

Abstract
The arachidonic acid derivative arachidonylethanolamide (anandamide) is an endogeneous ligand of cannabinoid receptors that induces pharmacological actions similar to those of cannabinoids such as Δ9-tetrahydrocannabinol (THC). We examined whether anandamide can influence excessive neuronal activity by investigating stimulation-induced population spikes and epileptiform activity in rat hippocampal slices. For this purpose, the effects of anandamide were compared with those of the synthetic cannabinoid agonist WIN 55,212-2 and its inactive S(−)-enantiomer WIN 55,212-3.
Both anandamide (1 and 10 μM) and WIN 55,212-2 (0.1 and 1 μM) decreased the amplitude of the postsynaptic population spike and the slope of the field excitatory postsynaptic potential (field e.p.s.p.) without affecting the presynaptic fibre spike of the afferents. At a concentration of 1 μM, WIN 55,212-2 completely suppressed the postsynaptic spike, whereas the S(−)-enantiomer WIN 55,212-3 produced only a slight depression. The CB1 receptor antagonist SR 141716 blocked the inhibition evoked by the cannabinoids. SR 141716 had a slight facilitatory effect on neuronal excitability by itself.
Anandamide shifted the input-output curve of the postsynaptic spike and the field e.p.s.p. to the right and increased the magnitude of paired-pulse facilitation indicating a presynaptic mechanism of action.
Anandamide and WIN 55,212-2, but not WIN 55,212-3, attenuated both stimulus-triggered epileptiform activity in CA1 elicited by omission of Mg2+ and spontaneously occurring epileptiform activity in CA3 elicited by omission of Mg2+ and elevation of K+ to 8 mM. The antiepileptiform effect of these cannabinoids was blocked by SR 141716.
In conclusion, cannabinoid receptors of the CB1 type as well as their endogeneous ligand, anandamide, are involved in the control of neuronal excitability, thus reducing excitatory neurotransmission at a presynaptic site, a mechanism which might be involved in the prevention of excessive excitability leading to epileptiform activity.
Keywords: Anandamide, cannabinoid CB1 receptors, hippocampus, paired-pulse facilitation, epileptiform activity
Introduction
Cannabis is known to produce euphoria leading to abuse and to cause cognitive and motor impairment. Beside these psychotropic effects, it is reported to possess antinociceptive, antiemetic and anticonvulsant effects (Carlini & Cunha, 1981; Karler & Turkanis, 1981; Abood & Martin, 1996). The effects of the major psychoactive component of cannabis, Δ9-tetrahydrocannabinol (THC) are mediated by CB1 receptors (Devane et al., 1992; Howlett, 1995) which are coupled to pertussis toxin-sensitive G proteins (Matsuda et al., 1990) and distributed widely throughout the CNS with highest density in the hippocampus (Herkenham, 1992; Matsuda et al., 1993; Gatley et al., 1998). Activation of CB1 receptors causes inhibition of adenylate cyclase, blockade of N- and P/Q-type-Ca2+-channels and increase in A-type K+ currents (Bidaut-Russell et al., 1990; Deadwyler et al., 1993; Mackie et al., 1993; Pacheco et al., 1993; Shen & Thayler, 1998). The CB1 receptors are in part located on axonal terminals where their activation causes inhibition of the release of several neurotransmitters, such as glutamate (Shen et al., 1996), acetylcholine (Gifford & Ashby, 1996; Gessa et al., 1997; Gifford et al., 1997), dopamine (Cadogan et al., 1997) and noradrenaline (Schlicker et al., 1997).
There is strong evidence that arachidonylethanolamide (anandamide) is an endogeneous agonist for the brain cannabinoid receptor (Devane et al., 1992; Di Marzo et al., 1994; Felder et al., 1996) which, in conjunction with the cannabinoid receptor forms a neuromodulatory system (Mechoulam et al., 1994). The highest level of anandamide has been found in the hippocampus (Felder et al., 1996). Anandamide is synthesized in neurons, released on depolarization and rapidly inactivated (Cadas et al., 1997; Deutsch & Chin, 1993; Di Marzo et al., 1994; Evans et al., 1994). Although the physiological role of anandamide has not yet been completely clarified, it has been shown to exhibit similar properties as THC in pharmacological and behavioural assays (Smith et al., 1994; Romero et al., 1995; Stein et al., 1996). Anandamide binds with relatively high affinity to the cloned rat (Vogel et al., 1993) and human (Felder et al., 1993) CB1 receptors and reduces cyclic AMP production (Felder et al., 1993, Vogel et al., 1993) and the N-type Ca2+ current (Mackie et al., 1993). It inhibits the formation of hippocampal long-term potentiation via CB1 receptors (Terranova et al., 1995), and thus may be involved in the impairment of memory processes associated with cannabis abuse in humans.
Since the hippocampal formation is enriched with CB1 receptors (Herkenham, 1992; Gatley et al., 1998) and known to be involved in the generation of seizure activity, the question arises of whether anandamide is involved in the control of seizure activity in this brain region. The aim of the present study was to further elucidate a possible physiological role of anandamide as an endogeneous ligand at the cannabinoid CB1 receptor and its involvement in control of neuronal excitation. For this purpose, the effects of anandamide were compared with those of the synthetic cannabinoid agonist, WIN 55,212-2 and its inactive S(−)-enantiomer WIN 55,212-3, on spontaneously occurring and stimulus-triggered epileptiform activity in the rat hippocampal slice.
