

Abstract
The kinetics of shortening of action potential duration (APD) following an increase in pacing rate, from 2 to 3.3 Hz, was characterized in guinea-pig ventricular preparations. Terikalant (RP62719), an inhibitor of the inwardly rectifying K+ current (IK1), and dofetilide, a specific inhibitor of the rapidly activating delayed-rectifier current (IKr), were applied to determine the effect of inhibition of these ion currents on slow APD shortening.
Action potentials were recorded from isolated guinea-pig ventricular myocytes using the perforated-patch patch-clamp technique, and monophasic action potentials were recorded from Langendorff-perfused guinea-pig ventricles using a contact epicardial probe.
Under control conditions, after an increase in pacing rate, APD immediately decreased, and then shortened slowly with an exponential time course. In ventricular myocytes, the time constant of this exponential shortening was 28±4 s and the amount of slow shortening was 21.9±0.9 ms (n=8) for an increase in rate from 2 to 3.3 Hz. Similar values were observed in Langendorff-perfused ventricles.
Terikalant dose-dependently increased APD and the increase was enhanced by rapid pacing (‘positive' rate-dependence). The drug dose-dependently decreased the time constant of shortening and amount of slow APD shortening. In contrast, dofetilide, an inhibitor of IKr, which shows ‘reverse' rate-dependent APD widening, had no significant effect on the time constant or amount of slow shortening.
These observations suggest that IK1 plays a role in rate-dependent shortening of APD. The results appear to support the hypothesis that ‘reverse' rate-dependent effects of IKr blockers are due to these drugs not affecting the ion current(s) mediating intrinsic rate-dependent slow shortening of APD.
Keywords: Class III anti-arrhythmic agent, action potential duration, rate-dependence, restitution, inward rectifier potassium current, terikalant, dofetilide
Introduction
Cardiac action potential duration (APD) is strongly rate dependent. Previous studies have shown that APD shortening with an increase in rate (a decrease in cycle length (CL)) occurs in two phases; an immediate rapid shortening is followed by a slower shortening to steady state (Boyett & Jewell, 1978; Elharrar & Surawicz, 1983). The rapid APD shortening, known as electrical restitution, occurs when the preceding diastolic interval is reduced (Boyett & Jewell, 1978; Bass, 1975). The difference in the APD after restitution and the APD at the new steady state has been attributed to a cumulative component, or memory effect; the APD after restitution appears to ‘remember' the previous cycle length at which it was paced. The slower shortening to steady state, which we will call slow accommodation, has been attributed to a dissipation of memory and, at fixed CL, occurs concomitantly with an increase in diastolic interval (Elharrar & Surawicz, 1983; Surawicz, 1992).
Numerous studies have looked at the properties of electrical restitution and characterized the process in both mammalian Purkinje and ventricular cells (Boyett & Jewell, 1978; Elharrar & Surawicz, 1983; Bass, 1975; Robinson et al., 1987). Fewer groups have looked at the process of slow accommodation, in part due to the time consuming nature of measuring each action potential during a sustained train (Robinson et al., 1987; Boyett & Fedida, 1984; Saitoh et al., 1988). Both restitution and slow accommodation of APD appear to be related to changes in ion currents. Suggestions for which ion currents may be involved have been based on measurements of activation, inactivation and deactivation kinetics, as well as on knowledge of ion channel sensitivity to intracellular and extracellular ion concentrations (Kline & Morad, 1978; Boyett & Fedida, 1984; Surawicz, 1992; Carmeliet, 1993). Particular attention has been paid to K+ currents as possible determinants of rate-dependent shortening, since many of these currents contribute to membrane repolarization.
The intrinsic APD-rate relationship has important consequences for class III anti-arrhythmic drug action. Class III anti-arrhythmic drugs act primarily by prolonging APD through a reduction of outward currents without affecting conduction velocity (Vaughan Williams, 1970). Unfortunately, most available class III drugs display the property of reverse rate-dependence. That is, the effectiveness of class III drugs are reduced at rapid heart rates (Hondeghem & Snyders, 1990). Recent studies suggest that this is generally not a result of drug unbinding from open channels (reverse use-dependence; Carmeliet, 1992; Jurkiewicz & Sanguinetti, 1993) and leaves the suggestion that an intrinsic APD-rate relationship may be responsible. An attractive hypothesis is that drug-induced widening of APD is reduced by an increase in rate, if the drug does not affect the ion current mediating intrinsic rate-dependent changes in APD.
This present article tests the effect of terikalant (RP 62719) and dofetilide on the kinetics of slow accommodation of APD following an increase in pacing rate. Using in-house analysis software (Dickenson et al., 1997), we have been able to perform beat to beat analysis of action potentials during action potential trains, and thus, accurately measure the effect of inhibition of an ion current on the kinetics of APD shortening.
Methods
Cell isolation
Single ventricular myocytes were enzymatically dissociated from hearts of adult guinea-pigs (450–600 g) using a modified method of Powell et al. (1980). Briefly, animals were killed by cervical dislocation, and the heart was removed and washed free of blood using ice-cold zero Ca2+ solution (see Solutions) that contained 0.1 mM EGTA and heparin (10 u ml−1). The heart was then mounted on a Langendorff apparatus and perfused for up to 36 min at 37°C with zero Ca2+ solution to which was added 0.6 mg ml−1 collagenase (type II, Worthington Biochemical, Freehold, NJ, U.S.A.) and 0.14 mM CaCl2. The ventricles were removed and mechanically dissociated in the presence of the collagenase solution. Cells were collected through a 250-μm mesh at 5 min intervals up to a total of 20 min from the start of agitation. Cells were stored in Dulbecco's modified Eagle medium (GIBCO, Burlington, ON, Canada) at room temperature for up to 8 h before use.
Cellular electrophysiology
Action potentials were measured using the amphotericin perforated-patch configuration of the patch-clamp technique. Delayed-rectifier K+ currents (IK), inward rectifier K+ currents (IK1) and Ca2+ currents (ICa) were measured using the standard whole-cell configuration of the patch-clamp technique. Pipettes were pulled from borosilicate glass (Garner Glass, Claremont, CA, U.S.A.), and resistances were between 2 and 5 MΩ when filled with intracellular solution.
Cells were allowed to settle for 2–4 min on the glass bottom of a tissue bath (1–1.5 ml) before seal formation. Junction potentials were zeroed before formation of the membrane seal in the external solution. After seal formation and during recording, cells were superfused (3 ml min−1) with external solution warmed to 34–37°C. Recording of action potentials was started once the membrane potential was at a steady value and the cells responded to a stimulus pulse. Cells had membrane potentials between −74 and −84 mV when in the extracellular bath solution. Recording of whole-cell currents were started 1–3 min after formation of the whole-cell configuration.
Cells were stimulated to fire action potentials by applying a square stimulus pulse of 4–10 nA and 2–4 ms duration generated with pCLAMP software (version 6.03, Axon Instruments) connected to an Axopatch 200A amplifier and a Digidata 1200 interface (Axon Instruments, Foster City, CA, U.S.A.). pCLAMP was triggered by an external computer-controlled signal generator, in order to stimulate the cell at the required cycle lengths. Action potentials were recorded with the Axopatch 200A amplifier and acquired on-line using a DT2831-G interface with Global Lab Data Acquisition software (V2.2, Data Translation, Marlboro, MA, U.S.A.). Signals were low-pass filtered at 2 kHz (−3bB) and digitized at 5 kHz.
Whole-cell currents were recorded with the Axopatch 200A amplifier and acquired on-line using the Digidata 1200 interface with pCLAMP software (Axon Instruments). Currents were low-pass filtered at 1 kHz (−3dB) and digitized at 2–3 kHz. Command pulses were generated by pCLAMP software and converted to analogue form using the Digidata 1200.
