2− production from xanthine/xanthine oxidase (Table 1). However, it is unlikely that the observed [Ca2+]i elevation was due only to an effect of H2O2 because the concentrations of authentic H2O2 needed to provoke [Ca2+]i elevation was >300 μM (Table 3), which is apparently higher than the maximum concentration of H2O2 yielded by the xanthine oxidase-catalyzed oxidation of 100 μM xanthine (=100 μM). It has been reported that •O2− passes through anion channels in the cell membrane (see Discussion). Thus, the effect of an anion channel blocker, DIDS, on the cytosolic Ca2+ movements was evaluated. A 15 min preincubation of PAE cells with 300 μM DIDS potently inhibited the [Ca2+]i elevation provoked by xanthine/xanthine oxidase (Figure 1d), while it oppositely enhanced the increase in [Ca2+]i provoked by 5 mM H2O2 (Table 3) or by 10 nM bradykinin (data not shown). The latter enhancement of [Ca2+]i elevation may be explained by the direct action of DIDS to open Ca2+ channels demonstrated earlier by other investigators (Gögelein & Pfannmüller, 1989; Kawasaki & Kasai, 1989). These in turn suggested that DIDS inhibited the xanthine/xanthine oxidase-provoked [Ca2+]i elevation through a mechanism other than direct inhibition of Ca2+ channels. Taking these findings together, it is likely that •O2− and H2O2 yielded from xanthine/xanthine oxidase reciprocally provoked [Ca2+]i elevation, and that •O2− influx through anion channels in the cell membrane, but not that the presence of extracellular •O2−, was required for the [Ca2+]i elevation.
Table 3.
Maximum [Ca2+]i of cultured porcine aortic endothelial cells exposed to hydrogen peroxide

Involvement of intracellular transition metals in the cytosolic Ca2+ movements provoked by xanthine/xanthine oxidase
To further assess the precise mechanisms of xanthine/xanthine oxidase-provoked [Ca2+]i elevation, effects of a •OH scavenger and iron chelators on the cytosolic Ca2+ movements were then evaluated since the transition metal(s)-catalyzed cleavage of H2O2 in the presence of •O2− (i.e., Haber-Weiss cycle) has been implicated in the generation of •OH in endothelial cells (Az-ma et al., 1996; Zweier et al., 1988; 1994a, 1994b). A 5 min pre- and co-incubation of PAE cells with an oxygen free radical scavenger, DMPO (90 mM), inhibited the [Ca2+]i elevation (Figure 1e), suggesting that •OH generation was involved in the xanthine/xanthine oxidase-provoked cytosolic Ca2+ movements. Pre- (15 min) and co-incubation of PAE cells with 100 μM o-phenanthroline, an iron chelator that is known to pass through cell membrane (Gopalakrishna et al., 1994), inhibited the rise in [Ca2+]i (Figure 1f). The other cell membrane permeable iron chelator, deferoxamine (100 μM; Britigan et al., 1992), also significantly suppressed the [Ca2+]i elevation (Table 2). Because the incubation buffer of PAE cells used in the experiment was free from transition metals (see Methods and Az-ma et al., 1996), it is suggested that these agents decreased the xanthine/xanthine oxidase-provoked [Ca2+]i elevation by inhibiting •OH generation through the chelation of intracellular iron or other transition metals.
Effects of sulphydryl-reducing agents on the cytosolic Ca2+ movements provoked by xanthine/xanthine oxidase
The removal of xanthine/xanthine oxidase slighly enhanced the gradual decrease in [Ca2+]i following a prompt [Ca2+]i elevation provoked by the exposure of PAE cells to this exogenous oxidant generator (Figure 3a). However, a sustained increase in [Ca2+]i was still observed after the removal of xanthine/xanthine oxidase as shown in Figure 2a, indicating that Ca2+ influx from the extracellular space through Ca2+ channels continued after the exposure to xanthine/xanthine oxidase. Extracellular addition of sulphydryl (SH)-reducing agents, dithiothreitol (DTT; ⩾1.0 mM) or reduced form of glutathione (GSH; ⩾10 mM), accelerated the decrease in [Ca2+]i observed after the removal of xanthine/xanthine oxidase (Figure 3b and c). In contrast, addition of oxidant inhibitors (SOD, catalase, DMPO, and o-phenanthroline) failed to accelerate the decrease in [Ca2+]i (Figure 3c). These results suggested that xanthine/xanthine oxidase modulated the function of cell-membrane Ca2+ channels to stimulate Ca2+ influx from the extracellular space, and that SH-reducing agents reversed the function of Ca2+ channels modulated by oxidative stress.
