

Abstract
This study describes the pharmacological comparison of the muscarinic partial agonists sabcomeline, xanomeline and milameline at human cloned muscarinic receptor subtypes (hM1–5).
Radioligand binding studies at the hM1–5 muscarinic receptor subtypes were compared with functional studies using microphysiometry using carbachol as the standard full agonist.
In binding assays none of the compounds studied displayed preferential affinity for the M1,3,4 or M5 subtypes although carbachol was less potent at hM1 than hM3,4,5.
In functional studies, all of the compounds studied displayed similar levels of efficacy across the muscarinic receptors with the exception of M3, where there was a large apparent receptor reserve and the compounds behaved essentially as full agonists.
Sabcomeline was the most potent agonist in functional studies but also showed the lowest efficacy. In terms of potency, xanomeline showed some selectivity for M1 over M2 receptors and milameline showed some selectivity for M2 over M1 receptors.
These results show the value of microphysiometry in being able to compare receptor pharmacology across subtypes irrespective of the signal transduction pathway.
None of the partial agonists showed functional selectivity for M1 receptors, or indeed any muscarinic receptor, in the present study.
Keywords: Muscarinic receptor, partial agonists, selectivity, microphysiometry
Introduction
Muscarinic acetylcholine receptors (mAChR) are members of the G protein receptor superfamily and are widely distributed throughout the periphery and the central nervous system (CNS) (Caulfield, 1993). Genes encoding five receptor subtypes have been identified with distinct amino acid sequence and ligand binding properties (Bonner, 1989). This receptor diversity together with their differential distribution in the CNS has led to the search for selective agents with therapeutic utility, notably in Alzheimer's disease (Ehlert et al., 1994). Although the five muscarinic receptors (M1–M5) all couple to G proteins, stimulation of the M1, M3, and M5 mAChR subtypes leads to activation of phospholipase C whereas stimulation of the M2 and M4 subtypes leads to inhibition of adenylyl cyclase (Bonner, 1989). Although some studies have reported a differential potency for agonists at the mAChR subtypes, no comparison has been made between all five subtypes in the same response system due to the differing functional coupling.
The Cytosensor microphysiometer (Molecular Devices, Sunnyvale, CA, U.S.A.) measures the extracellular acidification rate as a result of the production of acid metabolites and has proved to be a useful tool to measure the integrated functional response to receptor activation in recombinant systems (Baxter et al., 1994). As such, microphysiometry can be used to determine receptor activation independent of the intracellular signal transduction pathway. Pharmacological properties such as agonist potency and agonist efficacy can vary at the same receptor expressed in different cell lines. Comparisons of such properties at different mAChRs has been confused by the use of different cell lines e.g. CHO, BHK and A9L (Shannon et al., 1994), Sf9 (Kukkonen et al., 1996) and by the use of different functional models e.g. phosphoinositide hydrolysis in intact cells and [35S]-GTP-γ-S binding in membranes (Lazareno & Birdsall, 1993). Such comparisons have led to reports of subtype selectivity for some agonists such as xanomeline (Shannon et al., 1994).
The aim of this study, therefore, was to compare the pharmacological profile of a series of muscarinic agonists at all five human mAChR subtypes expressed in the same cell line (CHO) using the same functional model (microphysiometry) in an attempt to circumvent the above issues. Further, the functional potency and selectivity of the agonists were compared with their radioligand binding affinity at the cloned muscarinic receptors in the same cell lines. The agonists studied have been claimed to show functional selectivity in in vitro and in vivo models for the M1 receptor, such as xanomeline (Shannon et al., 1994), milameline (Toja et al., 1991) and sabcomeline (SB 202026, Loudon et al., 1997). It was therefore important to determine whether this functional selectivity was reflected in selectivity for cloned muscarinic receptor subtypes.