Ex-vivo ventricular preparations
Langendorff-perfused ventricular preparations were used to compare with results from isolated cells. Whole hearts were removed from adult guinea-pigs (450–800 g) following cervical dislocation and bilateral thoracotomy. The hearts were washed with ice-cold Krebs solution (see Solutions) containing 10 u ml−1 heparin, then mounted on a Langendorff-perfusion apparatus and gradually warmed to 37°C by perfusion with standard oxygenated Krebs solution. Hearts were stimulated at 450 ms CL continuously during warming. The left and right atrium were removed and the A-V node ablated using a cauterizing probe. A custom designed recording setup, incorporating a contact epicardial monophasic action potential (MAP) probe, a pair of silver-wire epicardial stimulating electrodes, and a pair of ECG electrodes were placed in contact with the heart to deliver and acquire signals. Recording was begun once the temperature was constant at 37°C and the heart could be paced at the required CL. MAP signals were recorded with a Cyberamp 380 amplifier (Axon Instruments), or in some experiments with a custom-built amplifier (Arbre Engineering, Ottawa, ON, Canada), and acquired on-line using a DT2831-G interface with Global Lab Data Acquisition software (V2.2, Data Translation, Marlboro, MA, U.S.A.). Signals were digitized at 2 kHz, without filtering.
Pacing protocols
A basic CL (BCL) of 500 ms (2 Hz) was applied to isolated cells for a minimum of 1 min before recording. To measure APD accommodation, the following pacing protocol was applied: 500 ms CL (2 Hz) for 500 beats followed by 300 ms CL (3.33 Hz) for 600 beats and then returned to 500 ms CL. Drug was applied for 5 min during pacing at 500 ms CL before repeating the above protocol. Steady state APD curves were obtained by pacing downwards from 700 to 500 ms CL in 50 ms steps, and then 500 to 250 ms CL in 25 ms steps, returning to 500 ms CL at the end. Drug was perfused for 5 min during pacing at 500 ms CL before beginning the steady state protocol again.
Langendorff-perfused ventricles were paced at a BCL of 450 ms, or 500 ms if possible, for 5 min before recording. Guinea-pig ventricles seemed to be more easily paced at 450 ms or less, although in some cases, we were able to pace at 500 ms to enable better comparison with isolated cell data. Recording of MAP signals was begun 30 s before switching from the BCL to 300 ms CL, and continued for a further 5 min at the new CL before returning to BCL. Drug was added for at least 10 min while pacing at BCL before recording with the above protocol.
Solutions
For cell isolation, the zero Ca2+ solution contained (in mM): NaCl 137, KCl 5.4, glucose 5, NaHCO3 12, Na+ pyruvate 1, NaH2PO4 0.4, MgCl2 1, and NaOH 0.8. During recording of action potentials, the isolated cells were bathed in standard extracellular solution containing (in mM): NaCl 132, KCl 4, CaCl2 1.8, MgCl2 1.2, HEPES 10 and glucose 10 (pH 7.4 with NaOH). Intracellular solution contained (in mM): K+ aspartate 120, NaCl 10, MgCl2 1, K2ATP 5, EGTA 1, HEPES 10 (pH 7.2 with KOH), to which was added 180–240 μg ml−1 of amphotericin B.
For measurement of IK and IK1, the extracellular solution contained (in mM): NaCl 145, KCl 4, MgCl2 1, CaCl2 0.1, HEPES 10 and glucose 5 (pH 7.4 with NaOH) to which was added 0.1 mM CdCl2 to block ICa. Intracellular solution contained (in mM): KCl 60, K+ gluconate 60, MgCl2 2, CaCl2 1, EGTA 11, K2ATP 5, HEPES 10 (pH 7.2 with KOH). For measurement of ICa, the extracellular solution contained (in mM): Tris HCl 137, CaCl2 5.5, MgCl2 1, CsCl 20, glucose 5.5 (pH 7.3 with CsOH). The intracellular solution for ICa contained (in mM): CsCl 125, MgATP 5, EGTA 15, TEACl 20, HEPES 10 (pH 7.2 with CsOH). The Langendorff-perfused ventricular preparation was perfused with standard oxygenated Krebs solution containing (in mM): NaCl 119, KCl 3, CaCl2 1.4, MgCl2 1.2, KH2PO4 1.2, NaHCO3 25, glucose 10.
Terikalant (RP62719), obtained from Rhone-Poulenc Rorer, Vitry-sur-Seine, France, was dissolved at 1 or 0.1 mM in the appropriate extracellular solution on the day of the experiment. Test concentrations were obtained by serial dilution. Dofetilide (Pfizer Central Research, Sandwich, U.K.) was made up as a 1 mM stock solution in acidified distilled water (pH 3–4) and aliquots were added to the bath solution on the day of the experiment. K2ATP and MgATP (Sigma-Aldrich, Mississauga, ON, Canada) were dissolved directly in the appropriate intracellular solution on the day of the experiment, and the pH adjusted as required. Amphotericin B (Sigma-Aldrich, Mississauga, ON, Canada) was made up as a 6 mg 100 μl−1 stock solution in DMSO and kept in the freezer (−20°C) for up to 4 days before use.
Data analysis
Analysis of MAP signals and intracellular action potentials was performed using in-house analysis software (Dickenson et al., 1997). From this program, we obtained values for APD90 (APD at 90% repolarization), and APD75 (APD at 75% repolarization) as well as plateau values and baseline membrane potential. Quoted APD values are averages of 50 consecutive beats, taken at steady state. APD accommodation curves were fit using Sigmaplot software (version 2.0, Jandel Scientific, San Raphael, CA, U.S.A.). Statistical significance, for the effect of drug on APD and fit parameters, was determined using the Student's t-test.
Whole-cell currents were analysed using pCLAMP software (Axon Instruments). The magnitude of IK currents was determined by subtraction of the minimum current level measured at the beginning of the test depolarizing pulse, after the capacitive transient, from the current at the end of the test depolarizing pulse. Peak Ica current was measured relative to zero current after subtraction of leak and capacitive currents. Leak and capacitive currents were subtracted during ICa recording by applying four positive sub-episodes of 1/4 amplitude from a holding potential of −90 mV (P/4 protocol in pCLAMP). A 2 min time average of the peak IK and ICa currents was calculated at steady state, in control and in the presence of drug, to determine drug effect. Rundown of currents was taken into account by using the average of pre- and post-drug as control, since the effect of terikalant appeared to be reversible. When only pre-drug values were used as control, the values for per cent block were not significantly different from those when rundown was taken into account.
Group data are expressed as means±s.e.mean, and n represents the number of samples. The experiments conform to the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care.