Figure 3.
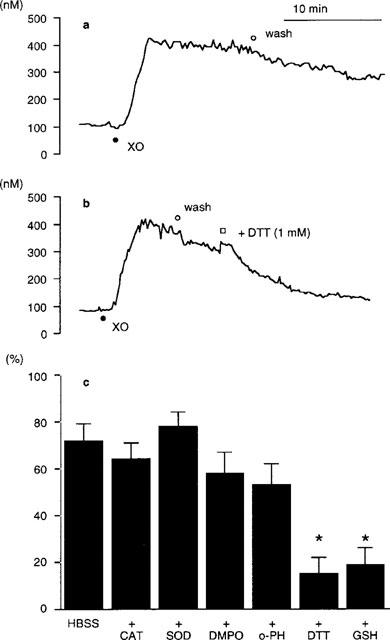
(a, b) Representative recordings of the change in [Ca2+]i of cultured porcine aortic endothelial (PAE) cells exposed to xanthine/xanthine oxidase followed by their removal in the absence or the presence of 1 mM dithiothreitol (DTT). [Ca2+]i was measured by using fura-2 fluorometry. PAE cells were exposed to 0.02 u ml−1 xanthine oxidase at 37°C under continuous perfusion at 1 ml min−1 with HBSS containing 100 μM xanthine (pH 7.4): (a) after the addition of xanthine oxidase, PAE cells were washed with HBSS to remove xanthine/xanthine oxidase; (b) after the addition of xanthine oxidase, PAE cells were washed with HBSS to remove xanthine/xanthine oxidase, followed by the addition of 1 mM DTT. (•) Addition of xanthine oxidase (XO). (○) Removal of xanthine/xanthine oxidase. (□) Addition of DTT. The traces shown were obtained from a batch of experiments using PAE cell monolayers split from the same origin. Similar results were obtained from at least three separate cell preparations cultured from different donors. (c) Effects of oxidant inhibitors and SH-reducing agents on the sustained increase in [Ca2+]i provoked by xanthine/xanthine oxidase. Each agent was added to PAE cells at approximately 5 min after the removal of xanthine/xanthine oxidase (see, Figure 3b). The [Ca2+]i of PAE cells at 15 min after the removal of xanthine/xanthine oxidase was shown as % decrease, assuming a basal [Ca2+]i of 0% and that obtained at the removal of xanthine/xanthine oxidase to be 100%, respectively. Data are expressed as mean±s.e.mean of four separate experiments. *Significant difference from HBSS (i.e., no agent was added after the removal of xanthine/xanthine oxidase) (P<0.01). CAT, catalase (300 u ml−1); SOD, superoxide dismutase (300 u ml−1); DMPO, 5,5-dimethyl-1-pyrroline-N-oxide (90 mM); o-PH. o-phenanthroline (100 μM); DTT, dithiothreitol (1 mM); GSH, reduced form of glutathione (10 mM).
Involvement of •OH in the Ca2+ release from intracellular Ca2+ stores
Effects of oxidant inhibitors on Ca2+ release from intracellular sources provoked by xanthine/xanthine oxidase were then evaluated in the presence of 2.0 mM Ni2+ (Table 2). Inhibitors of •OH generation from the Haber-Weiss cycle suppressed the [Ca2+]i elevation provoked by xanthine/xanthine oxidase (Figure 4), suggesting that the intracellular transition metal-dependent •OH generation was involved in the Ca2+ release from intracellular Ca2+ stores as well as the Ca2+ influx through the cell membrane Ca2+ channels. However, SOD (⩾30 u ml−1) also inhibited the [Ca2+]i elevation in contrast to the lack of its effect on the cytosolic Ca2+ movements in the absence of Ni2+ (Figure 1d), implicating that Ca2+ release from intracellular sources was regulated by •O2− directly and/or indirectly through unclear mechanisms other than the Haber-Weiss cycle.
Figure 4.