Methods
Cells
CHO cells stably expressing human mAChRs M1–M5 were obtained from National Institute of Mental Health (Bethesda, MD, U.S.A.; see Bonner, 1989). Cells were maintained in α-minimum essential medium (Gibco, Paisley, Scotland) containing 10% foetal bovine serum (Gibco, North American) at 37°C under 5% CO2/ 95% O2. Cells were grown to confluence and harvested by scraping in fresh medium.
For microphysiometry, cells were seeded into cytosensor cell capsules 24 h prior to experiments at a density of 300,000 cells per well. M2 and M4 cells were exposed to sodium butyrate to improve the response by synchronizing the cell cycle through arresting cell division and therefore allowing optimum protein/receptor expression. For butyrate treatment, cells were incubated with 5 mM sodium butyrate (sterilized by filtration) for 24 h on seeding into cytosensor capsules in medium containing 10% foetal bovine serum. For radioligand binding, cells were grown in 175 cm3 flasks and harvested by dispersal in calcium-free saline.
Radioligand binding
The binding of [3H]-Quinuclidinyl benzilate (QNB) to the muscarinic cloned receptors was performed as described elsewhere (Loudon et al., 1997). Briefly cells were harvested and homogenized in ice-cold Tris buffer (50 mM Tris-HCl, pH 7.4 at 37°C) and membranes obtained by centrifugation (24,000 ×g for 15 min). The membranes were washed twice by resuspension and centrifugation and then stored at −70°C in 1 ml aliquots at c 2×108 cells ml−1.
Concentration-inhibition curves were constructed and analysed as above to obtain IC50 values and the Ki determined using the Cheng-Prusoff equation where Ki=IC50/(1+[L]/KD) (Cheng & Prusoff, 1973). Data were expressed as pKi (−log10 (Ki))±standard error of the mean (s.e.m).
Microphysiometry
Changes in extracellular acidification were determined using the Cytosensor microphysiometer (Molecular Devices). Cells were perfused with media via a peristaltic pump, during which the pH of the microenvironment surrounding the sensor was kept constant. The removal of acid from the cells by the perfusate was periodically halted (pump turned off), allowing a build up of acid metabolites and, therefore, a change in chamber pH (acidification rate). This on–off cycle was repeated throughout the experiment and the effect of compounds determined by adding the compound to the chamber through a valve. An on–off cycle of 1 min on and 30 s pump off was employed for most of these experiments. Acidification rate measurements were optimized for the agonist exposure time, agonist addition time (within the cycle) and the time during the pump-off cycle when measurements were taken. Assay conditions were optimized for each individual cell line, CHO cells transfected with hM1, hM2, hM3, hM4 or hM5 receptor subtype, using carbachol as a control agonist. Cells were perfused at a flow rate of 100 μl min−1 with a low buffered, sterile filtered DMEM medium (bicarbonate-free DMEM, Gibco 52100-021), glutamine (2 mM), NaCl (44 mM), pH 7.4.
Concentration-effect curves were obtained by exposing the cells sequentially to increasing concentrations of agonist for periods of up to 1 min (as detailed in Results) at intervals of 21 min. No desensitization to administration of carbachol (100 μM) was observed using a 21 min cycle and no change in agonist potency or efficacy was seen using a longer interval of 30 min. The response was taken as the peak increase in acidification rate upon addition of agonist over basal taken immediately prior to agonist challenge. Data was normalized as a mean response to a maximal concentration (100%) of carbachol (100 μM) carried out at the start and end of the agonist concentration-effect curve. For antagonist studies, a control concentration-response curve to carbachol was conducted and the cells were then exposed to atropine for at least 42 min prior to construction of a further carbachol concentration-effect curve in the presence of atropine. Each chamber therefore acted as its own control. Drug additions were performed using the Cytosampler autosampler (Molecular Devices) from deep well blocks. Concentration-effect curves were constructed as the peak acidification response seen at increasing concentrations of the agonist and analysed using a 4-parameter logistic equation to give EC50, slope, minimum and maximum (Bowen & Jerman, 1995). The EC50 values were then expressed as pEC50 (−log10(EC50)). Antagonist data were analysed as the ability of the antagonist to shift the agonist concentration-effect curve and defined as KB [antagonist].M/concentration ratio−1, where concentration ratio is the EC50 obtained in the presence of the antagonist divided by that obtained in the absence of the antagonist (Arunlakshana & Schild, 1959). Data were expressed as pKB (−log10(KB)). Experiments were repeated and data expressed as the mean±s.e.mean.