Methods
Brain slice preparation
Experiments were performed on hippocampal slices from male Wistar rats (150–180 g). The rats were deeply anaesthetized with diethyl ether and killed by rapid decapitation. The brains were quickly removed from the skulls and the hippocampus of one hemisphere was isolated. Slices of 400 μm thickness were cut transversely to the longitudinal axis of the hippocampus by use of a McIlwain tissue chopper. Immediately after cutting, one slice was transferred into a submerged brain slice recording chamber, where it was continuously superfused with warmed (32°C) artificial cerebrospinal fluid (ACSF) at a flow rate of 3–4 ml min−1 and held down on a nylon net by a U-shaped piece of flattened platinum wire. The other slices were maintained at room temperature in an incubation chamber. The standard ACSF was continuously gassed with a mixture of 95% O2 and 5% CO2 and contained (in mM): NaCl 124, KCl 3, NaH2PO4 1.25, NaHCO3 26, CaCl2 2.5, MgSO4 2, glucose 10 at a pH of 7.4. In some experiments, a modified ACSF was used in which no MgSO4 was added (low Mg2+-ACSF) in order to evoke epileptiform activity. For recording of spontaneously occurring epileptiform activity, a low Mg2+/high K+-ACSF was superfused. This solution was nominally Mg2+-free, while the the concentration of KCl was elevated to 8 mM.
Stimulation and recording
The experimental protocol always included a recovery period of 1 h after slice preparation. For recordings of stimulus-evoked population spikes and field e.p.s.ps, the recording electrode was placed in stratum pyramidale and stratum radiatum of area CA1, respectively. The electrodes were pulled on a BB-CH-PC electrode puller (Mecanex S.A., Switzerland) from 1.5 mm borosilicate glass and filled with 3 M NaCl (resistance 5–10 MΩ). A concentric bipolar stainless steel electrode with 0.25 mm outer diameter (Rhodes Medical Instruments, U.S.A.) was positioned into the Schaffer collaterals (i.e. near the junction of CA1 and CA2 stratum radiatum) or in the alveus for orthodromic and antidromic activation of CA1 pyramidal neurons, respectively. Extracellular stimuli were rectangular current pulses of 60 μs in duration delivered every 15 s through a digitally controlled stimulus isolation unit (Axon Instruments, U.S.A.). At the beginning of each experiment, the stimulus intensity was adjusted until the responses to electrical stimulation were about 50% of the maximal response.
To construct stimulus-response curves, electrical stimuli of increasing intensity were applied to the Schaffer collaterals and the amplitude of the presynaptic fibre spike and the postsynaptic population spike as well as the maximum rate of change (i.e. slope) of the field e.p.s.p. was measured before (control) and at the end of a 60 min application of drug, and plotted as a function of stimulus intensity.
In paired-pulse stimulation experiments, stimulus pulses were delivered in pairs with an interval of 20, 40, 60, 80, 100, 150 and 200 ms between the stimuli and an interpair interval of 30 s. The response to the first stimulus of the pair was used to assess drug effects on synaptic transmission and the ratio of the second response to the first was used to assess their effect on paired-pulse facilitation. Responses evoked by ten consecutive stimulus pulses were averaged. The signal from the recording electrode was amplified by means of a DP 301 amplifier (Warner Instruments, U.S.A.). Analogue data were digitized and analysed using the data aquisition and analysis software TIDA (HEKA electronic, Germany).
Only the data of those hippocampal slices have been included into the present study which showed normal field potentials (i.e. no second population spike at maximal stimulation intensity) in response to electrical activation of Schaffer collaterals during the control in standard ACSF. Furthermore, the amplitudes of the population spikes had to be stable during a control period of at least 30 min prior to the application of drugs. During this control period differences in spike amplitude had to be below 5%.
Drugs
Anandamide, R(+)-WIN 55,212-2 and S(−)-WIN 55,212-3 were obtained from RBI (Cologne, Germany), and the CB1 receptor antagonist SR 141716 was a gift of Sanofi Recherche (Montpellier, France). Stock solutions (1–10 mM) of WIN 55,212-2, WIN 55,212-3, and SR141716 were prepared in DMSO and diluted with ACSF to the final concentration. Control experiments have revealed that the highest final DMSO concentration (0.1%) did not affect any of the measured parameters. All drugs were delivered through the superfusion medium. In all experiments, each drug application was preceded by a control period of at least 30 min.
Data analysis
All data are expressed as mean±standard deviation (s.d.). Comparisons of the effects of drug treatments (normalized as per cent of control) between groups of slices were performed using Student's t-test for differences between two independent means. The statistical significance of the difference of the amplitude of the electrophysiological responses prior to and following the administration of a drug was assessed with the paired Student's t-test. In both cases, differences were considered statistically significant when P⩽0.05. The amplitude of the population spike which appears as a large negative wave superimposed on a positive-going e.p.s.p. was determined as the length of a vertical line, drawn from the minimum of the population spike to the line that joined the two positive peaks of the field response. Drug effects on the field e.p.s.p. were determined as changes in the slope of the field e.p.s.p. to avoid contamination by the population spike. The e.p.s.p. slope was measured as the ascending gradient between 20 and 80% of the maximum field e.p.s.p.
Spontaneously occurring epileptiform discharges were quantified by coastline measurements which is implemented in the TIDA software system. The coastline index was determined by Apland & Cann (1995). In brief, the length of the line representing the epileptiform activity was measured at control (CIcon), after superperfusion of the slices by the Mg2+-free ACSF (CIepi) and after addition of the test compounds to the Mg2+-free ACSF (CIepi+test). The coastline index was determined by using the following formula
An index<100% indicates inhibition of burst activity, and index>100 indicates an enhancement of epileptiform activity.