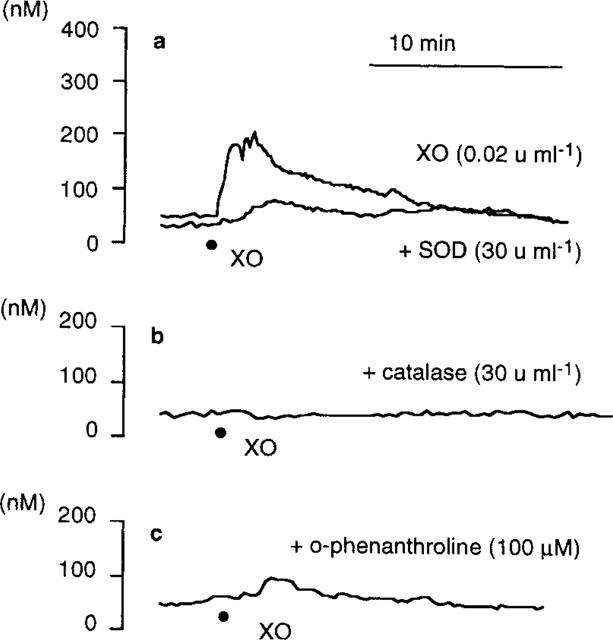
Representative recordings of the change in [Ca2+]i of cultured porcine aortic endothelial (PAE) cells exposed to 0.02 u ml−1 xanthine oxidase in the presence of Ni2+±various oxidant inhibitors at 37°C. [Ca2+]i was measured by using fura-2 fluorometry: (a) PAE cells were perfused at 1 ml min−1 with HBSS containing xanthine (100 μM), Ni2+ (2.0 mM)±superoxide dismutase (SOD; 30 u ml−1) (pH 7.4); (b) same condition as in (a) except that HBSS contained catalase (30 u ml−1) instead of SOD; (c) same condition as in (a) except that HBSS contained o-phenanthroline (100 μM) instead of SOD. (•) Addition of xanthine oxidase (XO). The shown traces were obtained from a batch of experiments using PAE cell monolayers split from the same origin. Similar results were obtained from at least three separate cell preparations cultured from different donors.
Effects of Ca2+ channel blockers and oxidant inhibitors on the cellular respiration of PAE cells exposed to xanthine/xanthine oxidase
In the presence of 100 μM xanthine, cellular respriation of PAE cells was concentration-dependently decreased by xanthine oxidase (Figure 5). Ni2+ (2.0 mM) significantly inhibited the xanthine/xanthine oxidase-provoked change in cellular respiration, suggesting that the increase in [Ca2+]i, especially due to Ca2+ influx through the cell membrane Ca2+ channels, was responsible for the xanthine/xanthine oxidase-provoked inhibition of cellular respiration. DTT (1 mM) also suppressed the change in cellular respiration. In contrast, SOD, which failed to decrease the Ca2+ influx provoked by xanthine/xanthine oxidase, did not interfere with the change in cellular respiration, also supporting the involvement of Ca2+ influx in the suppression of cellular respiration. However, inhibitors of •OH generation through the Haber-Weiss cycle (catalase, DMPO, o-phenanthroline, and deferoxamine) suppressed the change in cellular respiration more potently than Ni2+ or DTT, indicating that cytosolic Ca2+ movements did not perfectly account for the xanthine/xanthine oxidase-provoked change in cellular respiration.
Figure 5.
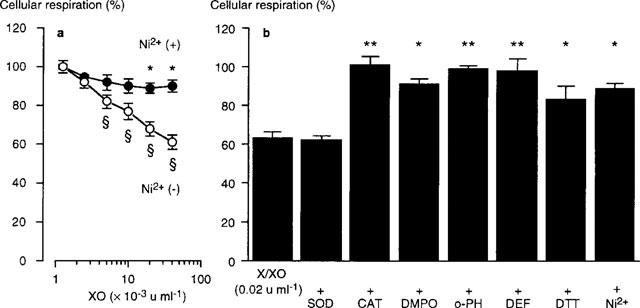
Effects of various agents on the change in cellular respiration of cultured porcine aortic endothelial (PAE) cells exposed to xanthine/xanthine oxidase for 30 min at 37°C (pH 7.4). The cellular respiration was measured by the conversion of 3-(4,5-dimethyl-2-thiazolyl)2-5,-diphenyl-2H tetrazolium bromide (MTT) to its formazan during the 4 h incubation, following the removal of xanthine/xanthine oxidase and other agents: (a) PAE cells were exposed to various concentrations of xanthine oxidase (XO) in the presence of 100 μM xanthine±2.0 mM Ni2+. *Significant difference from absent Ni2+ (P<0.01). ≈rcub;Significant difference from absent XO (P<0.01); (b) PAE cells were exposed to 100 μM xanthine and 0.02 u ml−1 xanthine oxidase (X/XO) for 30 min in the absence or the presence of various agents. *Significant difference from X/XO (P<0.05). **Significant difference from X/XO (P<0.01). Data are expressed as mean±s.e.mean of 4–6 separate experiments. SOD, superoxide dismutase (300 u ml−1); CAT, catalase (30 u ml−1); DMPO, 5,5-dimethyl-l-pyrroline-N-oxide (90 mM); o-PH, o-phenanthroline (100 μM); DEF, deferoxamine (100 μM); DTT, dithiothreitol (1 mM); Ni2+ (2.0 mM).