Drugs and solutions
All cell culture chemicals were from Gibco (Paisley, Scotland). Carbachol was from R.B.I. (Semat, U.K.), atropine from Sigma (Poole, U.K.) and xanomeline, milameline and sabcomeline were synthesized at SmithKline Beecham. [3H]-[QNB], 49 Ci mmol−1, was purchased from DuPont N.E.N.
Results
Radioligand binding
The potencies of the various agonists to inhibit [3H]-QNB binding to cloned M1, M3, M4 and M5 muscarinic receptors is shown in Table 1. The level of specific [3H]-QNB binding to membranes from hM2 expressing cells was too low to obtain quantifiable estimates of inhibitory potency. In the other cell lines, maximal [3H]-QNB binding (Bmax, pmole mg−1 protein) was hM1 5.81, hM3 5.42, hM4 0.92, hM5 0.92 with KD (nM) values for [3H]-QNB of 0.61, 0.57, 0.20 and 0.26 respectively (values are the mean of two separate determinations).
Table 1.
Binding profile of muscarinic agonists at human cloned muscarinic receptor subtypes

Microphysiometry optimization
Acidification rate measurements and agonist exposure times were optimized as: M1–pump cycle time was 1 min 30 s, pump on 1 min 13 s with a 32 s exposure to test compounds, commencing 15 s prior to pump off and data collection for 13 s commencing 2 s after pump off; M2 and M3–pump cycle time was 1 min 30 s, pump on 1 min with a 1 min 30 s exposure to test compound, data collection for 20 s commencing 8 s after pump off ; M4 and M5–pump cycle time 1 min 30 s, pump on 1 min with a 45 s exposure to test compound commencing 15 s prior to pump off, data collection for 20 s commencing 8 s after pump off. With longer agonist exposure times, a reduced functional response was seen presumably reflecting desensitization.
Acidification rate optimization was complex at the M1 receptor where the response to carbachol was rapid, large and associated with a non-linear increase in acidification rate (Figure 1). The acidification rate measurement was taken during the initial rapid phase (first 13 s) and with a reduced carbachol exposure time otherwise a reduced functional potency and efficacy to carbachol was seen. Thus the acidification rate to a sub-maximal concentration of carbachol (10 μM) was typically 476±32 μvolts s−1 (n=4) (corresponding to the initial phase seen in Figure 1) using an initial 13 s acidification rate measurement during a 17 s pump off cycle, compared with 138±5 μvolts s (n=4) (corresponding to the second phase in Figure 1) using a 20 s acidification rate measurement during a 30 s pump off cycle.
Figure 1.
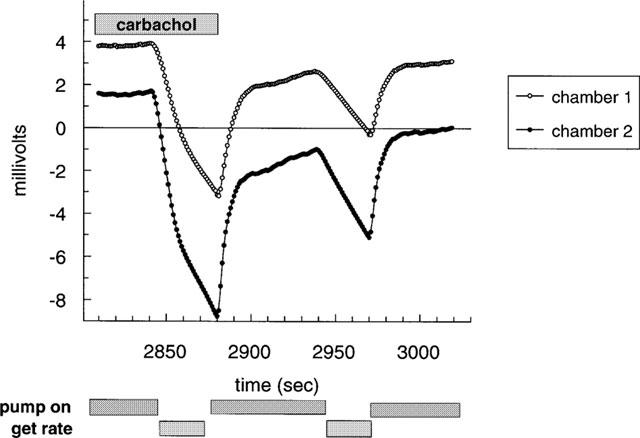
Biphasic change in acidification seen with carbachol at hM1 receptor. Shown are typical traces from two individual chambers where carbachol (10 μM) was introduced to the chambers 30 s prior to pump off and was present throughout the 30 s pump off cycle prior to wash out on resumption of perfusion. Time (s) is from start of experiment.