Results
Kinetics of action potential shortening
Figure 1 illustrates the effect on APD90 of decreasing CL from 500 to 300 ms, in an isolated guinea-pig ventricular myocyte. In this Figure, the two phases of APD shortening with an increase in rate are clearly shown. The rate was changed after 250 s of recording. At first, there was a sudden and rapid decrease in APD, known as restitution, when the diastolic interval was suddenly reduced. Restitution was followed by a slower exponential decrease in APD, and steady state was reached in approximately 100 s. All cells recorded in control conditions followed a similar time course. The slow shortening could be fit with a mono-exponential of the form shown in equation 1, where APD(t) is the action potential duration at
time t (t=0 at restitution), APD(t=∞) is the action potential duration at the new steady state, B is the amount of slow shortening, and τ is the time constant for APD to change from its value at restitution to its value at the new steady state. In half the cells tested, alternans occurred for the first 2–3 beats following the rate change. In these cases, the first APD after the rate change (the APD of restitution) was much smaller than subsequent APDs and appeared as a downward spike in the trace, as in Figure 1. The action potentials during the alternans were included with the rest of the data when calculating an exponential fit and did not significantly affect the fit results. The calculated APD of restitution, APD (t=0)[=APD(t=∞)+B], however, was sometimes slightly greater than the measured APD of restitution. Alternans were not prevented by the addition of terikalant. It has been shown previously that the time course of slow shortening is not affected by the presence of alternans (Saitoh et al., 1988), which appears consistent with our data, so we did not consider the alternans further. Under control conditions, in ventricular myocytes the time constant of slow APD shortening, τ, was 28±4 s and the amount of slow shortening, B was 21.9±0.9 ms (n=8) for a decrease of CL from 500 to 300 ms. In Langendorff-perfused ventricles, the slow shortening could also be fit by a mono-exponential, although in many cases a bi-exponential gave a better fit to the first 30 s of the shortening process (Figure 2). A good fit was determined both by eye and by comparing the coefficients of variation of the parameters, calculated by SigmaPlot. A mono-exponential gave a good fit for all data recorded during a switch from 500 to 300 ms CL; the time constant was 72±4 s and the amount of slow shortening, B was 51±4 ms (n=3). However, of the nine experiments paced from 450 to 300 ms CL, five were better fitted with a bi-exponential. For a switch from 450 to 300 ms CL, under control conditions, a mono-exponential fit gave τ=65±5 s, B=41±3 ms (n=9), and a bi-exponential fit gave τ1=3.2±0.7 s, B1=33±2 ms and τ2=77±8 s, B2=30±4 ms (n=9).
Figure 1.
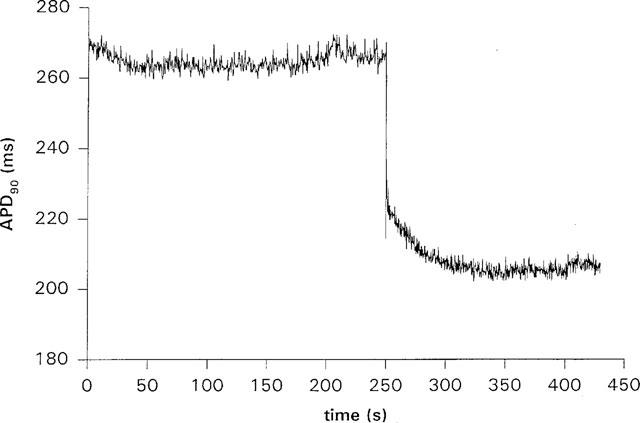
The time course of APD90 shortening during pacing in a guinea-pig ventricular myocyte. The cell was paced at 500 ms CL, and the cycle length was changed to 300 ms after 250 s from the start of recording. On changing the cycle length, APD90 shortened in two stages; a rapid decrease due to electrical restitution followed by a slower exponential decrease lasting about 100 s. The downward spike visible at the start of the slow shortening is due to alternans of APD over two to three beats.
Figure 2.
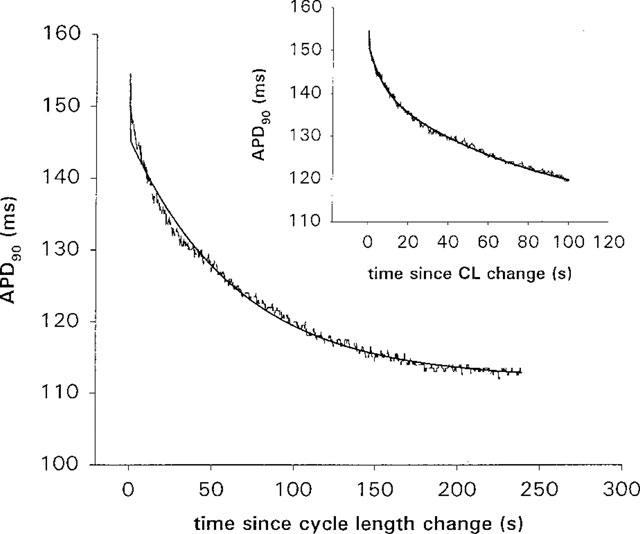
The time course of APD90 shortening following a cycle length change from 450 ms to 300 ms in control for a Langendorff-perfused ventricular preparation, showing the two types of exponential fit. Zero on the time axis is the time at which the cycle length was changed. In the main plot, the data was fitted with a mono-exponential (solid line) and, in the inset plot, the same data was fitted with a bi-exponential. The first 50 s of shortening are clearly better fitted with a bi-exponential. Of nine experiments, all could be fit with a mono-exponential, although in five experiments, the first 30–50 s of data were better fit with a bi-exponential.
Effects of terikalant on action potential duration
Consistent with Escande et al. (1992), terikalant dose-dependently increased APD in the range 1–10 μM (Figure 3a and b). There appeared to be a slight decrease in APD at 0.1 and 0.3 μM, at 500 ms CL, however, this decrease was not statistically significant. Under control conditions in isolated ventricular myocytes, APD90 was 196±9 ms at 300 ms CL and 250±13 ms at 500 ms CL (n=8). Addition of 10 μM terikalant to the bath solution significantly increased APD90 by 17±3% to 229±8 ms at 300 ms CL (n=8; P<0.001, one-tailed paired t-test), relative to control. At 500 ms CL, APD90 was also significantly increased, but only by 8±4% to 270±14 ms (n=8; P<0.05, one-tailed paired t-test), relative to control. The increase in APD90 at 300 ms CL was significantly greater than the increase at 500 ms CL (n=8; P<0.001, one-tailed paired t-test).
Figure 3.
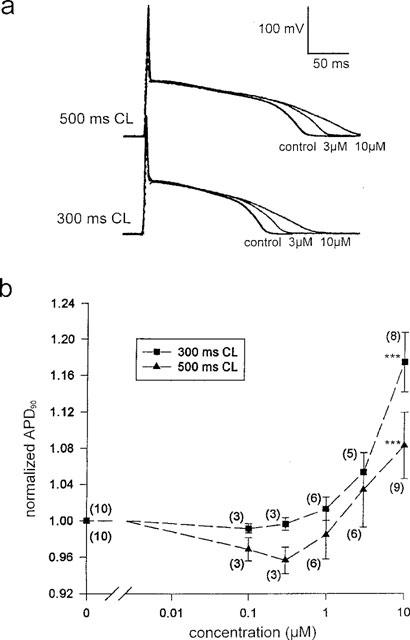
Terikalant dose-dependently increased action potential duration. (a) Intracellular action potentials measured at steady state, in control and two concentrations of terikalant, from an isolated ventricular myocyte at two different cycle lengths. The initial spikes in the action potentials are stimulus artifacts. (b) APD90 increases with terikalant concentration and the drug is more effective at 300 ms than at 500 ms CL. Values for APD90 have been normalized to the control value in the absence of terikalant, for each experiment. Action potentials were longer than the 300 ms cycle length at concentrations above 10 μM so that APD90 could not be measured. The number of samples is given by the number above each point. ***P<0.001 (one-tailed, paired t-test).
In the Langendorff-perfused ventricular preparations, the effect of terikalant could not be as easily measured; at concentrations above 5 μM, terikalant caused a large increase in refractory period followed by a reduction of contractility such that pacing at rapid rates could not be maintained and we were unable to record high quality MAPs. Under control conditions, APD90 was 154±33 ms at 300 ms CL, 221±39 ms at 450 ms CL (n=10) and 255±5 ms at 500 ms CL (n=3). Addition of 3 μM terikalant, increased APD90 to 167±19 ms at 300 ms (9±6%; n=4) and 230±21 ms at 450 ms CL (4±2%; n=4).