Results
Effect of the cannabinoids on Schaffer collateral-commissural synaptic transmission
Anandamide evoked an attenuation of the synaptic field potentials in a concentration-dependent manner. At a concentration of 1 and 10 μM, it decreased the slope of the field e.p.s.p. by 14.4±7% (n=5, P⩽0.05) and 28.3±4% of control (n=10, P⩽0.001, Figure 1), respectively. The amplitude of the orthodromic population spike was diminished by 10.0±5% of control (n=6, P⩽0.05) at 1 μM and by 24.2±6% of control (n=12, P⩽0.001) at 10 μM (Figure 2). The inhibitory effect of anandamide was reversible during washout with ACSF. The amplitude of the presynaptic population spike which precedes the postsynaptic population spike was not affected by anandamide. As shown in Figure 2B, the inhibitory effect of anandamide (10 μM) was suppressed by a 30 min pretreatment with the selective CB1 receptor antagonist, SR 141716 (1 μM, n=6). SR 141716 slightly increased the amplitude of the orthodromic population spike by 10.8±8% of control (P⩽0.05) and evoked a second population spike in about half of the slices. The synthetic cannabinoid receptor ligand R(+)-WIN 55,212-2 (100 nM) caused a reversible attenuation of the postsynaptic population spike by 42.6±10% of control (n=8, P⩽0.001). As shown in Figure 3A, this compound completely suppressed the orthodromic population spike at a concentration of 1 μM in all slices tested (n=10). In contrast to the lower concentration (100 nM), the effect of 1 μM WIN 55,212-2 was irreversible during washout with ACSF. However, it was partially reversed by application of 1 μM SR 141716 and completely reversed by 2 μM SR 141716. Moreover, as shown in Figure 3B, the inhibitory effect of 1 μM WIN 55,212-2 was prevented by pretreatment with SR 141716 (1 μM, n=7). At a concentration of 1 μM, the less active enantiomer S(−)-WIN 55,212-3, however, produced a decrease in spike amplitude of only 9.8±3% of control (n=6, P⩽0.01). Neither the presynaptic fibre spike (Figures 2 and 3) nor the antidromic population spike elicited by stimulation of the axons of the neurons recorded from (data not shown) was affected by the cannabinoids.
Figure 1.
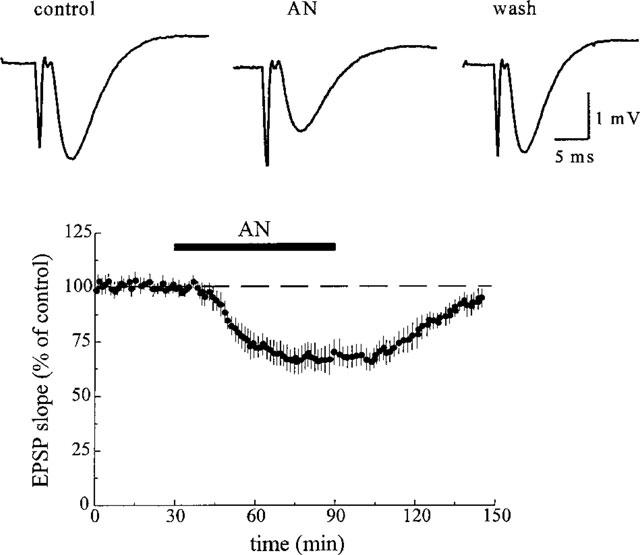
Inhibitory effect of anandamide (AN, 10 μM) on the field e.p.s.p. recorded in the CA1 stratum radiatum. The traces above the graph are the average of five subsequent responses from one representative experiment. The graph shows the time-course of the effect of anandamide on the slope of the field e.p.s.p. The data represent mean values±s.d. of six experiments.
Figure 2.
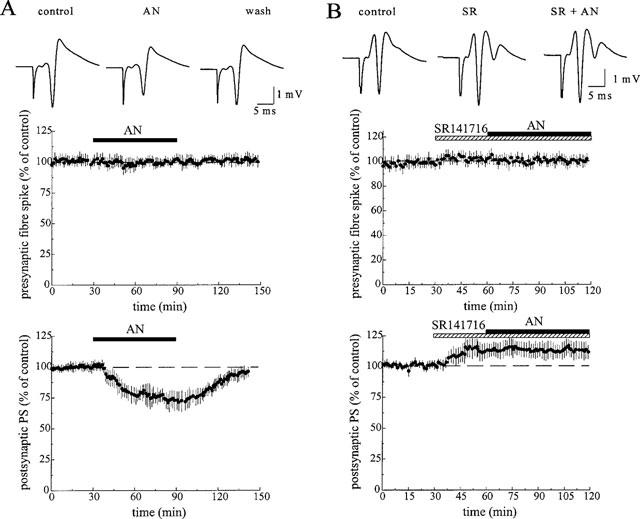
Effect of anandamide on the orthodromically evoked population spike (PS) in the hippocampal CA1 area. (A) Anandamide (AN, 10 μM) reversibly decreased the amplitude of postsynaptic population spike. (B) The anandamide-evoked inhibition was prevented by pretreatment with the CB1 receptor antagonist, SR 141716 (SR, 2 μM). The traces above the graphs derive from a representative experiment showing the average of five subsequent responses at the end of the control and at the end of drug-application. The graphs show the time-course of the amplitude of the presynaptic fibre spike and the postsynaptic population spike. The data represent mean values±s.d. of 9–12 experiments.
Figure 3.
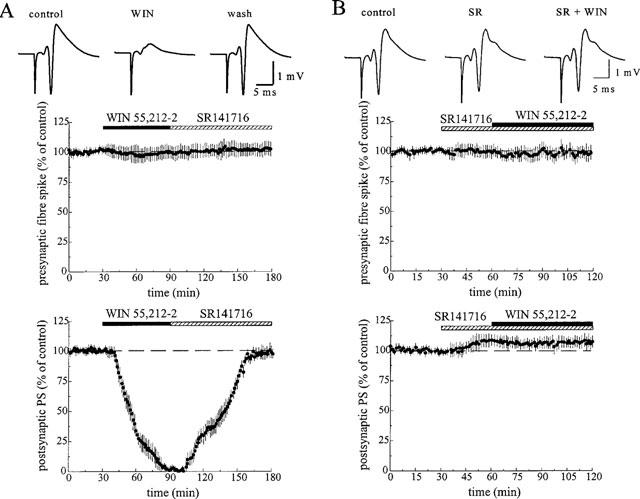
Effect of WIN 55,212-2 on the orthodromically evoked population spike (PS) in the hippocampal CA1 area. (A) WIN 55,212-2 (WIN, 1 μM) completely suppressed the orthodromic response, and this effect was reversed by SR 141716 (1 μM). (B) The suppression of the population spike by WIN 55,212-2 was prevented by pretreatment with the CB1 receptor antagonist, SR 141716 (SR, 1 μM). The traces above the graphs show the average of five subsequent responses at the end of the control and at the end of drug-application. The graphs show the time-course of the amplitude of the presynaptic fibre spike and the postsynaptic population spike. The data represent mean values±s.d. of 6–10 experiments.
