

Abstract
It was recently suggested that an endogenous cannabinoid could represent an endothelium-derived hyperpolarizing factor (EDHF). The aim of the present study was to clarify whether CB1 cannabinoid receptors are involved in the nitric oxide (NO)- and prostanoid-independent vasodilation produced by acetylcholine in rabbits.
Pithed rabbits received indomethacin. Noradrenaline was infused to raise blood pressure, and vasodilation was elicited by bolus injections of acetylcholine. The NO-synthase inhibitor Nω-nitro-L-arginine methylester inhibited the acetylcholine-evoked vasodilation by about 40%. The remaining vasodilation was unaffected by the CB1 cannabinoid receptor antagonist SR141716A, but was inhibited by the potassium channel blocker tetraethylammonium. In addition, the mixed CB1/CB2 cannabinoid receptor agonist WIN55212-2 did not elicit vasodilation.
No CB1 cannabinoid receptors were involved in the prostanoid- and NO-independent vasodilation produced by acetylcholine. An exogenous cannabinoid also did not cause vasodilation. Therefore, it is unlikely that an endogenous cannabinoid serves as an EDHF acting at smooth muscle CB1 cannabinoid receptors in the rabbit.
Keywords: Acetylcholine, cannabinoid receptor, EDHF, endothelium, pithed rabbit, smooth muscle relaxation
Introduction
Activation of muscarinic receptors in vascular endothelial cells elicits vasodilation. The endothelium releases nitric oxide (NO) and in some vessels also prostacyclin which in turn relax vascular smooth muscle. However, endothelium-dependent vasodilation is still observed in vessels in which NO- and prostanoid synthesis is inhibited: this residual vasodilation is attributed to an endothelium-derived hyperpolarizing factor (EDHF). The chemical identity of EDHF is not known (for review see Garland et al., 1995; Edwards & Weston, 1998).
It was recently suggested that anandamide (N-arachidonylethanolamide), an endogenous cannabinoid, could represent an EDHF; it would cause vasodilation by activating CB1 cannabinoid receptors on vascular smooth muscle cells (for review see Randall & Kendall, 1998). This hypothesis was based on observations in isolated rat mesenteric and coronary arteries: carbachol and bradykinin elicited vasodilation in the presence of NO and prostanoid synthesis inhibitors; the vasodilation was antagonized by the CB1 cannabinoid receptor antagonist SR141716A; anandamide elicited direct smooth muscle relaxation; and this latter effect was also antagonized by SR141716A (Randall et al., 1996; Randall & Kendall, 1997; and see White & Hiley, 1997, 1998).
We recently observed that the CB1/CB2 cannabinoid receptor agonist WIN55212-2 (an aminoalkylindole, structurally unrelated to anandamide) does not elicit vasodilation in pithed rabbits (Niederhoffer & Szabo, 1999; Figure 1A in the present paper). This is in contrast with the hypothesis that an endogenous cannabinoid (an EDHF) elicits vasodilation by activating vascular CB1 receptors. The aim of the present study was to clarify whether CB1 cannabinoid receptors–activated by an endogenous cannabinoid–are involved in the NO- and prostanoid-independent vasodilation produced by acetylcholine in rabbits. Pithed rabbits were used, a preparation in which the function of small resistance arteries–in which EDHF plays a major role–can be effectively studied.
Figure 1.
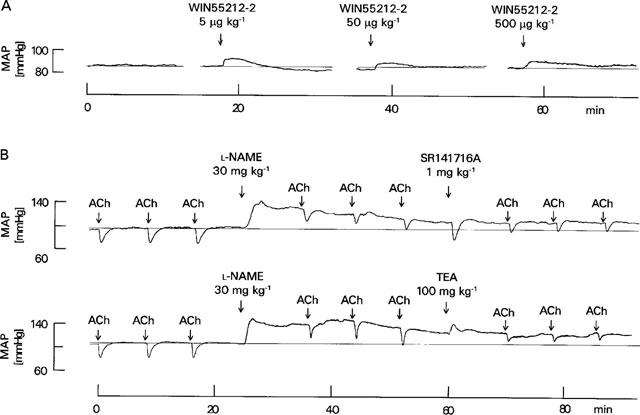
(A) Effect of WIN55212-2 on mean arterial pressure (MAP) in a pithed rabbit in which blood pressure was raised by an infusion of noradrenaline (2 μg kg−1 min−1)–original tracing representing four experiments with similar results (the average values are shown in Niederhoffer & Szabo, 1999). WIN55212-2 was injected as indicated by arrows; the slight increase in blood pressure after injection of WIN55212-2 is due to the solvent. (B) Effects of L-NAME, SR141716A and TEA on the acetylcholine (ACh, 1 μg kg−1)-evoked decrease in mean arterial pressure (MAP) in pithed rabbits–original tracings. Blood pressure was raised by an infusion of noradrenaline (2 μg kg−1 min−1) and indomethacin (5 mg kg−1) was administered 10 min before the first acetylcholine injection. Other drugs were injected as indicated by arrows.