From our results, it appeared that the effects of terikalant were enhanced by rapid pacing. In order to test this further, we measured APD90 at steady state in isolated cells over the range 700–240 ms CL in the presence and absence of terikalant. Figure 4a illustrates the result of one experiment, and Figure 4b shows the average prolongation measured in four cells. Terikalant appears to be positive rate-dependent in the physiological range, 300–550 ms. At CL <300 ms, in the presence of terikalant, APD90 was approaching the duration of the CL, leading to an apparent reduction in APD widening by terikalant; this effect was only observed in isolated cells. Strangely, in three of four cells APD90 was shorter in the presence of terikalant compared to control at long cycle lengths: in one cell at CL >475 ms, in the other two at CL >575 ms and CL >700 ms. In a fourth cell, APD90 was prolonged at all CL's up to 2000 ms. This effect may be similar to that observed at low concentrations in Figure 3 (see Discussion).
Figure 4.
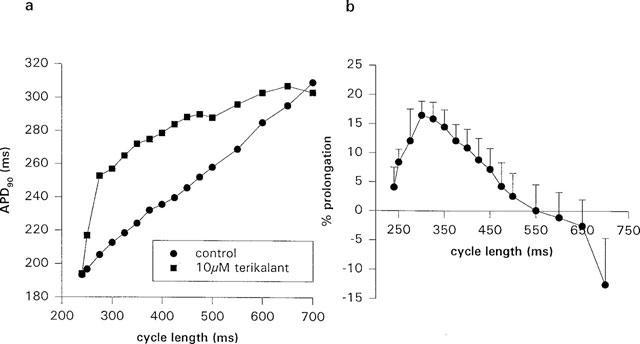
APD90 widening by terikalant is positive rate-dependent and increases with a decrease in cycle length, at cycle lengths between 550 and 300 ms. When the cycle length duration was similar in value to the APD90 value (<300 ms CL), there was a gradual reduction in the effect of terikalant. (a) Steady state APD90 plotted against cycle length in the presence and absence of 10 μM terikalant for one ventricular myocyte. (b) Per cent prolongation of steady state APD90 by 10 μM terikalant for a range of cycle lengths. The data shown is an average of four cells. At cycle lengths greater than 475 ms, there was an apparent shortening of APD90 in some cells, in the presence of terikalant.
In all experiments, the amount of widening at 300 ms CL was greater than at 500 ms CL. The average per cent APD90 widening was 8±2, 17±2, 14±3, 11±3, 7±4 and 0±4% during pacing at 250, 300, 350, 400, 450 and 500 ms CL respectively, while at 700 ms CL, average APD90 appeared to shorten by 13±8% (n=4).
The effect of terikalant on APD was entirely reversible and following washout of the drug, APD90 at 300 ms CL was 195±7 ms (n=7) and at 500 ms CL was 240±12 ms, compared to 196±9 and 250±13 ms, respectively, in control. Terikalant (10 μM) had no significant effect on the resting membrane potential (−77±2 mV (n=8) in control compared to −78±2 mV (n=8) in 10 μM terikalant and −80±2 mV (n=7) after washout). There was also no significant difference between the average of the baseline in control and after washout (=−80±1 mV) and that in 10 μM terikalant, (P=0.059, one-tailed paired t-test). The drug did not affect the action potential plateau value.
Effect of terikalant on kinetics of slow APD shortening
In the presence of terikalant, the fit parameters, τ and B (Figures 5 and 6) were decreased dose dependently. Indeed, in four of ten experiments in ventricular myocytes, the slow APD shortening at 10 μM terikalant was almost completely abolished. At 10 μM, B and τ were decreased significantly by 63±11 and 63±10% (to 11±4 s and 8±2 ms; n=8) respectively, relative to that in control (P<0.01, one-tailed, unpaired t-test). The effect of terikalant on the slow shortening process was reversible and on re-application of 10 μM of the drug, the fit parameters were again decreased. The parameters, τ and B, after washout were 30±5 s and 21±2 ms, respectively, which were not significantly different from that in control (29±4 s and 21.9±0.9 ms, respectively).
Figure 5.
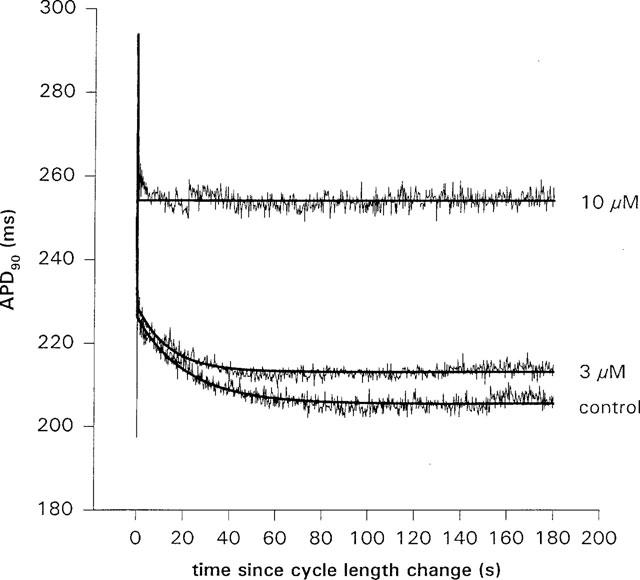
The time course of APD90 shortening following a cycle length change from 500 to 300 ms CL in control and in the presence of two concentrations of terikalant, for a single myocyte. Zero on the time axis is the time at which the cycle length was changed. The smooth lines are fits of a mono-exponential, equation 1, to the data. Both the amount of slow shortening and the time constant was reduced by terikalant. In this particular cell (and in 40% of experiments), terikalant at 10 μM almost completely abolished the slow shortening.
Figure 6.
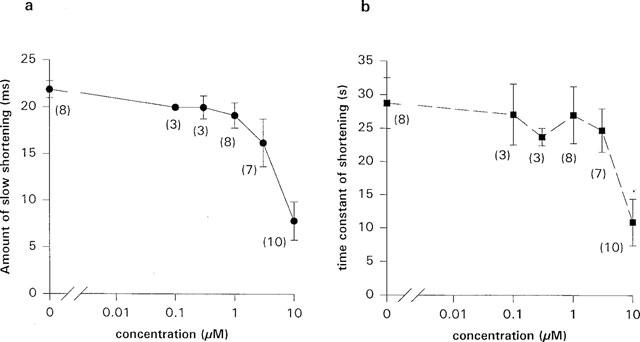
(a and b) The amount of slow shortening and the time constant, obtained by fitting equation 1 to the data, were reduced dose-dependently by terikalant. The amount of slow shortening was decreased by 63±9% and time constant by 62±12% (n=10) (or 63±10 and 63±11%, respectively, for paired experiments (n=8)) in 10 μM terikalant. The number given beside each point gives the number of experiments performed.
The total change in steady state APD90 on switching from 500 to 300 ms CL (ΔAPD90) was significantly reduced by terikalant; in control, ΔAPD90=53±7 ms and in 10 μM terikalant, ΔAPD90=29±4 ms (n=8, P<0.01, one-tailed, unpaired t-test). By contrast, the change in APD on restitution, calculated by subtraction of the amount of slow shortening, B, from ΔAPD90 was not significantly changed by the addition of terikalant (P=0.23; 31±6 ms in control compared to 21±4 ms in 10 μM terikalant, n=8).
In the Langendorff perfused ventricular preparations, the effect of terikalant was not as clear, due to the fact that concentrations above 5 μM could not be applied. At 3 μM, for a switch from 450 to 300 ms CL, the time constant for a mono-exponential fit was 57±9 s, which was not significantly different from control (56±8, n=4). The parameter, B, decreased to 34±3 ms (14±5% decrease; n=4). For a bi-exponential fit, the time constants were not significantly changed (2.3±0.4 and 65±6 s with terikalant; n=3), and the parameter, B1, became 28±3 ms (9±7% decrease; n=4), and B2 became 27±4 ms (9±2% decrease; n=4).