Discussion
In the present study, we demonstrated that xanthine/xanthine oxidase-provoked cytosolic Ca2+ movements in endothelial cells consisted of two separate processes: Ca2+ release from the intracellular sources was involved in the initial increase in [Ca2+]i provoked by xanthine/xanthine oxidase. However, Ca2+ influx from the extracellular space through Ca2+ channels appeared to be the principal source of Ca2+ movements because the xanthine/xanthine oxidase-provoked rise in [Ca2+]i continued after the removal of this oxidant generator as well as during the exposure, while Ca2+ channel blockers decreased [Ca2+]i to basal levels after an initial transient [Ca2+]i elevation. Similar results have been reported from other investigators using canine venous endothelial cells (Doan et al., 1994) or A7r5 rat aortic smooth muscle cells (Roveri et al., 1992) exposed to H2O2. Therefore, we first emphasize the discussion concerning the mechanisms involved in the xanthine/xanthine oxidase-provoked Ca2+ influx through Ca2+ channels existing in the endothelial cell membrane. The choice of this enzymic, thus, exogenous oxidant generating system, which concurrently yields •O2− and H2O2, also made it possible to analyse the sites of action at which these oxidant species influence Ca2+ channels and the involvement of •OH generation in Ca2+ movements.
We demonstrated that the cell-membrane permeable iron chelators, o-phenanthroline (Gopalakrishna et al., 1994) and deferoxamine (Britigan et al., 1992) as well as a •OH scavenger, DMPO, inhibited the xanthine/xanthine oxidase-provoked [Ca2+]i elevation, leading to our idea that the generation of •OH through intracellular iron or other transition metal(s)-catalyzed Haber-Weiss cycle (equations 1,2,3) is implicated in the Ca2+ influx provoked by xanthine/xanthine oxidase.
The inhibitory effect of catalase on this Ca2+ movement also supports the involvement of the Haber-Weiss cycle since the reaction of H2O2 with the reduced form of transition metals is the final step for •OH generation (Equation 2). It is also noteworthy that the anionic action of •O2− appears to influence its cell-membrane transport, which differs from those of the other reactive oxygen metabolites (H2O2 and •OH). Growing evidence from several types of cells shows that anion channel inhibitors decrease •O2− movements across the cell membrane (Ikebuchi et al., 1991; Kontos et al., 1985; Masumoto et al., 1992; Nozik-Grayck et al., 1997; Terada et al., 1992; Terada, 1996). In the present study, we confirmed that an anion channel blocker, DIDS, potently inhibited the xanthine/xanthine oxidase-provoked [Ca2+]i elevation, suggesting that •O2− influx through anion channels is responsible for the xanthine/xanthine oxidase-provoked Ca2+ movements. Since •OH generation through the Haber-Weiss cycle requires a reciprocal action of •O2− and H2O2 catalyzed by transition metals, the inhibition by DIDS of exogenous oxidants-provoked [Ca2+]i elevation also supported our concept that the generation of •OH occurs intracellularly.
In contrast to the inhibitory effect of DIDS on xanthine/xanthine oxidase-provoked cytosolic Ca2+ movements, extracellular addition of SOD did not inhibit the Ca2+ influx from the extracellular space. The mechanism for the lack of effect of SOD against the Ca2+ influx was unclear in the present study. However, we and other investigators have previously demonstrated that SOD increases Fe3+-dependent •OH generation from xanthine/xanthine oxidase through the enhanced generation of H2O2 (Az-ma et al., 1996; Britigan et al., 1986). This may explain why a single application of SOD cannot block the xanthine/xanthine oxidase-provoked Ca2+ influx, in which the generation of •OH catalyzed by intracellular transition metals is suggested to be involved.