To examine whether the effect of the cannabinoids depends on stimulus-intensity, the stimulus-response relationship for the orthodromic population spike and the field e.p.s.p. was determined. Both anandamide and WIN55,212-2 elicited a rightward shift in the relationship between stimulus intensity vs population spike amplitude and between stimulus intensity vs field e.p.s.p. slope, with no effect on the relationship between stimulus intensity vs presynaptic fibre spike amplitude (Figure 4), indicating that there was no obvious change in the axonal excitability. The relationship between the presynaptic fibre spike vs e.p.s.p. slope (not shown) indicated that a given fibre spike evoked a smaller e.p.s.p.
Figure 4.
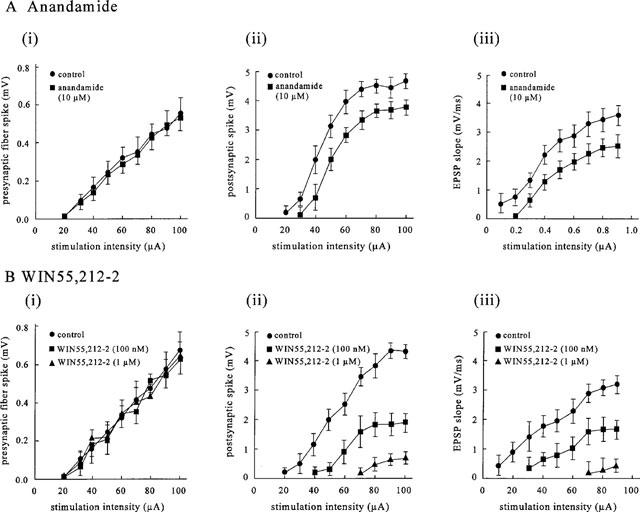
The effects of anandamide and WIN 55,212-2 on the stimulus-response relationship. The slices were stimulated with intensities ranging from subthreshold to maximal. For each response, the amplitude of the presynaptic fibre spike (i), the amplitude of the postsynaptic population spike (ii) and the slope of the field e.p.s.p. (iii) were measured and plotted as function of stimulus-intensity. The data represent mean values±s.d. of 5–6 experiments.
Paired-pulse facilitation
Since both the decrease of the maximum and the shift to the right of the input-output curve for the postsynaptic population spike and the field e.p.s.p. are not due to changes in excitability of the afferents, the question raises of whether they are induced by presynaptic or postsynaptic mechanisms. For this purpose, paired-pulse experiments were performed. If the anandamide-induced attenuation of the orthodromic response involves a presynaptic mechanism of action, it will be associated with an increase in the ration of the second to the first pulse response (P2/P1). In order to test this hypothesis, the magnitude of the facilitation quotient was determined at control prior to the application of anandamide and 60 min after starting the application of 10 μM anandamide. Paired-pulse induced field e.p.s.ps were evoked with various interstimulus intervals ranging from 20–200 ms. It has been shown recently (Debanne et al., 1996) that the magnitude of paired-pulse facilitation is also affected by changes in the amplitude of the first response. That is, an attenuation of the first response will cause per se an increase in the second response. In order to investigate if the enhancement of the paired-pulse facilitation by anandamide is due to the inhibition of the first response, the stimulus-intensity was increased so as to counteract the direct depressant effect of anandamide. At this condition, an enhancement effect of anandamide on paired-pulse facilitation was significantly higher than at the control. Figure 5A shows synaptic field responses to a pair of stimuli with an interstimulus interval of 40 ms. The field e.p.s.ps displayed a facilitation of 141.5±8% (n=7) when evoked in this manner. After superfusion of anandamide (10 μM), the response to the first stimulus was decreased, but the magnitude of paired-pulse facilitation significantly increased to reach a mean value of 172.3±8% (n=7). Figure 5B shows the increase in paired-pulse facilitation by anandamide (10 μM) at various interstimulus intervals.
Figure 5.
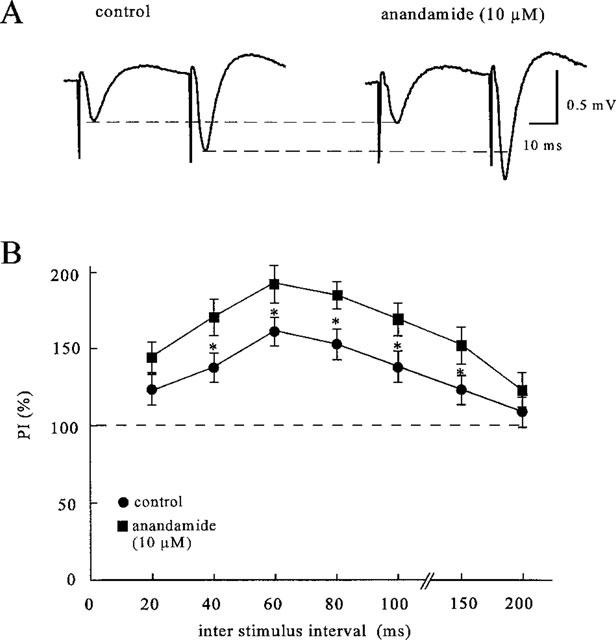
Enhancement of paired-pulse facilitation by anandamide. (A) Average of five consecutive field e.p.s.ps evoked by paired stimuli with an interstimulus interval of 40 ms at control and 60 min after starting the superfusion of anandamide (10 μM). At the end of the anandamide application, the stimulation intensity was increased to counteract a direct depressant effect of anandamide on the first field e.p.s.p. (B) Effects of anandamide (10 μM) on the paired-pulse index (PI) calculated from responses to paired-pulse stimulation of different intervals (20–200 ms) in the CA1 stratum radiatum. Anandamide significantly increased the PI values at all intervals (at least P⩽0.05, n=8). (C) The PI was calculated according to the formula PI=P2/P1×100% with P1 being the average of five responses to the first stimulus and P2 being the average of five responses to the second stimulus. Data are mean values±s.d.
