

Abstract
UK-78,282, a novel piperidine blocker of the T lymphocyte voltage-gated K+ channel, Kv1.3, was discovered by screening a large compound file using a high-throughput 86Rb efflux assay. This compound blocks Kv1.3 with a IC50 of ∼200 nM and 1 : 1 stoichiometry. A closely related compound, CP-190,325, containing a benzyl moiety in place of the benzhydryl in UK-78,282, is significantly less potent.
Three lines of evidence indicate that UK-78,282 inhibits Kv1.3 in a use-dependent manner by preferentially blocking and binding to the C-type inactivated state of the channel. Increasing the fraction of inactivated channels by holding the membrane potential at −50 mV enhances the channel's sensitivity to UK-78,282. Decreasing the number of inactivated channels by exposure to ∼160 mM external K+ decreases the sensitivity to UK-78,282. Mutations that alter the rate of C-type inactivation also change the channel's sensitivity to UK-78,282 and there is a direct correlation between τh and IC50 values.
Competition experiments suggest that UK-78,282 binds to residues at the inner surface of the channel overlapping the site of action of verapamil. Internal tetraethylammonium and external charybdotoxin do not compete UK-78,282's action on the channel.
UK-78,282 displays marked selectivity for Kv1.3 over several other closely related K+ channels, the only exception being the rapidly inactivating voltage-gated K+ channel, Kv1.4.
UK-78,282 effectively suppresses human T-lymphocyte activation.
Keywords: Potassium channels, C-type inactivation, Kv1.3, T lymphocytes, immune suppression, piperidines, benzhydryl moiety, verapamil, charybdotoxin
Introduction
The type n voltage-gated K+ channel in T lymphocytes plays a vital role in regulating the membrane potential and in modulating calcium signalling (reviewed in Lewis & Cahalan, 1995; Cahalan & Chandy, 1997). Selective blockers of this channel depolarize the membrane, attenuate the calcium signalling response, inhibit T-cell activation in vitro, and suppress immune responses in vivo (Price et al., 1989; Leonard et al., 1992; Freedman et al., 1992; Lin et al., 1993; Hill et al., 1995; Rader et al., 1996; Nguyen et al., 1996; Koo et al., 1997; Kath et al., 1997; Kalman et al., 1998). The native channel is a homotetramer of Kv1.3 subunits (Grissmer et al., 1990; Aiyar et al., 1995; Spencer et al., 1997). Electron microscopic images of the purified Kv1.3 tetramer show that it has x-y dimensions of 65×65 Å (Spencer et al., 1997). Besides T lymphocytes, Kv1.3 is expressed faintly in the brain, as well as macrophages, osteoclasts, platelets, B-lymphocytes, and the kidney (for reviews see Chandy & Gutman 1995; Kath et al., 1997; Cahalan & Chandy 1997). In the brain, Kv1.3 is associated with Kv1.1 and Kv1.2 in heterotetramers (Shamotienko et al., 1997; Koschak et al., 1998). In macrophages, osteoclasts and platelets, Kv1.3 is one of many channels controlling the membrane potential, and selective blockers of this channel do not affect cell signalling (for reviews see Cahalan & Chandy, 1997; Kath et al., 1997). Because of its demonstrated role in T-lymphocyte activation, and its functionally restricted expression to lymphocytes, Kv1.3 is widely recognized as a potential target for therapeutic immunosuppressive agents (see reviews Lewis & Cahalan, 1995; Kath et al., 1997; Cahalan & Chandy, 1997).