Microphysiometry optimisation
In non-transfected cells, none of the agonists at concentrations up to 100 μM, had any significant effect on acidification rates. In transfected cells, all of the agonists induced concentration-dependent increases in acidification rates at the cloned muscarinic receptors. Typically basal acidification rates were 100 μvolts s−1 and this was increased by maximal concentration of carbachol to 880, 220, and 210 μvolts s−1 at hM1, hM3, and hM5 receptors respectively. Acidification responses at hM2 and hM4 receptor were too small to quantify in normal cells but increased with butyrate treatment (data shown from butyrate treated cells) such that typical response rates at hM2 and hM4 to carbachol were 150 and 180 μvolts s−1 respectively. At the hM1 receptor, sabcomeline was more potent than milameline and carbachol, but also had the lowest efficacy (Emax). Milameline and xanomeline were also partial agonists (Table 2 and Figure 2). Comparable results were seen at the hM2 and hM5 receptor (Table 2). A similar trend was seen at the hM3 receptor, where sabcomeline was the most potent and had the lowest efficacy, but here xanomeline and milameline were full agonists with respect to carbachol (Figure 3). At the hM4 receptor, xanomeline appeared to be a full agonist whilst milameline and sabcomeline were partial agonists (Table 2). In general, sabcomeline was the most potent agonist, xanomeline showed some selectivity for hM1 over hM2 whereas milameline showed functional selectivity for hM2 over hM1. Carbachol was markedly less potent at hM1 receptors than the other mAChR subtypes. The functional efficacy for carbachol at the muscarinic receptors was expressed as the ratio between concentration of carbachol producing half-maximal response(EC50) and that needed for half maximal receptor occupancy (Ki), which gives an indication of receptor reserve (Kenakin, 1993) and which was 40 at hM1, 290 at hM3, 2 at hM4 and 20 at hM5.
Table 2.
Functional profile of muscarinic agonists at human cloned muscarinic receptor subtypes using microphysiometry
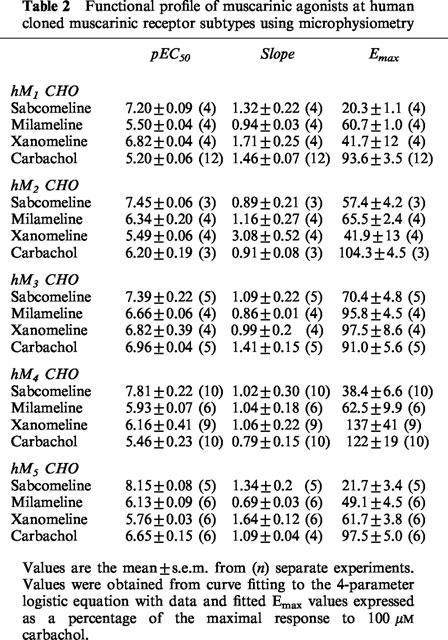
Figure 2.
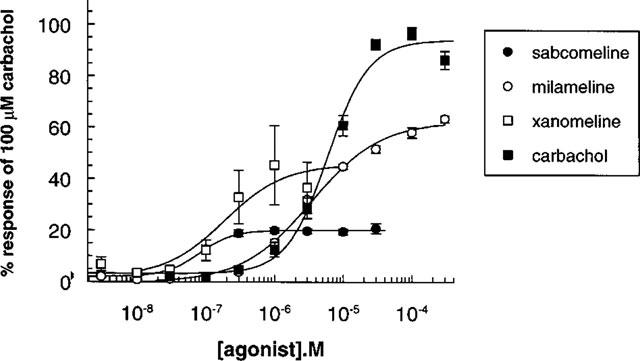
Stimulation of extracellular acidification in CHO cells stably expressing the hM1 receptor by muscarinic agonists. Data points with error bars represent the mean±s.e.m. of three separate experiments.