Effect of dofetilide on kinetics of slow APD shortening
It has been reported that terikalant inhibits IKr in cat and guinea-pig ventricular myocytes (Bridal et al., 1996; Jurkiewicz et al., 1996). We therefore tested the effect of dofetilide, a specific inhibitor of IKr (Carmeliet, 1992; Jurkiewicz & Sanguinetti, 1993) on the rate-dependent shortening of APD. It has been shown that dofetilide inhibits IKr in guinea-pig ventricular myocytes with an IC50 of 30 nM, and widens APD90 at 1 Hz with an EC50 of 30 nM in canine ventricular tissue (Jurkiewicz & Sanguinetti, 1993; Gwilt et al., 1991). We therefore tested the effect of dofetilide at 0.01, 0.1 and 1 μM on APD. In the absence of dofetilide, ADP90 was 200±11 ms at 300 ms CL and 242±20 ms at 500 ms CL (n=4). The mean increase in APD, at 300 ms CL, was 4±4% in 0.1 μM and 1±4% in 1 μM dofetilide (n=4). At 500 ms CL, the mean increase was 7±7% in 0.1 μM, and 6±6% in 1 μM dofetilide at 500 ms (n=4). In two of the four cells, dofetilide produced limited APD widening. In the cells in which significant APD widening occurred, the widening was greater at 500 than at 300 ms, consistent with ‘reverse' rate-dependent effects previously described for dofetilide (Jurkiewicz & Sanguinetti, 1993). An example of the effect of dofetilide on slow APD accommodation is shown in Figure 7. There was no significant change in B, which was 21±1 ms in control compared to 21.7±0.7 in 0.1 μM and 22.8±0.8 in 1 μM dofetilide (n=4). There was also no significant effect on the time constant with dofetilide dose; the average time constant was 21±2 s in control compared to 20±3 s in 0.1 μM and 22.9±4 s in 1 μM dofetilide (n=4).
Figure 7.
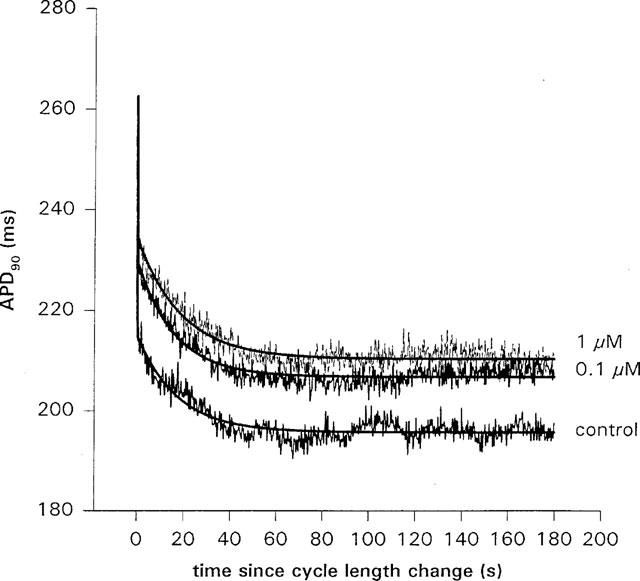
The effect of dofetilide on the slow shortening of APD90 following a cycle length change from 500 to 300 ms, in an isolated ventricular myocyte. In this cell, the amount of slow shortening, B, obtained from a fit of equation 1 to the data, increased with dose. In control, B=19.5±0.5 ms compared to 23.5±0.5 ms in 0.1 μM, and 24.3±0.5 ms in 1 μM dofetilide. There was no consistent effect on the time constant; τ=17.1±0.7 s in control, compared to 15.9±0.5 s in 0.1 μM and 18.8±0.6 s in 1 μM dofetilide. When the average of four cells was taken, there was no significant change in B or τ with dofetilide dose. Zero on the time axis is the time at which the cycle length was changed.
Specificity of terikalant inhibition
Terikalant has been reported to be a specific inhibitor of the inward rectifier K+ current (IK1) in cardiac tissue (Escande et al., 1992). However, more recently, other groups have suggested that terikalant also inhibits the rapidly activating delayed-rectifier K+ current (IKr) (Bridal et al., 1996; Jurkiewicz et al., 1996). In this paper, the reduction of slow accommodation by terikalant appears to be most effective at 10 μM. We therefore tested terikalant (10 μM) on IK, IKr, ICa and IK1 in order to test its specificity. Consistent with the results of Escande et al. (1992), but in contrast to the results of Jurkiewicz et al. (1996), we found that only IK1 was significantly affected by 10 μM terikalant (Figure 8).
Figure 8.
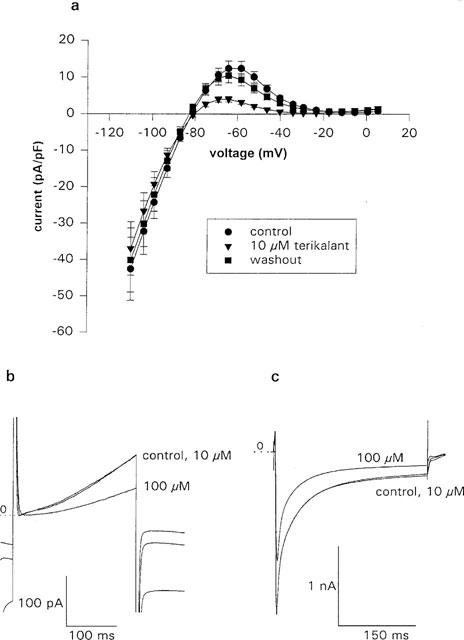
Terikalant at 10 μM inhibits IK1 without affecting IK and ICa. (a) IK1 was measured by applying a 500-ms ramp from 10 mV to −110 from a potential of −40 mV. Terikalant (10 μM) inhibited the outward current more than the inward current, although in both cases inhibition was significant (P<0.05, one-tailed paired t-test on the individual points). The current was maximally reduced by 89±4% at −30 mV. The effect of terikalant was reversible and following 10 min of washout of the drug, the current had almost returned to control levels (n=10 for control and 10 μM terikalant, and n=9 for washout). (b) The average, over 2 min, of 60 consecutive IKr current traces in control, and in the presence of 10 and 100 μM terikalant. At 10 μM, IKr was unaffected but at 100 μM the current was reduced by 51%. IKr was measured by stepping to −40 mV for 50 ms from a holding potential of −70 mV and then applying a 250-ms duration pulse to +20 mV. The zero IKr current level is shown by the dotted line, marked 0. The current at −40 mV, shown before and after the 250-ms pulse, is reduced by both 10 and 100 μM terikalant (lower most trace), due to inhibition of IK1. (c) The average over 2 min of 24 consecutive ICa current traces in control, and in the presence of 10 and 100 μM terikalant. ICa was also unaffected by 10 μM terikalant, although 100 μM inhibits the current by 26%. The dotted line, marked 0, is the zero current level. ICa was measured by applying a 300-ms duration pulse to +10 mV from a holding potential of −70 mV.
To measure IK1, the cell was stepped, from a holding potential of −70 to −40 mV for 50 ms, then a 500-ms ramp from 10 to −110 mV was applied. A series of ten ramps were applied at a rate of one per s, and the average of the ten ramp currents calculated. From Figure 8a, it can be seen that terikalant was more effective at inhibiting outward than inward IK1 currents, although in both cases inhibition was significant (P<0.05, one-tailed paired t-test). Indeed, maximum inhibition of outward currents was 89±4% at −30 mV, compared to a maximum inhibition of inward currents of 24±4% at −90 mV (n=10). Peak outward current, at −65 mV, was inhibited by 67±3%. Terikalant did not inhibit the current at potentials positive to −17 mV. After washing out the drug for 10 min, the IK1 current returned to control levels.