Methods
Rabbits of a local mixed breed (derived from `Deutscher Riesenscheck') and weighing 1.8–3.7 kg were used. Animals were anaesthetized with pentobarbitone (75 mg kg−1, i.v.) and artificially ventilated. The left carotid artery was cannulated for recording arterial pressure with a Statham P23Db transducer coupled to a bridge amplifier (Hugo Sachs Elektronik, Hugstetten, Germany). The right carotid artery was ligatured. Both jugular veins were cannulated for administration of drugs (all drugs were given i.v.). After relaxation of skeletal muscles by gallamine triethiodide (5 mg kg−1) the animals were pithed. To increase vascular tone, an infusion of noradrenaline (2 μg kg−1 min−1) was started. This infusion increased mean arterial pressure from 57±4 mmHg to 100±5 mmHg (n=15). The cyclo-oxygenase inhibitor indomethacin (5 mg kg−1) was injected 30 min after the beginning of noradrenaline infusion; it did not change blood pressure. Forty minutes after the beginning of noradrenaline infusion, the first acetylcholine injection (1 μg kg−1) was given (t=0 min in Figure 1B). After three successive acetylcholine injections (at 8 min intervals), the NO-synthase inhibitor L-NAME (30 mg kg−1) was administered. After three further acetylcholine injections, the solvent for SR141716A (66% DMSO), the CB1 cannabinoid receptor antagonist SR141716A (1 mg kg−1) or the potassium channel blocker TEA (100 mg kg−1) was injected. Three final acetylcholine injections followed. For each acetylcholine injection, the decrease in blood pressure was expressed as a percentage of the blood pressure value preceding the respective acetylcholine injection.
Means±s.e.mean of n experiments are given throughout. The paired t-test was used to evaluate statistical differences and P<0.05 was taken as the limit of significance.
Drugs were obtained from the following sources: acetylcholine iodide, indomethacin, Nω-nitro-L-arginine methylester hydrochloride (L-NAME), (−)-noradrenaline (+)-bitartrate and tetraethylammonium chloride (TEA) from Sigma (Deisenhofen, Germany); N-piperidino-5-(4-chlorophenyl)- 1 - (2,4 - dichlorophenyl) - 4 - methyl - 3 - pyrazole - carboxamide (SR141716A) from Sanofi (Montpellier, France); R(+)-[2,3-dihydro - 5 - methyl - 3 -[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl)methanone mesylate (WIN-55212-2) from RBI (Köln, Germany). Acetylcholine, L-NAME, TEA and noradrenaline were dissolved in saline. Indomethacin was dissolved in 1.2% wv−1 NaHCO3 in saline. SR141716A was dissolved in 66% vv−1 dimethylsulphoxide (DMSO) in distilled water. WIN55212-2 was dissolved in 19% wv−1 2-hydroxypropyl-β-cyclodextrin in distilled water. Doses refer to the salts. Injections had a volume of 0.5 ml kg−1.
Results
Bolus injections of acetylcholine (1 μg kg−1) elicited transient decreases in blood pressure (Figures 1B and 2). Immediately after injection of the NO synthase inhibitor L-NAME (30 mg kg−1), resting blood pressure increased by 30±4% (n=15; P<0.05; see Figure 1B); blood pressure declined thereafter slowly. The second effect of L-NAME was an inhibition, by about 40%, of the acetylcholine-evoked hypotension (Figure 2). A higher dose of L-NAME (100 mg kg−1) did not inhibit further the vasodilatory effect of acetylcholine (not shown).
Figure 2.

Effects of L-NAME (30 mg kg−1), solvent (SOL; 66% DMSO; 0.5 ml kg−1), SR141716A (1 mg kg−1) and TEA (100 mg kg−1) on the acetylcholine (1 μg kg−1)-evoked decrease in mean arterial pressure in pithed rabbits. Means±s.e.mean; n=5 in each group. Difference from the last value before L-NAME: *P<0.05 (the horizontal arrow indicates that in all groups all values measured after L-NAME [18 values] were significantly different); difference from the last value before administration of SOL, SR141716A or TEA: +P<0.05.