Figure 3.
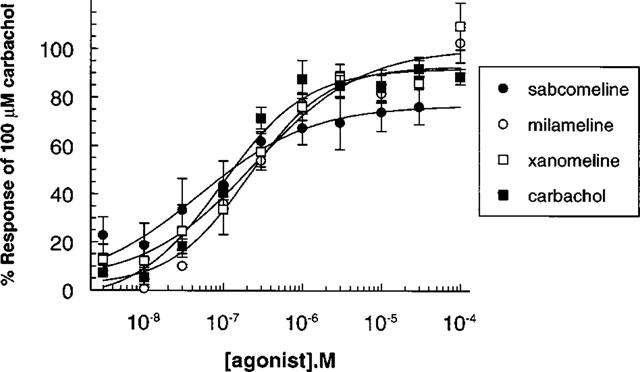
Stimulation of extracellular acidification in CHO cells stably expressing the hM3 receptor by muscarinic agonists. Data points with error bars represent the mean±s.e.m. of three separate experiments.
Atropine (10 nM) was a potent antagonist of the carbachol induced acidification response at M1, M3, M4 and M5 receptors shifting the curve to the right in a competitive manner. Reliable shift data could not be generated at the M2 receptor because of the low level of the response. Affinity estimates (pKB) for atropine at M1, M3, M4 and M5 receptors were 9.17±0.04 (8), 9.70±0.04 (7), 9.29±0.09 (8) and 8.99±0.02(8) respectively (results are mean±s.e.m. from n separate chambers).
Discussion
The present study is the first to compare the functional properties of a series of muscarinic receptor agonists at all five human muscarinic receptor subtypes in the same assay system. Previous studies have compared the pharmacology at selected muscarinic receptors, usually as a result of differences in the intracellular signalling pathways employed by the muscarinic receptors. Thus M1, M3 and M5 preferentially couple to phospholipase C while M2 and M4 preferentially couple to inhibition of adenylyl cyclase. By using the Cytosensor microphysiometer, we have been able to measure activation of the muscarinic receptors irrespective of the signal transduction mechanism. The acidification response seen at the hM2 and hM4 receptors was small and the cells required pre-treatment with butyrate in order to increase this response (see Kassis et al., 1984). At all receptors prolonged carbachol exposure times resulted in a reduced functional response, requiring optimization of agonist exposure times. This desensitization was particularly marked at the hM1 such that carbachol exposure time was kept to 32 s.
The marked increase in functional potency compared to binding affinity for carbachol at the muscarinic receptor subtypes (with the exception of the hM4 receptor) suggests the presence of receptor reserve. This is particularly so at the hM3 receptor, suggesting a large receptor reserve, which may explain why milameline and xanomeline appear as full agonists and sabcomeline as an almost full agonist at this receptor. Differential increases in functional potency compared to binding potency may also reflect differences in amplification factors (see Kenakin, 1993). This is unlikely to be the case in the present experiments as previous studies have found that different functional models (phosphoinositide hydrolysis and intracellular calcium mobilization) give similar potency estimates to microphysiometry at hM1 and hM3 receptors and, further, show that microphysiometry functional potency estimates vary with receptor density (Baxter et al., 1994). The study of Baxter et al. (1994) also suggests that promiscuous signalling is unlikely to be responsible for this difference between functional potency and binding affinity, in that similar potency estimates were obtained using different functional models at different levels of receptor expression e.g. intracellular calcium mobilisation which would reflect Gq activation and microphysiometry which would reflect total G-protein activation. The marked increase in functional potency compared to binding affinity may also reflect signal amplification processes as the events measured using microphysiometry are downstream of the initial binding event. Also, for many G-protein coupled receptors, different agonist and antagonist affinity states exist such that it is difficult to correlate agonist binding affinity with functional potency (see Richards, 1991).