To measure IK, cells were held at −70 mV and then stepped to −40 mV for 25–50 ms, to inactivate Na+ currents, before the test depolarizing pulse was applied. A 250-ms depolarizing pulse to +20 mV was applied every 2 s for measurements of terikalant block of the rapidly activating component (IKr). A pulse to +20 mV was chosen since we have found that at this potential, IKr is close to maximum (Williams & Beatch, 1997). A 5-s depolarizing pulse to +40 mV was applied every 10 s to measure the effect of terikalant on the total IK. In several cells, depolarizing pulses were applied in 10 mV steps between −20 and +70 mV, and a current-voltage relation obtained in the presence and absence of terikalant.
To measure ICa, a 300-ms depolarizing pulse to +10 mV was applied every 5 s, from a holding potential of −70 mV. The protocols for IK and ICa recording were applied continuously for 6 min before adding drug, then recording was continued for another 6 min in the presence of drug before washing out.
Figure 8b and c shows that terikalant at 10 μM did not have a significant effect on IKr, or ICa. There was also no consistent effect of terikalant on the current-voltage relations (data not shown). Similarly, there was no significant effect of terikalant on total IK. Delayed-rectifier and calcium currents are known to decline with time following formation of the whole-cell configuration, a phenomenon known as rundown (Duchatelle-Gourdon et al., 1989; Kameyama et al., 1988). Rundown was taken into account in our experiments by calculating the average of pre- and post-drug currents and using this value as the control current. The average decrease on addition of 10 μM terikalant was 5±6% (n=4) for IK, −4±15% (n=12) for IKr, and −1±7% (n=8) for ICa. When rundown was not taken into account, the values were 15±6% (n=5) for IK, 9±7% (n=12) for IKr and 4±11% for ICa. At 100 μM, terikalant was non-specific and blocked IK by 34% (n=2), IKr by 46±5% (n=5), and ICa by 24±7% (n=5). In addition, at 100 μM, there was an obvious reduction in amplitude of the Na+ current transient on stepping to −40 from −70 mV, but there was no obvious effect at lower concentrations (data not shown).
Discussion
Role of IK1
Our results seem to suggest that IK1 may play a role in the intrinsic rate-dependent shortening of APD with an increase in pacing rate. Several groups have shown that terikalant is less specific than originally suggested (Bridal et al., 1996; McLarnon & Xu, 1995; Jurkiewicz et al., 1996). Despite this evidence, we believe, for several reasons, that the effect of terikalant on APD accommodation, observed in this paper, is most likely due to the inhibition of IK1 and not IKr: (1) Dofetilide did not produce the same effect as terikalant on the kinetics of APD shortening; (2) We observed a change in kinetics at concentrations which coincided with IK1 blockade and (3) In our preparation, terikalant did not produce a consistent inhibition of ICa, IK or IKr at concentrations ⩽10 μM. Jurkiewicz et al. (1996) found that IKr was inhibited by terikalant with an IC50 of 31 nM and that APD90 was increased by <1 μM terikalant. We observed little or no APD widening until concentrations were greater than 1 μM terikalant (Figure 3b). Except for the possibility that there was a reduced amount of IKr in our cells, we are unable to explain the difference between our results on IKr and those of Jurkiewicz et al. (1996). Our measurement of the inhibition of IK1 by terikalant is, however, consistent with the results of Jurkiewicz et al. (1996); at 10 μM terikalant they found an inhibition of approximately 65% at −60 mV (Figure 4 in their paper), whereas we found an inhibition of 72±4% at −60 mV.
There are several possible mechanisms by which terikalant blockade of IK1 may lead to the reduction of APD accommodation. The kinetics of IK1 blockade by terikalant may be such that with each action potential, at the faster rate, inhibition of the outward current increases. The drug would then be acting in a voltage- and use-dependent manner; however, from our measurements of the effect of terikalant on IK1 ramps, use-dependence appears unlikely. (Note that use-dependence refers to a change in effect of the drug on the current with open time (use), whereas rate-dependence of the drug refers to a change in effect on the action potential with pacing rate). Ramps were applied at a rate of 1 Hz. If the positive rate-dependence of the drug were due to use-dependence, then one would expect that at 1 Hz there would be little block of the IK1 ramp current, since at rates greater than 500 ms CL (2 Hz), APD90 prolongation was minimal (Figure 4). The ramp IK1 current, however, was inhibited almost maximally (90% at −30 mV) and did not increase with each consecutive ramp.
Another possibility is that, under normal conditions, outward IK1 may increase gradually, over several seconds, as APD adjusts to a new more rapid rate. This seems an odd suggestion since, until recently, IK1 was assumed not to exhibit significant time-dependence during the final repolarization of the action potential, it having been shown to be time- and voltage-dependent only at potentials negative to the reversal potential (Surawicz, 1992; Kurachi, 1985; Biermans et al., 1987). However, Shimoni et al. (1992) using rabbit ventricular myocytes, found that IK1 was inactivated during the up-stroke and plateau of the action potential, and that inactivation required 100–300 ms to develop. Removal of inactivation was equally slow, particularly at more depolarized potentials. Thus, more IK1 may remain active to contribute to shortening when the action potential plateau is shorter, and thereby contribute to the intrinsic rate-dependent shortening. In this case, terikalant would not have to block in a use-dependent manner in order to block rate-dependent APD shortening.
Rate-dependent changes in IK1 could also be due to an indirect effect of changes in intracellular Ca2+ (Bassingthwaighte et al., 1976; Colatsky & Hogan, 1980) or extracellular K+ (Kline & Morad, 1978; Boyett & Jewell, 1980); however, from more recent studies, this appears unlikely. Our results and those of Robinson et al. (1987) were performed on isolated single cells, making the previously suggested hypothesis of accumulation of K+ in restricted extracellular spaces unlikely. Intracellular calcium has been shown to inhibit outward IK1 in chick ventricular cells (Mazzanti & DeFrancesco, 1989), and in contrast to our results, Delmar et al. (1991) using guinea-pig ventricular myocytes, have attributed a decrease in outward IK1 following repetitive stimulation to an increase in intracellular Ca2+. Delmar et al. (1991), however, applied a series of 300 ms duration square pulses as opposed to action potentials, so that the timing of ion channel activation and inactivation may have been quite different.
Thus, it appears that the effect of terikalant on APD may be due largely to a change in the amount of IK1 with pacing, but the factor which leads to this change in IK1 remains to be identified. The effect of terikalant on slow APD accommodation at cycle lengths greater than 500 ms was not tested; however, since the drug did not greatly increase APD90 at these cycle lengths, it would seem likely that the kinetics of slow accommodation would be little affected.
Kinetics of APD shortening
We have found that slow APD accommodation occurs with an exponential time course in both isolated single cells, and Langendorff-perfused ventricles. A fit of a mono-exponential function to the data gave a time constant of approximately 30 s for isolated myocytes, and a time constant of 72 s for intact ventricles, paced from 500 to 300 ms CL. For pacing intact ventricles from 450 to 300 ms, the time constant was approximately 65 s. These values are consistent with the results of Robinson et al. (1987) who found that the slow APD shortening occurred with a time constant of 71 s, for a 4500 to 500 ms transition in canine Purkinje cells, and with Boyett & Fedida (1984) who obtained a time constant of 64 s, for a 3000 s to 300 ms transition in canine Purkinje strands. It appears that the rate of slow APD accommodation occurs with time constants between 30–70 s. The differences in these mono-exponential time constants are quite small given that these are from different species. Given the ubiquitous nature of IK1, however, it is perhaps not surprising, if IK1 is involved, that there is little interspecies variation. However, the effects of the magnitude of the cycle length transition on slow accommodation is unknown.