Effects of the cannabinoids on epileptiform activity in hippocampal area CA1 and CA3
Epileptiform activity in response to electrical stimulation was induced after a control period of 30 min in standard ACSF by omission of Mg2+ from the ACSF. After recording 15–20 min in absence of Mg2+, the orthodromic response in CA1 stratum pyramidale was changed into an epileptiform bursting, made up by an increase in amplitude of the primary potential and by the building up of additional multiple epileptiform potentials evoked by the electrical stimulation of the afferents (Figure 6A and B). The amplitudes of these spikes became stable after another 15–20 min and were observed in control experiments to persist during the entire recording time of up to 6 h.
Figure 6.

Effect of the cannabinoids on stimulus-triggered epileptiform activity elicited by omission of Mg2+ from the superfusate. (A) Extracellularly recorded population spikes from one representative experiment out of eight similar ones showing the effect of anandamide. (B) Effects on the first, second and third population spike (PS) of the burst by anandamide alone and together with SR 141716, by WIN 55,212-2 and by WIN 55,212-3. The amplitudes of the spikes were normalized with respect to the amplitudes measured during superfusion with the nominal Mg2+-free bathing medium. The anandamide-evoked reduction of the spike amplitudes was significant (*P⩽0.001, n=8). Each column shows the mean±s.d.
As shown in Figure 6A, anandamide (10 μM) reversibly attenuated the stimulus-triggered epileptiform burst discharges elicited by omission of Mg2+ from the superfusate (n=8). The antiepileptiform effect of anandamide manifested itself as both a significant decrease in the amplitudes of the multiple population spikes (Figure 6B) and as a significant decrease in the number of the additional spikes from 8.3±1 at control to 5.9±1 after 60 min of drug-application (P⩽0.01). The antiepileptiform effect was already significant 15 min after starting the superfusion. The CB1 receptor antagonist SR 141716 (1 μM) abolished the antiepileptiform action of anandamide (n=7) indicating that it is mediated by CB1 receptors. SR 141716 evoked a slight but not significant increase in the number of additional population spikes elicited by the low Mg2+-ACSF.
At a concentration of 500 nM (n=8), WIN 55,212-2 evoked an antiepileptiform action similar to that of anandamide (Figure 6B) which was completely prevented by pretreatment with the antagonist SR 141716 (1 μM). In contrast, the S(−)-enantiomer WIN 55,212-3 failed to affect the stimulus-triggered epileptiform activity (n=5).
In area CA3, the low Mg2+/high K+-ACSF caused recurrent epileptiform discharges with a regular repetition rate about 20–40 min after starting the superfusion which occurred in absence of electrical stimulation. The epileptiform field potentials were monophasic or biphasic with a duration of 168.5±13 ms and occurred with a regular repetition rate of 28.4±6 min−1 (n=46). After the stabilization of the activity (20–30 min after onset of the epileptiform discharges) anandamide was applied for a period of 60 min at a concentration of 1 or 10 μM. The frequency of the spontaneously occurring recurrent discharges was decreased concentration-dependently to 81.5±8% (n=7, P⩽0.01) and to 57.9±13% of the initial value (n=9, P⩽0.001) after application of 1 and 10 μM anandamide, respectively (Figure 7A). The anticonvulsant index, determined as described in Methods was reduced to 89.3±11% (n=7, P⩽0.05) and to 63.8±15% (n=9, P⩽0.01) after application of 1 and 10 μM anandamide, respectively. The antiepileptiform effect of anandamide was abolished by the antagonist SR 141716 (1 μM). Moreover, SR 141716, had facilatory effects. The frequency of burst discharges was increased, accompanied by a significant enhancement of the anticonvulsant index to 115.3±9% (n=18, P⩽0.05)
Figure 7.
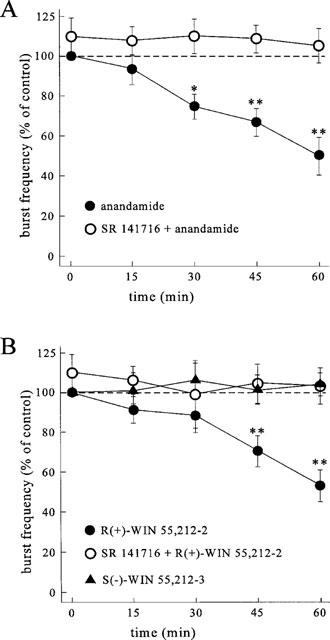
Time-course of the effect of the cannabinoids on the frequency of spontaneously occurring epileptiform burst discharges in the stratum pyramidale of the CA3 region. Epileptiform activity was elicited by omission of Mg2+ and elevation of K+ to 8 mM. (A) Antiepileptic effect of anandamide (10 μM) and blockade of the effect by simultaneous application of SR 141716 (1 μM). (B) Antiepileptiform effect of WIN 55,212-2 (500 nM) and its suppression by SR 141716 (1 μM). Note that the S(−)-enantiomer WIN 55,212-2 failed to alter the frequency of the epileptiform burst discharges. Data points represent the mean±s.d. of 6–9 experiments.
The synthetic cannabinoid receptor agonist WIN 55,212-2 (500 nM) reduced the repetition rate of the epileptiform field potentials as revealed by an anticonvulsant index of 43.7±12% of the initial value (n=9, P⩽0.001), with a time-course and SR 141716-sensitivity similar to those of anandamide (Figure 7B). In contrast, the S(−)-enantiomer WIN 55,212-3 failed to significantly affect the frequency of discharges (Figure 7B).