Sabcomeline was the most potent agonist tested on function but displayed similar binding affinities to xanomeline. For sabcomeline, in general there was a good correlation between functional potency and binding affinity. Sabcomeline was a low efficacy partial agonist at all the muscarinic receptor subtypes with the exception of the hM3 where there was a large apparent receptor reserve. This therefore confirms in vitro studies (Loudon et al., 1997) and shows the partial agonist activity of sabcomeline is retained at human mAChR subtypes.
Xanomeline was less potent on function using microphysiometry than on radioligand binding. Anomalous kinetics have been observed with xanomeline, suggesting a slow off-rate (Christopoulos & El-Fakahany, 1997), which may have a differential effect on binding and function because of the different agonist incubation times. In terms of functional potency, xanomeline also showed some selectivity for the hM1 receptor compared to the hM2 receptor. Due to low levels of specific binding (presumably reflecting a lower receptor density) binding data comparing hM1 to hM2 are not available in this study and the effect of butyrate treatment on radioligand binding was not investigated. This data does not appear to be available in the literature, but binding studies to homogenates from different rat brain regions (cortex, M1 rich; brain stem, M2 rich) supports this separation (Shannon et al., 1994).
Xanomeline had a higher intrinsic activity than sabcomeline and was a full receptor agonist at hM3 and hM4 subtypes. In this respect, milameline appeared to be similar to xanomeline with the exception of a lower functional potency at hM1 receptors. In functional studies using the microphysiometer, milameline therefore appeared to show selectivity for the hM2 receptor over the hM1 receptor. It should be noted that all results have compared efficacy values to carbachol assuming that carbachol is a full agonist at all five receptors.
It has been reported that some of the receptor agonists tested display functional selectivity for the hM1 receptor (e.g. sabcomeline, Loudon et al., 1997; xanomeline, Shannon et al., 1994). This was based on functional selectivity in in vitro and in vivo models rather than receptor subtype selectivity. In radioligand binding studies, with the exception of carbachol which shows a low affinity for the hM1 receptor, all of the agonists displayed similar affinities across the muscarinic receptor subtypes, i.e. they did not demonstrate receptor subtype binding selectivity.
The present study shows that the recently developed muscarinic partial agonists sabcomeline, milameline and xanomeline do not exhibit significant selectivity for muscarinic receptor subtypes expressed in cell lines. They have similar affinity across the muscarinic receptor subtypes and, in general, they have similar efficacy across the muscarinic receptor subtypes, although this is difficult to verify at the hM3 receptor due to the high level of receptor reserve. The tissue response to an agonist is a function of different factors including affinity, efficacy, receptor number and receptor-response coupling (Ringdahl et al., 1987). Partial agonists may therefore exhibit in vivo functional selectivity due to tissue differences and in vivo studies will reveal if this is so. This has been suggested for sabcomeline and related to potential utility in cognition (Loudon et al., 1997). The reported functional selectivity of sabcomeline and xanomeline is consistent with their partial agonist activity at mAChRs but is not consistent with mAChR subtype selectivity. The present study also shows the value of microphysiometry in being able to carry out functional studies irrespective of the signal transduction pathway, allowing comparative studies on different receptor subtypes to be conducted.
Acknowledgments
The authors would like to Frances Jewitt for her technical support in the provision of cell culture work.