The presence of a second exponential in the paced intact ventricle is interesting and it is possible that it is due to a gradual synchronization of these syncytial cells to the new rate. Alternatively, it may be a reflection of the multi-cellular nature of MAP recording. The data which were fitted by the rapid time constant of slow shortening, in the intact ventricles, were such a small proportion of the entire data trace, that they were difficult to fit, and indeed, appeared not always to be present. Quantification of the affects of drugs and other modulators on this component in order to determine its cause is therefore intrinsically difficult.
The observed shortening of APD in the presence of terikalant at long cycle lengths (Figure 4), and at low concentrations (Figure 3b), in some cells, is curious. One explanation is that terikalant may have blocked a small as-yet-undefined inward current (or activated an outward current) in a reverse rate-dependent manner, so that its effect was only observed when block of IK1 was small and the cycle length long. However, as we also observed a small decrease in APD at low dofetilide concentrations (0.01 μM) in some cells, this suggestion appears unlikely. Another possibility is that, when the effect of terikalant was small, small shifts in APD, unrelated to the presence of terikalant, led to an apparent decrease in APD. Such shifts in APD were sometimes observed under control conditions during the 5 min pacing done before recording. In many cells, APD would recover to the initial steady state values after 30–60 s. The observation that dofetilide had limited effects on APD tested at 500 and 300 ms CL may have also been due to small shifts in APD; since dofetilide shows reverse rate-dependent effects, it is possible that the pacing rate was too fast for significant effects to always be observed. We have previously tested our dofetilide preparation on IKr on guinea-pig ventricular myocytes and found consistent and reproducible inhibition of IKr (Williams & Beatch, 1997). Since the APD90 values under control conditions at the start of the experiment, and after washout of terikalant at the end of the experiment were not significantly different, it is evident that ‘rundown' of APD over time did not generally occur, and therefore, cannot be used to explain the apparent ineffectiveness of the drugs.
In contrast to the effect on slow accommodation, terikalant appears not to have altered restitution at 300 ms. This suggests that slow accommodation and restitution may be separate processes; however, the effect of terikalant on restitution was obtained indirectly using the fit parameter, B, and at only one cycle length. Further studies are required to verify the effect of terikalant on restitution; in particular, restitution should be measured directly over a wider range of cycle lengths. It seems probable, however, that currents which are more time dependent, such as IK or ICa, may contribute more to restitution than IK1 (Carmeliet, 1993; Hauswirth et al., 1972; Gettes & Reuter, 1974).
Positive rate-dependence
Terikalant, unlike other class III anti-arrhythmic agents, was found to be positive rate-dependent, in the physiological range of cycle lengths. The effect of RP58866, a racemate of terikalant, is also not reduced by rapid pacing at 5 Hz (200 ms CL), in Langendorff-perfused rat hearts (Rees & Curtis, 1993). The fact that terikalant inhibited the slow shortening of APD, and was also positive rate-dependent, whereas dofetilide had no effect on the slow shortening, and is reverse rate-dependent, supports the hypothesis that the reverse rate-dependence of an IKr blocker may be due to such a drug not inhibiting the ion current(s) mediating intrinsic rate-dependent slow shortening of APD. We must await the development of other IK1 inhibitors, or other positive rate-dependent agents, to confirm this hypothesis.
From our data, and those of Rees & Curtis (1993) using RP58866, it appears that drugs which block IK1 possess several properties believed to be important for effective anti-arrhythmic therapy. APD prolongation, by terikalant and RP58866, are positive rate-dependent, and furthermore, RP58866 has been shown to be effective at preventing ischaemia-induced ventricular fibrillation, as has tacrine, another non-selective IK1 blocking agent (Rees & Curtis, 1994). It remains controversial, however, whether IK1 blockade will be a useful anti-arrhythmic mechanism (Rees & Curtis, 1994; Opthof, 1994). The results from research on these drugs, however, suggest an avenue along which more effective therapeutic agents may be developed.
Conclusions
Terikalant, an inhibitor of IK1, increased the rate and reduced the amount of slow shortening during an increase in pacing rate, whereas dofetilide, a specific inhibitor of IKr, had no significant effect on the rate and the amount of slow shortening. We therefore suggest that IK1 may play a role in intrinsic rate-dependent slow shortening of APD.
Acknowledgments
We would like to thank the Heart and Stroke Foundation of Ontario for financial support and Rhone-Poulenc Rorer for the donation of terikalent.
Abbreviations
- APD90, ADP75
action potential duration at 90% repolarization, or at 75% repolarization
- APD(t)
action potential duration (at time t)
- B
amount of slow APD shortening
- BCL
basic cycle length
- CL
cycle length
- ICa
calcium current
- IK
delayed-rectifier K+ current
- IK1
inward rectifier K+ current
- IKr
rapidly activating delayed-rectifier K+ current; MAP monophasic action potential
- n
number of samples
- τ
time constant for slow APD shortening
References
- BASS B.G. Restitution of the action potential in cat papillary muscle. Am. J. Physiol. 1975;228:1717–1724. doi: 10.1152/ajplegacy.1975.228.6.1717. [DOI] [PubMed] [Google Scholar]
- BASSINGTHWAIGHTE J.B., FRY C.H., MCGUIGAN J.A.S. Relationship between internal calcium and outward current in mammalian ventricular muscle; a mechanism for the control of the action potential duration. J. Physiol. (Lond.) 1976;262:15–37. doi: 10.1113/jphysiol.1976.sp011583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIERMANS G., VEREECKE J., CARMELIET E. The mechanism of the inactivation of the inward-rectifying K current during hyperpolarization steps in guinea-pig ventricular myocytes. Pflügers Arch. 1987;410:604–613. doi: 10.1007/BF00581320. [DOI] [PubMed] [Google Scholar]
- BOYETT M.R., FEDIDA D. Changes in the electrical activity of dog cardiac Purkinje fibres at high heart rates. J. Physiol. (Lond.) 1984;350:361–391. doi: 10.1113/jphysiol.1984.sp015206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOYETT M.R., JEWELL B.R. A study of the factors responsible for rate-dependent shortening of the action potential in mammalian ventricular muscle. J. Physiol. (Lond.) 1978;285:359–380. doi: 10.1113/jphysiol.1978.sp012576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOYETT M.R., JEWELL B.R. Analysis of the effects of changes in rate and rhythm upon electrical activity in the heart. Prog. Biophys. Mol. Bio. 1980;36:1–52. doi: 10.1016/0079-6107(81)90003-1. [DOI] [PubMed] [Google Scholar]