Discussion
The present study has two major findings: firstly, it provides electrophysiological evidence that anandamide and WIN 55,212-2 produced an attenuation of the postsynaptic population spike and the field e.p.s.p. Secondly, both compounds had an depressant effect on epileptiform discharges evoked by omission of Mg2+. These effects were abolished by the CB1 receptor antagonist SR 141716 (Rinaldi-Carmona et al., 1994). Although the synthetic cannabinoid WIN 55,212-2 was more potent than anandamide, it should be emphasized that the effect of anandamide is probably underestimated due to the concomitant degradation by fatty acid amidase (Di Marzo et al., 1994).
Axonal excitability was unaffected by the investigated cannabinoids as revealed by the lack of effect on the presynaptic fibre spike, indicating that changes in axonal excitability cannot account for the inhibitory action of these compounds. Therefore, the depressant and antiepileptiform effect of these two compounds may be a result of either a decrease in neurotransmitter release or a result of a postsynaptic action. However, since both compounds failed to affect the antidromic population spike, a reduction of pyramidal cell excitability also does not account for their depressant effect on the field potentials. It is consistent with the increased magnitude of the paired-pulse facilitation to propose that there is a decrease in synaptic efficiency during the application of anandamide and WIN 55,212-2. These results suggest that anandamide might act on the presynaptic site to modulate the transmitter release mechanisms in the CA1 region of the rat hippocampus. This conclusion is consistent with previous findings in other test systems. It has been shown that CB1 receptors on presynaptic terminals inhibit the release of several neurotransmitters including the excitatory aminoacid glutamate (Shen et al., 1996). Since paired-pulse facilitation can depend on presynaptic Ca2+ accumulation (Zucker, 1989), disinhibition due to GABAB receptor-mediated blockade of GABA release from inhibitory interneurons (Davies et al., 1990; Nathan et al., 1990) and by extracellular K+ accumulation reducing GABAB receptor-mediated inhibitory postsynaptic potentials (Rausche et al., 1989), it is at present unclear which of these mechanisms is involved in the increase of paired-pulse facilitation. However an effect on GABAB receptors or an inhibition GABA release seems less likely (Shen et al., 1996).
A cannabinoid-induced decrease in the glutamate release could also explain the antiepileptiform effect of these compounds. Excessive glutamatergic transmission, in turn, has been implicated in the pathology of epilepsy (Löscher, 1993). In the present experiments, epileptiform activity has been elicited by omission of Mg2+ from the ACSF, which leads to demasking of NMDA receptor-mediated responses (Coan & Collingridge, 1985; Anderson et al., 1986). Inhibition of Ca2+ channels such as N- and P/Q-type Ca2+ channels by cannabinoids (Twitchell et al., 1997; Shen & Thayler, 1998) is expected to reduce excessive transmitter release, thereby preventing spread of neuronal excitation. Thus, the action of anandamide on excitatory neurotransmission responsible for decreasing cell firing in response to synaptic activation is a powerful effect which may be relevant to its antiepileptiform effect reported in the present study. It is unlikely that the depressant and antiepileptiform effect of anandamide and WIN 55,212-2 reported in the present study is mediated by an enhancement of GABA-mediated inhibitory neurotransmission, because Shen et al. (1996) have shown that GABAergic transmission in rat hippocampal cells is not susceptible to modulation by cannabimimetics. However, in contrast to the hippocampus, the cannabinoids inhibit GABAergic transmission in the striatum (Szabo et al., 1998).
Alternatively, the antiepileptiform effect of anandamide might be mediated by an inhibition of gap junctions. Previously, anandamide has been reported to inhibit gap junction conductance in striatal astrocytes (Venance et al., 1995). Electrotonic coupling of neurons by gap junctions is involved in high frequency network oscillitations in the hippocampus slice which occur as brief series of repetitive population spikes (Draguhn et al., 1998). However, although this alternative mechanism of action cannot be excluded, it seems to be unlikely that the antiepileptiform effect of anandamide is mediated by an inhibition of gap junction coupling, because this effect is neither mimicked by WIN 55,212-2 nor prevented by SR141716 (Venance et al., 1995). In contrast, the present findings have clearly demonstrated that (1) WIN 55,212-2 was more potent than anandamide in suppressing antiepileptiform activity, (2) WIN 55,212-3 was inactive indicating stereoselectivity, and (3) the antiepileptiform action of both cannabinoids was antagonized by SR 141716.
The question remains of whether the inhibitory action is the biological role of anandamide. Several observations support this concept. First, the CB1 receptor antagonist SR 141716 increases the frequency of spontaneously occurring epileptiform discharges, suggesting that blockade of a tonic inhibition by anandamide increases the excitatory tone in the hippocampus. However, since SR 1417161 has been reported to exert inverse agonistic effects at CB1 receptors (Bouaboula et al., 1997; MacLennan et al., 1998), it cannot be excluded that the facilatory effect of this compound is partially due to a constitutive activation of CB1 receptors. Second, the hippocampus has a high level of anandamide (Felder et al., 1996) as well as a high density of CB1 receptors which are situated next to glutamatergic presynaptic terminals (Twitchell et al., 1997). Isolation and quantitation of anandamide by liquid chromatography and mass spectrometry revealed the highest level in the human and rat hippocampus with 29 pmol g−1 and 148 pmol g−1, respectively (Felder et al., 1996). The endogeneous cannabinoid and its receptor are thus matched to the task of inhibiting glutamatergic transmission. Furthermore, the present data show that the CB1 receptor antagonist SR 141716 increases the frequency of spontaneously occurring epileptiform discharges, suggesting that blockade of the tonic inhibition by anandamide increases the excitatory tone in the hippocampus. It is probable that anandamide subserves a more subtle physiological role in the hippocampus than the prevention of seizures. However, the present results support the hypothesis that, within its biological role, endogeneous anandamide may well act to limit excitability within the hippocampus. On the basis of the present results, hippocampal CB1 receptors may also be considered as potential targets for anticonvulsant therapy.