Abbreviations
- CNS
central nervous system
- DMEM
Dulbecco's modification of Eagle's medium
- mAChR
muscarinic acetylcholine receptor
- QNB
quinuclidinyl benzilate
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAXTER G.T., YOUNG M-L., MILLER D.L., OWICKI J.C. Using microphysiometry to study the pharmacology of exogenously expressed m1 and m3 muscarinic receptors. Life Sci. 1994;55:573–583. doi: 10.1016/0024-3205(94)00483-8. [DOI] [PubMed] [Google Scholar]
- BONNER T.I. The molecular basis for muscarinic receptor diversity. Trends Neurosci. 1989;12:148–150. doi: 10.1016/0166-2236(89)90054-4. [DOI] [PubMed] [Google Scholar]
- BOWEN W. P., JERMAN J.C. Nonlinear regression using spreadsheets. Trends Pharmacol. Sci. 1995;16:413–417. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P. Muscarinic receptors–characterization, coupling and function. Pharmacol. Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- CHENG Y., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of an inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CHRISTOPOULOS A., EL-FAKAHANY E.E. Novel persistent activation of muscarinic M1 receptors by xanomeline. Eur. J. Pharmacol. 1997;334:R3–R4. doi: 10.1016/s0014-2999(97)01162-x. [DOI] [PubMed] [Google Scholar]
- EHLERT F.J., ROESKE W.R., YAMAMURA H.I. Muscarinic receptors and novel strategies for the treatment of age-related brain disorders. Life. Sci. 1994;55:2135–2145. doi: 10.1016/0024-3205(94)00394-7. [DOI] [PubMed] [Google Scholar]
- KASSIS S., HENNEBERRY R.C., FISHMAN P.H. Induction of catecholamine-responsive adenylate cycalse in HeLa cells by sodium butyrate. J. Biol. Chem. 1984;259:4910–4916. [PubMed] [Google Scholar]
- KENAKIN T.P. Pharmacologic analysis of drug-receptor interaction. New York: Raven Press; 1993. [Google Scholar]
- KUKKONEN J.P., NASMAN J, , OJALA P., OKER-BLOM C., AKERMAN K.E.O. Functional properties of muscarinic receptor subtypes Hm1, Hm3 and Hm5 expressed in Sf9 cells using the baculovirus expression system. J. Pharmacol. Exp. Ther. 1996;279:593–601. [PubMed] [Google Scholar]
- LAZARENO S., BIRDSALL N.J.M. Pharmacological characterization of acetylcholine-stimulated [35S]-GTPγS binding mediated by human muscarinic m1-m4 receptors: antagonist studies. Br. J. Pharmacol. 1993;109:1120–1127. doi: 10.1111/j.1476-5381.1993.tb13738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOUDON J.M., BROMIDGE S.M., BROWN F., CLARK M.S.G., HATCHER J.P., HAWKINS J., RILEY G.J., NOY G., ORLEK B.S. SB 202026:A novel muscarinic partial agonist with functional selectivity for M1 receptors. J. Pharmacol. Exp. Ther. 1997;283:1059–1068. [PubMed] [Google Scholar]
- RICHARDS M.H. Pharmacology and second messenger interactions of cloned muscarinic receptors. Biochem. Pharmacol. 1991;42:1645–1653. doi: 10.1016/0006-2952(91)90498-t. [DOI] [PubMed] [Google Scholar]
- RINGDAHL B., ROCH M., JENDEN D.J. Regional differences in receptor reserve for analogs of oxotremorine in vivo: Implications for development of selective muscarinic agonists. J. Pharmacol. Exp. Ther. 1987;242:464–471. [PubMed] [Google Scholar]
- SHANNON H.E., BYMASTER F.P., CALLIGARO D.O., GREENWOOD B., MITCH C.H., SAWYER B.D., WARD J.S., WONG D.T., OLESEN P.H., SHEARDOWN M.J., SWEDBERG M.D.B., SUZDAK P.D., SAUERBERG P. Xanomeline: a novel muscarinic receptor agonist with functional selectivity for M1 receptors. J. Pharmacol. Exp. Ther. 1994;269:271–281. [PubMed] [Google Scholar]
- TOJA E., BONETTI C., BUTTI A., HUNT P., FORTIN M., BARZAGHI F., FORMENTO M.L., MAGGIONI A., NENCIONI A., GALLIANIN G. 1-Alkyl-1,2,5,6-tetrahydropyridine-3-carboxaldehyde-O-alkyl-oximes: A new class of potent orally active muscarinic agonists related to arecoline. Eur. J. Med. Chem. 1991;26:853–868. [Google Scholar]