It has been reported that oxidant species alter the time constant of Ca2+ channel gating to activate Ca2+ transport through the sarcoplasmic reticulum Ca2+ channel (Boraso & Williams, 1994) or the L-type Ca2+ channel (Coetzee & Opie, 1992) in cardiac myocytes using patch clamp techniques. Boraso & Williams (1994) further demonstrated that oxidant-provoked change in Ca2+ channel gating is inhibited by a SH-reducing agent, DTT, suggesting that the redox state of SH-residue(s) in Ca2+ channel proteins influences the function of these channels. The reversible effect of SH-reducing agents on the H2O2-provoked [Ca2+]i elevation in smooth muscle cells was also reported by the other investigators (Roveri et al., 1992; Krippeit-Drews et al., 1995). From this point of view, it is interesting in the present study that the extracellular addition of DTT or GSH counteracted the xanthine/xanthine oxidase-provoked Ca2+ influx into endothelial cells, suggesting that Ca2+ channels existing in the endothelial cell membrane possess functionally important SH-groups, the redox state of which can be accessible from outside of the cell membrane, although xanthine/xanthine oxidase-provoked Ca2+ influx appeared to be regulated by •OH generated through intracellular mechanisms as discussed above.
It is suggested that the xanthine/xanthine oxidase-provoked Ca2+ release from intracellular pools was also regulated by the intracellular transition metal-dependent •OH generation, because the inhibitors of Haber-Weiss cycle potently suppressed the [Ca2+]i elevation in the presence of Ni2+. The involvement of the Haber-Weiss cycle in the xanthine/xanthine oxidase-provoked cytosolic Ca2+ movements from both intracellular and extracellular sources was thus strongly indicated, while several of our results further implied that •O2− regulates these cytosolic Ca2+ movements directly and/or indirectly through unknown mechanisms other than Haber-Weiss cycle: (1) DIDS potently suppressed the Ca2+ influx through the cell membrane Ca2+ channels although iron chelators incompletely inhibited it; (2) a single application of SOD potently decreased the Ca2+ release from intracellular sources. The •NO production in endothelial cells may be considered as an alternative pathway to interfere with the xanthine/xanthine oxidase-provoked cytosolic Ca2+ movements because •NO is known to react with •O2− to yield peroxynitrite anion (ONOO−), a reactive intermediate exhibiting •OH-like oxidant activity (Az-ma et al., 1996; Beckman et al., 1990). However, it is unlikely that ONOO− is responsible for the xanthine/xanthine oxidase-provoked increase in [Ca2+]i because the production of •NO/ONOO− follows the establishment of [Ca2+]i elevation, which is required for the stimulation of constitutive form of •NO synthase in endothelial cells (Moncada et al., 1991; Pollock et al., 1991). Indeed, we confirmed that a L-arginine/•NO pathway antagonist (NG-monomethyl L-arginine) did not influence the cytosolic Ca2+ movements provoked by xanthine/xanthine oxidase (data not shown). Further evaluation using other intracellular probes is thus required to clarify the proximal action of •O2− to influence the cytosolic Ca2+ movements.
The pathophysiological interpretation of oxidant-provoked cytosolic Ca2+ elevation remains to be established. However, we demonstrated in the present study that a Ca2+ channel blocker, Ni2+, suppressed the cytotoxicity of xanthine/xanthine oxidase in endothelial cells defined as the inhibition of cellular respiration. Inhibitors of the Haber-Weiss cycle, by which xanthine/xanthine oxidase-provoked Ca2+ influx was decreased, also suppressed the changes in cellular respiration provoked by xanthine/xanthine oxidase, suggesting that [Ca2+]i elevation, as well as •OH generation itself, is involved in the cytotoxicity of oxidative stress in endothelial cells. Several other reports supported that the blockade of [Ca2+]i elevation decreases oxidant-associated cytotoxicity in a variety of types of cells (Josephson et al., 1991; Murata et al., 1994; Rojanasakul et al., 1993; Ueda & Shah, 1992). It has been reported that proteolytic activities of cysteine proteases existing in mammalian cells (e.g., calpain) is regulated by [Ca2+]i (Mirabelli et al., 1989; Nicotera et al., 1986; Tsujinaka et al., 1988). Thus, the stimulation of these proteases has been suggested for a possible explanation of Ca2+-dependent cytotoxicity (Nicotera et al., 1986; Murata et al., 1994). Murata et al. (1994) have demonstrated that a calpain inhibitor decreases oxidant-provoked and Ca2+-associated cytotoxicity in hepatic cells. Again, the regulation of •NO production by [Ca2+]i in endothelial cells raises a question concerning the Ca2+-associated cytotoxicity; i.e., does the •NO/ONOO−-pathway interfere with the oxidant-provoked cytotoxicity? However, the roles of •NO in the cytotoxicity of oxidative stress are additionally complex: we have previously observed that the enhancement of •NO production promotes acute endothelial cell death provoked by xanthine/xanthine oxidase (Az-ma et al., 1996), while endothelial cell-derived •NO has been reported to inhibit the adhesion of leukocytes (Niu et al., 1994), the most important oxidant generator in vivo.