- BRIDAL T.R., REES S.A., SPINELLI W., COLATSKY T.J.Terikalant (RP62719) is not a selective blocker of inward rectifier current in cat ventricular myocytes Circulation 199694I–529.(Abstract) [Google Scholar]
- CARMELIET E. Voltage- and time-dependent block of the delayed K+ current in cardiac myocytes by dofetilide. J. Pharmacol. Exp. Ther. 1992;262:809–817. [PubMed] [Google Scholar]
- CARMELIET E. K+ channels and control of ventricular repolarization in the heart. Fundam. Clin. Pharmacol. 1993;7:19–28. doi: 10.1111/j.1472-8206.1993.tb00214.x. [DOI] [PubMed] [Google Scholar]
- COLATSKY T.J., HOGAN P.M. Effects of external calcium, calcium channel-blocking agents, and stimulation frequency on cycle length-dependent changes in canine cardiac action potential duration. Circ. Res. 1980;46:543–552. doi: 10.1161/01.res.46.4.543. [DOI] [PubMed] [Google Scholar]
- DELMAR M., IBARRA J., DAVIDENKO J., LORENTE P., JALIFE J. Dynamics of the background outward current of single guinea pig ventricular myocytes. Circ. Res. 1991;69:1316–1326. doi: 10.1161/01.res.69.5.1316. [DOI] [PubMed] [Google Scholar]
- DICKENSON D.R., DAVIS D.R., BEATCH G.N. Development and Evaluation of a Fully Automated Monophasic Action Potential Analysis Program. Med. Biol. Eng. Comp. 1997;35:653–660. doi: 10.1007/BF02510974. [DOI] [PubMed] [Google Scholar]
- DUCHATELLE-GOURDON I., HARTZELL H.C., LAGRUTTA A.A. Modulation of the delayed rectifier K+ current in frog cardiomyocytes by β-adrenergic agonists and Mg2+ J. Physiol. (Lond.) 1989;415:251–274. doi: 10.1113/jphysiol.1989.sp017721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELHARRAR V., SURAWICZ B. Cycle length effect on restitution of action potential duration in dog cardiac fibers. Am. J. Physiol. 1983;244:H782–H792. doi: 10.1152/ajpheart.1983.244.6.H782. [DOI] [PubMed] [Google Scholar]
- ESCANDE D., MESTRE M., CAVERO I., BRUGADA J., KIRCHHOF C. RP58866 and its active enantiomer RP62719 (Terikalant) blockers of the inward rectifier K+ current acting as pure class III antiarrhythmic agents. J. Cardiovasc. Pharmacol. 1992;20 Suppl. 2:S106–S113. [PubMed] [Google Scholar]
- GETTES L.S., REUTER H. Slow recovery from inactivation of inward currents in mammalian myocardial fibres. J. Physiol. (Lond.) 1974;240:703–724. doi: 10.1113/jphysiol.1974.sp010630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GWILT M., ARROWSMITH J.E., BLACKBURN K.J., BURGES R.A., CROSS P.E., DALRYMPLE H.W., HIGGINS A.J. UK-68,798: A novel, potent and highly selective class III antiarrhythmic agent which blocks potassium channels in cardiac cells. J. Pharmacol. Exp. Ther. 1991;256:318–324. [PubMed] [Google Scholar]
- HAUSWIRTH O., NOBLE D., TSIEN R.W. The dependence of plateau currents in cardiac Purkinje fibres on the interval between action potentials. J. Physiol. (Lond.) 1972;222:27–49. doi: 10.1113/jphysiol.1972.sp009786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONDEGHEM L.M., SNYDERS D.J. Class III antiarrhythmic agents have a lot of potential but a long way to go: Reduced effectiveness and dangers of reverse use dependence. Circulation. 1990;81:686–690. doi: 10.1161/01.cir.81.2.686. [DOI] [PubMed] [Google Scholar]
- JURKIEWICZ N.K., SANGUINETTI M.C. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent: Specific block of a rapidly activating delayed rectifier K+ current by dofetilide. Circ. Res. 1993;72:75–83. doi: 10.1161/01.res.72.1.75. [DOI] [PubMed] [Google Scholar]
- JURKIEWICZ N.K., WANG J., FERMINI B., SANGUINETTI M.C., SALATA J.J. Mechanism of action potential prolongation by RP58866 and its active enantiomer terikalant. Circulation. 1996;94:2938–2946. doi: 10.1161/01.cir.94.11.2938. [DOI] [PubMed] [Google Scholar]
- KAMEYAMA M., KAMEYAMA A., NAKAYAMA T., KAIBARA M. Tissue extract recovers cardiac calcium channels from ‘run-down'. Pflügers Arch. 1988;412:328–330. doi: 10.1007/BF00582516. [DOI] [PubMed] [Google Scholar]
- KLINE R.P., MORAD M. Potassium efflux in heart muscle during activity: Extracellular accumulation and its implications. J. Physiol. (Lond.) 1978;280:537–558. doi: 10.1113/jphysiol.1978.sp012400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURACHI Y. Voltage-dependent activation of the inward rectifier potassium channel in the ventricular cell membrane of guinea-pig heart. J. Physiol. (Lond.) 1985;366:365–385. doi: 10.1113/jphysiol.1985.sp015803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAZZANTI M., DEFRANCESCO D. Intracellular Ca modulates K-inward rectification in cardiac myocytes. Pflügers Arch. 1989;413:322–324. doi: 10.1007/BF00583549. [DOI] [PubMed] [Google Scholar]
- MCLARNON J.G., XU R. Actions of the benzopyran compound terikalant on macroscopic currents in rat ventricular myocytes. J. Pharmacol. Exp. Ther. 1995;275:389–396. [PubMed] [Google Scholar]
- OPTHOF T. IK1 blockade is unlikely to be a useful antiarrhythmic mechanism. Cardiovasc. Res. 1994;28:420. doi: 10.1093/cvr/28.3.420. [DOI] [PubMed] [Google Scholar]
- POWELL T., TERRAR D.A., TWIST V.W. Electrical properties of individual cells isolated from adult rat ventricular myocardium. J. Physiol. (Lond.) 1980;302:131–153. doi: 10.1113/jphysiol.1980.sp013234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REES S.A., CURTIS M.J. Specific IK1 blockade: A new antiarrhythmic mechanism? Effect of RP58866 on ventricular arrhythmias in rat, rabbit, and primate. Circulation. 1993;87:1979–1989. doi: 10.1161/01.cir.87.6.1979. [DOI] [PubMed] [Google Scholar]
- REES S.A., CURTIS M.J. IK1 blockade is a potentially useful antiarrhythmic mechanism. Cardiovasc. Res. 1994;28:421. doi: 10.1093/cvr/28.3.421. [DOI] [PubMed] [Google Scholar]
- ROBINSON R.B., BOYDEN P.A., HOFFMAN B.A., HEWETT K.W. Electrical restitution process in dispersed canine cardiac Purkinje and ventricular cells. Am. J. Physiol. 1987;253:H1018–H1025. doi: 10.1152/ajpheart.1987.253.5.H1018. [DOI] [PubMed] [Google Scholar]
- SAITOH H., BAILEY J.C., SURAWICZ B. Alternans of action potential duration after abrupt shortening of cycle length: Differences between dog Purkinje and ventricular muscle fibers. Circ. Res. 1988;62:1027–1040. doi: 10.1161/01.res.62.5.1027. [DOI] [PubMed] [Google Scholar]
- SHIMONI Y., CLARK R.B., GILES W.R. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J. Physiol. (Lond.) 1992;448:709–727. doi: 10.1113/jphysiol.1992.sp019066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SURAWICZ B. Role of potassium channels in cycle length dependent regulation of action potential duration in mammalian cardiac Purkinje and ventricular muscle fibers. Cardiovasc. Res. 1992;26:1021–1029. doi: 10.1093/cvr/26.11.1021. [DOI] [PubMed] [Google Scholar]
- VAUGHAN WILLIAMS E.M.Classification of antiarrhythmic drugs Symposium on Cardiac Arrhythmias 1970449–472.ed. Sandoe, E., Flensted-Jensen, E., Olesen, K.H.
- WILLIAMS B.A., BEATCH G.N. Magnesium shifts voltage dependence of activation of delayed rectifier IK in guinea-pig ventricular myocytes. Am. J. Physiol. 1997;272 (Heart Circ. Physiol. 41):H1292–H1301. doi: 10.1152/ajpheart.1997.272.3.H1292. [DOI] [PubMed] [Google Scholar]