Acknowledgments
The generous gift of SR 141716 from Sanofi Recherche, Montpellier, France, is greatly appreciated.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CB1
cannabinoid-CB1-receptor
- NMDA
N-methyl-D-aspartate
- SR 141716
N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorphenyl)-4-methyl-3-pyrazole-carboxamide
- THC
Δ9-tetrahydroncannabinol
- WIN 55,212-2
R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolol[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl)methanone
References
- ABOOD M.E., MARTIN B.R. Molecular neurobiology of the cannabinoid receptor. Int. Rev. Neurobiol. 1996;39:197–221. doi: 10.1016/s0074-7742(08)60667-4. [DOI] [PubMed] [Google Scholar]
- ANDERSON W.W., LEWIS D.V., SWARTZWELDER H.S., WILSON W.A. Magnesium-free medium activates seizure events in rat hippocampal slices. Brain Res. 1986;398:215–219. doi: 10.1016/0006-8993(86)91274-6. [DOI] [PubMed] [Google Scholar]
- APLAND J.P., CANN F. Anticonvulsant effects of memantine and MK-801 in guinea pig hippocampal slices. Brain Res. Bull. 1995;37:311–316. doi: 10.1016/0361-9230(95)00038-g. [DOI] [PubMed] [Google Scholar]
- BIDAUT-RUSSELL M., DEVANNE W.A., HOWLETT A.C. Cannabinoid receptors and modulation of cyclic AMP in the rat brain. J. Neurochem. 1990;55:21–26. doi: 10.1111/j.1471-4159.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- BOUABOULA M., PERRACHON S., MILLIGAN L., CANAT X., RINALDI-CARMONA M., PORTIER M., BARTH F., CALANDRA B., PECCEU F., LUPKER J., MAFFRAND J.-P., LE FUR G., CASELLAS P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J. Biol. Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- CADAS H., DI TOMASO E., PIOMELLI D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonyl phosphatidylethanolamine, in rat brain. J. Neurosci. 1997;17:1226–1242. doi: 10.1523/JNEUROSCI.17-04-01226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CADOGAN A.K., ALEXANDER S.P.H., BOYD E.A., KENDALL D.A. Influence of cannabinoids on electrically evoked dopamine release and cyclic AMP generation in the rat striatum. J. Neurochem. 1997;69:1131–1137. doi: 10.1046/j.1471-4159.1997.69031131.x. [DOI] [PubMed] [Google Scholar]
- CARLINI E.A., CUNHA J.M. Hypnotic and antiepileptic effects of cannabidiol. J. Clin. Pharmacol. 1981;21:417–427. doi: 10.1002/j.1552-4604.1981.tb02622.x. [DOI] [PubMed] [Google Scholar]
- COAN E.J., COLLINGRIDGE G.L. Magnesium ions block an N-methyl-D-aspartate receptor-mediated component of synaptic transmission in rat hippocampus. Neurosci. Lett. 1985;53:21–26. doi: 10.1016/0304-3940(85)90091-6. [DOI] [PubMed] [Google Scholar]
- DAVIES C.H., DAVIES S.N., COLLINGRIDGE G.L. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J. Physiol. 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEADWYLER S.A., HAMPSON R.E., BENNET B.A., EDWARDS T.A., MU J., PACHECO M.A., WARD S.J., CHILDERS S.R. Cannabinoids modulates potassium currents in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- DEBANNE D., GUERINEAU N.C., GÄHWILER B.H., THOMPSON S.M. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuations affects subsequent release. J. Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEUTSCH D.G., CHIN S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- DEVANE W.A., DYSARZ F.A., JOHNSON M.R., MELVIN L.S., HOWLETT A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1992;34:605–613. [PubMed] [Google Scholar]
- DI MARZO V., FONTANA A., CADAS H., SCHINELLI S., CIMINO G., SCHWARZ J.C., PIOMELLI D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- DRAGUHN A., TRAUB R.D., SCHMITZ D., JEFFERYS J.G.R. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature. 1998;394:189–192. doi: 10.1038/28184. [DOI] [PubMed] [Google Scholar]
- EVANS D.M., LAKE J.T., JOHNSON M.R., HOWLETT A.C. Endogenous cannabinoid receptor binding activity released from rat brain slices by depolarization. J. Phamacol. Exp. Ther. 1994;268:1271–1277. [PubMed] [Google Scholar]
- FELDER C.C., BRILEY E.M., AXELROD J., SIMPSON J.T., MACKIE K., DEVANE W.A. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELDER C.C., NIELSON A., BRILEY E.M., PALKOVITS, PRILLER J., AXELROD J., NGUYEN D.N., RICHARDSON J.M., RIGGIN R.M., KOPPEL G.A., PAUL S.M., BECKER G.W. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393:231–235. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- GATLEY S.J., LAN R., VOLKOW N.D., PAPPAS N., KING P., WONG C.T., GIFFORD A.N., PYATT B., DEWEY S.L., MAKRIYANNIS A. Imaging the brain marijuana receptor: development of a radioligand that binds to cannabinoid CB1 receptors in vivo. J. Neurochem. 1998;70:417–423. doi: 10.1046/j.1471-4159.1998.70010417.x. [DOI] [PubMed] [Google Scholar]
- GESSA G.L., MASCIA M.S., CASU M.A., CARTA G. Inhibition of hippocampal acetylcholine release by cannabinoids: reversal by SR141716A. Eur. J. Pharmacol. 1997;327:R1–R2. doi: 10.1016/s0014-2999(97)89683-5. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., ASHBY C.R. Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN55121-2, and is potentiated by the cannabinoid antagonist SR 141716A. J. Pharmacol. Exp. Ther. 1996;27:1431–1436. [PubMed] [Google Scholar]