In conclusion, Ca2+ influx through the cell membrane Ca2+ channels as well as Ca2+ release from intracellular Ca2+ stores is modulated by intracellular transition metals-catalyzed •OH generation in endothelial cells exposed to xanthine/xanthine oxidase. The change in cellular respiration provoked by oxidative stress is partly regulated by cytosolic Ca2+ movements.
Acknowledgments
This study was supported in part by a Grant-in-aid for Scientific Research from the Ministry of Education, Science and Culture of Japan (No. 09771160). We thank Ms C. Uenodo and M. Sumiyama for their assistance in preparing this manuscript.
Abbreviations
- DIDS
disodium 4, 4′-diisothiocyano-2, 2′-stilbenedisulphonic acid
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
- DMSO
dimethyl sulphoxide
- DTPA
diethylenetriaminepentaacetic acid
- DTT
dithiothreitol
- GSH
reduced form of glutathione
- H2O2
hydrogen peroxide
- HBSS
Hanks Balanced Salt Solution
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide
- •NO
nitric oxide
- •O2−
superoxide anion
- •OH
hydroxyl radical
- ONOO−
peroxynitrite anion
- PAE
procine aortic endothelial
References
- AZ-MA T., FUJII K., YUGE O. Inhibitory effect of sevoflurane on nitric oxide release from cultured endothelial cells. Eur. J. Pharmacol. 1995a;289:33–39. doi: 10.1016/0922-4106(95)90165-5. [DOI] [PubMed] [Google Scholar]
- AZ-MA T., FUJII K., YUGE O. Self-limiting enhancement by nitric oxide of oxygen free radical-induced endothelial cell injury: Evidence against the dual action of NO as hydroxyl radical donor/scavenger. Br. J. Pharmacol. 1996;119:455–462. doi: 10.1111/j.1476-5381.1996.tb15694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZ-MA T., HARDIAN, YUGE O. Inhibitory effect of lidocaine on cultured porcine aortic endothelial cell dependent anti-aggregation of platelets. Anesthesiology. 1995b;83:374–381. doi: 10.1097/00000542-199508000-00018. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., BECKMAN T.W., CHEN J., MARSHALL P.A., FREEMAN B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORASO A., WILLIAMS A.J. Modification of the gating of the cardiac sarcoplasmic reticulum Ca2+-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 1994;267:H1010–H1016. doi: 10.1152/ajpheart.1994.267.3.H1010. [DOI] [PubMed] [Google Scholar]
- BRITIGAN B.E., COFFMAN T.J., BUETTNER G.R. Spin trapping evidence for the lack of significant hydroxyl radical production during the respiratory burst of human phagocytes using a spin adduct resistant to superoxide-mediated destruction. J. Biol. Chem. 1990a;265:2650–2656. [PubMed] [Google Scholar]
- BRITIGAN B.E., POU S., ROSEN G.M., LILLEG D.M., BUETTNER G.R. Hydroxyl radical is not a product of the reaction of xanthine oxidase and xanthine: the confounding problem of adventitious iron bound to xanthine oxidase. J. Biol. Chem. 1990b;265:17533–17538. [PubMed] [Google Scholar]
- BRITIGAN B.E., ROEDER T.L., SHASBY D.M. Insight into the nature and site of oxygen-centered free radical generation by endothelial cell monolayers using a novel spin trapping technique. Blood. 1992;79:699–707. [PubMed] [Google Scholar]
- BRITIGAN B.E., ROSEN G.M., CHAI Y., COHEN M.S. Do human neutrophils make hydroxyl radical? Determination of free radicals generated by human neutrophils activated with a soluble or particulate stimulus using electron paramagnetic resonance spectrometry. J. Biol. Chem. 1986;261:4426–4431. [PubMed] [Google Scholar]