Administration of either solvent (66% DMSO; 0.5 ml kg−1), the CB1 cannabinoid receptor antagonist SR141716A (1 mg kg−1) or the potassium channel blocker TEA (100 mg kg−1) caused only transient changes in resting blood pressure (Figure 1B; not shown for solvent). The hypotension produced by acetylcholine after blockade of NO synthesis was not altered by solvent (Figure 2). SR141716A also did not change the acetylcholine-evoked hypotension (Figures 1B and 2). In contrast, injection of TEA inhibited the vasodilatory response to acetylcholine by more than 50% (Figures 1B and 2).
Discussion
In rabbits in which prostanoid synthesis was inhibited by indomethacin, the acetylcholine-evoked vasodilation was only partly inhibited by the NO synthase inhibitor L-NAME (30 mg kg−1). This dose of L-NAME completely inhibits NO synthesis in rats and rabbits (Rees et al., 1990; Persson et al., 1991); indeed, a 3 fold higher dose of L-NAME did not cause further inhibition of the acetylcholine-evoked vasodilation in the present study. The non-selective potassium channel blocker TEA inhibited the residual acetylcholine-evoked prostanoid- and NO-independent vasodilation. TEA blocked EDHF-mediated vasodilation in several blood vessels (e.g., Cowan et al., 1993; Randall et al., 1997). It is, thus, very probable that the prostanoid- and NO-independent vasodilation produced by acetylcholine in the present experiments is due to EDHF, confirming previous observations on the significant contribution of EDHF to vasodilation in rabbit blood vessels (Persson et al., 1991; Mügge et al., 1991; Cowan et al., 1993; Lischke et al., 1995; Dong et al., 1997).
Two observations argue against the hypothesis that in the rabbit, EDHF is a cannabinoid which activates CB1 cannabinoid receptors on vascular smooth muscle. First, the mixed CB1/CB2 cannabinoid receptor agonist WIN55212-2 did not lower blood pressure in pithed rabbits in which vascular tone was maintained by an infusion of noradrenaline (Niederhoffer & Szabo, 1999; Figure 1A in the present paper). The dose of WIN55212-2 was sufficient to produce a CB1-receptor-mediated, strong peripheral presynaptic inhibition of noradrenaline release from postganglionic sympathetic neurons (Niederhoffer & Szabo, 1999). It is, therefore, unlikely that vasodilatory CB1 or CB2 (the affinity of WIN55212-2 is higher for CB2 than for CB1 receptors) cannabinoid receptors exist in rabbit resistance vessels (on endothelial or smooth muscle cells). Second, the selective CB1 cannabinoid receptor antagonist SR141716A did not modify the indomethacin- and L-NAME-resistant vasodilation produced by acetylcholine. The dose of SR141716A used in the present study was sufficient to block peripheral CB1 cannabinoid receptors in rabbits: already a 2 fold lower dose of SR141716A prevented the peripheral presynaptic inhibition produced by WIN55212-2 (Niederhoffer & Szabo, 1999).
Several recent studies using isolated rat, porcine and bovine blood vessels also came to the conclusion that EDHF is not an endogenous cannabinoid: either SR141716A did not antagonize the EDHF-mediated vasodilation, or cannabinoids did not cause direct smooth muscle relaxation or hyperpolarization (Plane et al., 1997; Zygmunt et al., 1997; Chataigneau et al., 1998; Fulton & Quilley, 1998; Pratt et al., 1998). Our study confirms these observations, and extends them to another species, the rabbit, and to a whole animal preparation.
What is EDHF if not an endogenous cannabinoid? A recent publication suggests it is potassium released from endothelial cells (Edwards et al., 1998).
Acknowledgments
The authors gratefully acknowledge the advice and support of Klaus Starke. We thank Sanofi (France) for generous supply of SR141716A. The study was supported by the Alexander von Humboldt Foundation (Germany), the INSERM (France) and the Deutsche Forschungsgemeinschaft (Sz 72/2-2; Germany).