- GIFFORD A.N., SAMIIAN L., GALEY S.J., ASHLEY C.R. Examination of the effects of the cannabinoid receptor agonist, CP 55,940, on electrically evoked transmitter release from rat brain slices. Eur. J. Pharmacol. 1997;324:187–192. doi: 10.1016/s0014-2999(97)00082-4. [DOI] [PubMed] [Google Scholar]
- HERKENHAM M. Cannabinoid receptor localization in brain: relationship to motor and reward system. Ann. N.Y. Acad. Sci. 1992;654:19–32. doi: 10.1111/j.1749-6632.1992.tb25953.x. [DOI] [PubMed] [Google Scholar]
- HOWLETT A.C. Pharmacology of cannabinoid receptors. Annu. Rev. Pharmacol. Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- KARLER R., TURKANIS S.A. The cannabinoids as potential antiepileptics. J. Clin. Pharmacol. 1981;21:437–448. doi: 10.1002/j.1552-4604.1981.tb02624.x. [DOI] [PubMed] [Google Scholar]
- LÖSCHER W. Basic aspects of epilepsy. Curr. Opin. Neurol. Neurosurg. 1993;6:223–232. [PubMed] [Google Scholar]
- MACKIE K., DEVANE W.A., HILLE B. Ananandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol. Pharmacol. 1993;44:498–503. [PubMed] [Google Scholar]
- MACLENNAN S.J., REYNEN P.H., KWAN J., BONHAUS D.W. Evidence for inverse agonism of SR141716 at a human recombinant cannabinoid CB1 and CB2 receptors. Br. J. Pharmacol. 1998;124:619–622. doi: 10.1038/sj.bjp.0701915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUDA L.A., BONNER T.L., LOLAIT S.J. Localization of cannabinoid receptor mRNA in rat brain. J. Comp. Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- MATSUDA L.A., LOLAIT S.J., BROWNSTEIN M.J., YOUNG A.C., BONNER T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- MECHOULAM R., HANUS L., MARTIN B.R. Search for endogenous ligands of the cannabinoid receptor. Biochem. Pharmacol. 1994;48:1537–1544. doi: 10.1016/0006-2952(94)90197-x. [DOI] [PubMed] [Google Scholar]
- NATHAN T., JENSEN M.S., LAMBERT J.D.C. GABAB receptors play a major role in paired-pulse facilitation in area CA1 of the rat hippocampus. Brain Res. 1990;531:55–65. doi: 10.1016/0006-8993(90)90757-3. [DOI] [PubMed] [Google Scholar]
- PACHECO M.A., WARD S.J., CHILDERS S.R. Identification of cannabinoid receptors in cultures of rat cerebellar granule cells. Brain Res. 1993;603:102–110. doi: 10.1016/0006-8993(93)91304-b. [DOI] [PubMed] [Google Scholar]
- RAUSCHE G., SARVEY J.M., HEINEMANN U. Slow synaptic inihibition in relation to frequency habituation in dentate granule cells of rat hippocampal slices. Exp. Brain Res. 1989;78:233–242. doi: 10.1007/BF00228895. [DOI] [PubMed] [Google Scholar]
- RINALDI-CARMONA M., BARTH F., HÉAULME M., SHIRE D., CALANDRA B., CONGY C., MARTINEZ S., MARUANI J., NÉLIAT G., CAPUT D., FERRARA P., SOUBRIÉ P., BRELIÈRE J.C., LE FUR G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- ROMERO J., DE MIGUEL R., GARCIA-PALOMERO E., FERNANDEZ-RUIZ J.J., RAMOS J.A. Time-course of the effects of anandamide, the putative endogenous cannabinoid receptor ligand, on extrapyramidal function. Brain. Res. 1995;694:223–232. doi: 10.1016/0006-8993(95)00835-e. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., TIMM J., ZENTNER J., GÖTHERT M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- SHEN M., PISAR T.M., SEYBOLD V.S., THAYLER S.A. Cannabinoid receptor agonist inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J. Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN M., THAYLER S.A. The cannabinoid agonist WIN 55,212-2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- SMITH P.B., COMPTON D.R., WELCH S.P., RAZDAN R.K., MECHOULAM R., MARTIN B.R. The pharmacological activity of anandamide, a putative endogenous cannabinoid, in mice. J. Pharmacol. Exp. Ther. 1994;270:219–227. [PubMed] [Google Scholar]
- STEIN E.A., FULLER S.A., EDGEMOND W.S., CAMPBELL W.B. Physiological and behavioural effects of the endogenous cannabinoid, arachidonylethanolamide (anandamide), in the rat. Br. J. Pharmacol. 1996;119:107–114. doi: 10.1111/j.1476-5381.1996.tb15683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZABO B., DÖRNER L., PFREUNDTNER C., NÖRENBERG W., STARKE K. Inhibition of GABAergic inhibitory postsynaptic currents by cannabinoids in rats corpus striatum. Neuroscience. 1998;85:395–403. doi: 10.1016/s0306-4522(97)00597-6. [DOI] [PubMed] [Google Scholar]
- TERRANOVA J.P., MICHAUD J.C., LE FUR G., SOUBRIÉ P. Inhibition of long-term potentiation in rat hippocampal slices by annandamide and WIN 55212-2: reversal by SR 141716A, a selective antagonist of CB1 cannabinoid receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1995;352:576–579. doi: 10.1007/BF00169393. [DOI] [PubMed] [Google Scholar]
- TWITCHELL W., BROWN S., MACKIE K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- VENANCE L., PIOMELLI D., GLOWINSKI J., GIAUME C. Inhibition by anandamide of gap junctions and intracellular calcium signalling in striatal astrocytes. Nature. 1995;376:590–594. doi: 10.1038/376590a0. [DOI] [PubMed] [Google Scholar]
- VOGEL Z., BARG J., LEVY R., SAYA D., HELDMAN E., MECHOULAM R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J. Neurochem. 1993;61:352–355. doi: 10.1111/j.1471-4159.1993.tb03576.x. [DOI] [PubMed] [Google Scholar]
- ZUCKER R. Short-term plasticity. Annu. Rev. Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]