- COETZEE W.A., OPIE L.H. Effects of oxygen free radicals on isolated cardiac myocytes from guinea-pig ventricle: electrophysiological studies. J. Mol. Cell. Cardiol. 1992;24:651–663. doi: 10.1016/0022-2828(92)91049-b. [DOI] [PubMed] [Google Scholar]
- DOAN T.N., GENTRY D.L., TAYLOR A.A., ELLIOTT S.J. Hydrogen peroxide activates agonist-sensitive Ca2+-flux pathways in canine venous endothelial cells. Biochem. J. 1994;297:209–215. doi: 10.1042/bj2970209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOX R.B. Prevention of granulocyte-mediated oxidant lung injury in rats by a hydroxyl radical scavenger, dimethylthiourea. J. Clin. Invest. 1984;74:1456–1464. doi: 10.1172/JCI111558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GÖGELEIN H., PFANNMÜLLER B. The nonselective cation channel in the basolateral membrane of rat exocrine pancreas. Pflügers Arch. 1989;413:287–298. doi: 10.1007/BF00583543. [DOI] [PubMed] [Google Scholar]
- GOPALAKRISHNA R., CHEN Z.H., GUNDIMEDA U. Tobacco smoke tumor promoters, catechol and hydroquinone, induce oxidative regulation of protein kinase C and influence invasion and metastasis of lung carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12233–12237. doi: 10.1073/pnas.91.25.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IKEBUCHI Y., MASUMOTO N., TASAKA K., KOIKE K., KASAHARA K., MIYAKE A., TANIZAWA O. Superoxide anion increases intracellular pH, intracellular free calcium, and arachidonate release in human amnion cells. J. Biol. Chem. 1991;266:13233–13237. [PubMed] [Google Scholar]
- JOSEPHSON R.A., SILVERMAN H.S., LAKATTA E.G., STERN M.D., ZWEIER J.L. Study of the mechanisms of hydrogen peroxide and hydroxyl free radical-induced cellular injury and calcium overload in cardiac myocytes. J. Biol. Chem. 1991;266:2354–2361. [PubMed] [Google Scholar]
- KAWASAKI T., KASAI M. Disulfonic stilbene derivatives open the Ca2+ release channel of sarcoplasmic reticulum. J. Biochem. 1989;106:401–405. doi: 10.1093/oxfordjournals.jbchem.a122865. [DOI] [PubMed] [Google Scholar]
- KONTOS H.A., WEI E.P., ELLIS E.F., JENKINS L.W., POVLISHOCK J.T., ROWE G.T., HESS M.L. Appearance of superoxide anion radical in cerebral extracellular space during increased prostaglandin synthesis in cats. Circ. Res. 1985;57:142–151. doi: 10.1161/01.res.57.1.142. [DOI] [PubMed] [Google Scholar]
- KRIPPEIT-DREWS P., HABERLAND C., FINGERLE J., DREWS G., LANGE F. Effects of H2O2 on membrane potential and [Ca2+]i of cultured rat arterial smooth muscle cells. Biochem. Biophis. Res. Commun. 1995;209:139–145. doi: 10.1006/bbrc.1995.1481. [DOI] [PubMed] [Google Scholar]
- KURODA M., MURAKAMI K., ISHIKAWA Y. Role of hydroxyl radicals derived from granulocytes in lung injury induced by phorbol myristate acetate. Am. Rev. Respir. Dis. 1987;136:1435–1444. doi: 10.1164/ajrccm/136.6.1435. [DOI] [PubMed] [Google Scholar]
- MASUMOTO N., TASAKA K., MIYAKE A., TANIZAWA O. Superoxide anion increases intracellular free calcium in human myometrial cells. J. Biol. Chem. 1990;265:22533–22536. [PubMed] [Google Scholar]
- MASUMOTO N., TASAKA K., MIZUKI J., MIYAKE A., TANIZAWA O. Regulation of intracellular Mg2+ by superoxide in amnion cells. Biochem. Biophys. Res. Commun. 1992;182:906–912. doi: 10.1016/0006-291x(92)91818-b. [DOI] [PubMed] [Google Scholar]
- MIRABELLI F., SALIS A., VAIRETTI M., BELLOMO G., THOR H., ORRENIUS S. Cytoskeletal alterations in human platelets exposed to oxidative stress are mediated by oxidative and Ca2+-dependent mechanisms. Arch. Biochem. Biophys. 1989;270:478–488. doi: 10.1016/0003-9861(89)90529-8. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- MURATA M., MONDEN M., UMESHITA K., NAKANO H., KANAI T., GOTOH M., MORI T. Role of intracellular calcium in superoxide-induced hepatocyte injury. Hepatology. 1994;19:1223–1228. [PubMed] [Google Scholar]