Abbreviations
- CB
cannabinoid
- EDHF
endothelium-derived hyperpolarizing factor
- NO
nitric oxide
References
- CHATAIGNEAU T., FELETOU M., THOLLON C., VILLENEUVE N., VILAINE J-P., DUHAULT J., VANHOUTTE P.M. Cannabinoid CB1 receptor and endothelium-dependent hyperpolarization in guinea-pig carotid, rat mesenteric and porcine coronary arteries. Br. J. Pharmacol. 1998;123:968–974. doi: 10.1038/sj.bjp.0701690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COWAN C.L., PALACINO J.J, , NAJIBI S., COHEN R.A. Potassium channel-mediated relaxation to acetylcholine in rabbit arteries. J. Pharmacol. Exp. Ther. 1993;266:1482–1489. [PubMed] [Google Scholar]
- DONG H., WALDRON G.J., GALIPEAU D., COLE W.C., TRIGGLE C.R. NO/PGI2-independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br. J. Pharmacol. 1997;120:695–701. doi: 10.1038/sj.bjp.0700945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDWARDS G., DORA K.A., GARDENER M.J., GARLAND C.J., WESTON A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. Endothelium-derived hyperpolarizing factor–a critical appraisal. Prog. Drug Res. 1998;50:107–133. doi: 10.1007/978-3-0348-8833-2_2. [DOI] [PubMed] [Google Scholar]
- FULTON D., QUILLEY J. Evidence against anandamide as the hyperpolarizing factor mediating the nitric oxide-independent coronary vasodilator effect of bradykinin in the rat. J. Pharmacol. Exp. Ther. 1998;286:1146–1151. [PubMed] [Google Scholar]
- GARLAND C.J., PLANE F., KEMP B.K., COCKS T.M. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol. Sci. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- LISCHKE V., BUSSE R., HECKER M. Selective inhibition by barbiturates of the synthesis of endothelium-derived hyperpolarizing factor in the rabbit carotid artery. Br. J. Pharmacol. 1995;115:969–974. doi: 10.1111/j.1476-5381.1995.tb15905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MÜGGE A., LOPEZ J.A.G., PIEGORS D.J., BREESE K.R., HEISTAD D.D. Acetylcholine-induced vasodilatation in rabbit hindlimb in vivo is not inhibited by analogues of L-arginine. Am. J. Physiol. 1991;260:H242–H247. doi: 10.1152/ajpheart.1991.260.1.H242. [DOI] [PubMed] [Google Scholar]
- NIEDERHOFFER N., SZABO B. Effect of the cannabinoid receptor agonist WIN55212-2 on sympathetic cardiovascular regulation. Br. J. Pharmacol. 1999;126:457–466. doi: 10.1038/sj.bjp.0702337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERSSON M.G., WIKLUND N.P., GUSTAFSSON L.E. Nitric oxide requirement for vasomotor nerve-induced vasodilatation and modulation of resting blood flow in muscle microcirculation. Acta Physiol. Scand. 1991;141:49–56. doi: 10.1111/j.1748-1716.1991.tb09043.x. [DOI] [PubMed] [Google Scholar]
- PLANE F., HOLLAND M., WALDRON G.J., GARLAND C.J., BOYLE J.P. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. Br. J. Pharmacol. 1997;121:1509–1511. doi: 10.1038/sj.bjp.0701361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRATT P.F., HILLARD C.J., EDGEMOND W.S., CAMPBELL W.B. N-arachidonylethanolamide relaxation of bovine coronary artery is not mediated by CB1 cannabinoid receptor. Am. J. Physiol. 1998;274:H375–H381. doi: 10.1152/ajpheart.1998.274.1.H375. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., ALEXANDER S.P.H., BENNETT T., BOYD E.A., FRY J.R., GARDINER S.M., KEMP P.A., MCCULLOCH A.I., KENDALL D.A. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem. Biophys. Res. Comm. 1996;229:114–120. doi: 10.1006/bbrc.1996.1766. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. Eur. J. Pharmacol. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., KENDALL D.A. Endocannabinoids: a new class of vasoactive substances. Trends Pharmacol. Sci. 1998;19:55–58. doi: 10.1016/s0165-6147(97)01161-9. [DOI] [PubMed] [Google Scholar]
- RANDALL M.D., McCULLOCH A.I., KENDALL D.A. Comparative pharmacology of endothelium-derived hyperpolarizing factor and anandamide in rat isolated mesentery. Eur. J. Pharmacol. 1997;333:191–197. doi: 10.1016/s0014-2999(97)01137-0. [DOI] [PubMed] [Google Scholar]
- REES D.D., PALMER R.M.J., SCHULZ R., HODSON H.F., MONCADA S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE R., HILEY C.R. A comparison of EDHF-mediated and anandamide-induced relaxations in the rat isolated mesenteric artery. Br. J. Pharmacol. 1997;122:1573–1584. doi: 10.1038/sj.bjp.0701546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE R., HILEY C.R. The actions of some cannabinoid receptor ligands in the rat isolated mesenteric artery. Br. J. Pharmacol. 1998;125:533–541. doi: 10.1038/sj.bjp.0702111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZYGMUNT P.M., HÖGESTÄTT E.D., WALDECK K., EDWARDS G., KIRKUP A.J., WESTON A.H. Studies on the effects of anandamide in rat hepatic artery. Br. J. Pharmacol. 1997;122:1679–1686. doi: 10.1038/sj.bjp.0701601. [DOI] [PMC free article] [PubMed] [Google Scholar]