- NICOTERA P., HARTZELL P., BALDI C., SVENSSON S.-A., BELLOMO G., ORRENIUS S. Cystamine induces toxicity in hepatocytes through the elevation of cytosolic Ca2+ and the stimulation of a nonlysosomal proteolytic system. J. Biol. Chem. 1986;261:14628–14635. [PubMed] [Google Scholar]
- NIU X.-F., SMITH C.W., KUBES P. Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 1994;74:1133–1140. doi: 10.1161/01.res.74.6.1133. [DOI] [PubMed] [Google Scholar]
- NOZIK-GRAYCK E., PIANTADOSI C.A., VAN-ADELSBERG J., ALPER S.L., HUANG Y.C. Protection of perfused lung from oxidant injury by inhibitors of anion exchange. Am. J. Physiol. 1997;273:L296–L304. doi: 10.1152/ajplung.1997.273.2.L296. [DOI] [PubMed] [Google Scholar]
- POLLOCK J.S., FORSTERMANN U., MITCHELL J.A., WARNER T.D., SCHMIDT H.H., NAKANE M., MURAD F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POU S., COHEN M.S., BRITIGAN B.E., ROSEN G.M. Spin-tapping and human neutrophils: limits of detection of hydroxyl radical. J. Biol. Chem. 1989;264:12299–12302. [PubMed] [Google Scholar]
- ROJANASAKUL Y., WANG L., HOFFMAN A.H., SHI X., DALAL N.S., BANKS D.E., MA J.K.H. Mechanisms of hydroxyl free radical-induced cellular injury and calcium overload in alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 1993;8:377–383. doi: 10.1165/ajrcmb/8.4.377. [DOI] [PubMed] [Google Scholar]
- ROVERI A., COASSIN M., MAIORINO M., ZAMBURLINI A., AMSTERDAM F.T.M.V., RATTI E., URSINI F. Effect of hydrogen peroxide on calcium homeostasis in smooth muscle cells. Arch. Biochem. Biophys. 1992;297:265–270. doi: 10.1016/0003-9861(92)90671-i. [DOI] [PubMed] [Google Scholar]
- TERADA L.S. Hypoxia-reoxygenation increases O2− efflux which injures endothelial cells by an extracellular mechanism. Am. J. Physiol. 1996;270:H945–H950. doi: 10.1152/ajpheart.1996.270.3.H945. [DOI] [PubMed] [Google Scholar]
- TERADA L.S., GUIDOT D.M., LEFF J.A., WILLINGHAM I.R., HANLEY M.E., PIERMATTEI D., REPINE J.E. Hypoxia injures endothelial cells by increasing endogenous xanthine oxidase activity. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3362–3366. doi: 10.1073/pnas.89.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSUJINAKA T., KAJIWARA Y., KAMBAYASHI J., SAKON M., HIGUCHI N., TANAKA T., MORI T. Synthesis of a new cell penetrating calpain inhibitor (calpeptin) Biochem. Biophys. Res. Commun. 1988;153:1201–1208. doi: 10.1016/s0006-291x(88)81355-x. [DOI] [PubMed] [Google Scholar]
- UEDA N., SHAH S.V. Role of intracellular calcium in hydrogen peroxide-induced renal tubular cell injury. Am. J. Physiol. 1992;263:F214–F221. doi: 10.1152/ajprenal.1992.263.2.F214. [DOI] [PubMed] [Google Scholar]
- ZWEIER J.L., BRODERICK R., KUPPUSAMY P., THOMPSON-GORMAN S., LUTTY G.A. Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J. Biol. Chem. 1994a;269:24156–24162. [PubMed] [Google Scholar]
- ZWEIER J.L., KUPPUSAMY P., LUTTY G.A. Measurement of endothelial cell free radical generation: evidence for a central mechanism of free radical injury in postischemic tissues. Proc. Natl. Acad. Sci. U.S.A. 1988;85:4046–4050. doi: 10.1073/pnas.85.11.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZWEIER J.L., KUPPUSAMY P., THOMPSON-GORMAN S., KLUNK D., LUTTY G.A. Measurement and characterization of free radical generation in reoxygenated human endothelial cells. Am. J. Physiol. 1994b;266:C700–C708. doi: 10.1152/ajpcell.1994.266.3.C700. [DOI] [PubMed] [Google Scholar]