Several groups have been actively engaged in the discovery, design, and characterization of Kv1.3 inhibitors. Many peptides isolated from scorpion venoms and sea anemone potently block Kv1.3 and also inhibit T-lymphocyte activation (Sands et al., 1989; Price et al., 1989; Deutsch et al., 1991; Leonard et al., 1992; Lin et al., 1993; Aiyar et al., 1995; 1996; Rader et al., 1996; Koo et al., 1997; Kalman et al., 1998). Although these peptide inhibitors are very potent and highly selective, clinical application may be limited by their need to be administered parenterally (Koo et al., 1997; Kath et al., 1997). Over a decade ago, a variety of non-peptide Kv1.3 inhibitors were shown to inhibit T-cell mitogenesis and block Kv1.3 channels, but were limited in their potency and selectivity (DeCoursey et al., 1984; Chandy et al., 1984; Sabath et al., 1986; reviewed in Kath et al., 1997). More recently, two groups described immunosuppressive dihydroquinoline compounds that block Kv1.3 at submicromolar concentrations by interacting with the C-type inactivated conformation of the channel (Hill et al., 1995; Nguyen et al., 1996). This finding raised the possibility of using high-throughput screening strategies to discover novel Kv1.3 inhibitors. Here we report the characterization of a piperidine blocker of Kv1.3, UK-78,282, which was discovered by screening a compound collection with a high-throughput 86Rb efflux assay (Burgess et al., 1997).
Methods
Synthesis of UK-78,282 and CP-190,325
UK-78,282 (4-[diphenylmethoxymethyl]-1-[3-(4-methoxyphenyl)-propyl]-piperidine) and the analogue, CP-190,325 (4-[benzylmethoxymethyl]-), were synthesized as previously described (Burgess et al., 1997). Purification of these compounds was accomplished by recrystallization from ethanol and/or column chromatography. All compounds were fully characterized by proton NMR, carbon NMR, and mass spectroscopy.
Chemicals and solutions
For patch-clamp recording studies, cells were bathed in mammalian Ringer solution (290–320 mOsm) containing (in mM): NaCl 160, KCl 4.5, CaCl2 2, MgCl2 1, HEPES 5 (4-2-hydroxyethyl-1-piperazineethanesulphonic acid) adjusted to pH 7.4 with NaOH. The internal pipette solution (290–320 mOsm) contained (in mM): KF 134, MgCl2 2, HEPES 5, CaCl2 1, and EGTA 10 (ethylenebis[oxyethylene-nitrilo]tetracetic acid), adjusted to pH 7.2 with KOH. A syringe-driven perfusion system was used to exchange the bath solution. The following reagents were purchased from the vendors indicated in parentheses: kaliotoxin (KTX; Peptides International, Louisville, KY, U.S.A.), phorbol myristate acetate (PMA; Sigma Chemicals, St. Louis, MO, U.S.A.), ionomycin, (Calbiochem, San Diego, CA, U.S.A.), phytohemagglutinin (PHA; Murex Diagnostics, Dartford, England), anti-CD25 antibody (Immunotech, Westbrook, ME, U.S.A.), 3H-thymidine and 86RbCl (NEN, Cambridge, MA, U.S.A.), and Ficoll-Paque (Pharmacia, Piscataway, NJ, U.S.A.). For the KCa channel studies, the pipette solution (290–320 mOsm) contained (in mM): K+-aspartate 150, MgCl2 2, HEPES 5, EGTA 10, CaCl2 (free pCa ∼6) 8.7, final pH 7.2.
Cell lines
Rat basophilic leukaemia (RBL) cells, maintained in EMEM supplemented with 10% heat-inactivated foetal calf serum and 1% L-glutamine, were plated onto glass coverslips 1 day prior to use for electrophysiological experiments. Louckes cells were grown in RPMI-1640 with 10% heat-inactivated foetal bovine serum, 100 units ml−1 penicillin, 100 μg ml−1 streptomycin, 100 μM MEM non-essential amino acids, 25 mM HEPES, and 2 mM L-glutamine. The COS-7 and L929 cell lines were grown in DMEM (high glucose) with the same additives as above. Foetal bovine serum was obtained from Hyclone (Logan, UT, U.S.A.), and media and additives were obtained from Biowhittaker (Walkersville, MD, U.S.A.). Cell lines stably expressing Kv1.1, Kv1.2, Kv1.3, Kv1.5, and Kv3.1 have been previously described (Grissmer et al., 1994; Nguyen et al., 1996) and were maintained in DMEM containing 10% foetal calf serum and G418 (1 mg ml−1) (GIBCO, Grand Island, NY, U.S.A.).
Purification of T cells
Human T lymphocytes, separated as described previously (Nguyen et al., 1996), were resuspended in medium (RPMI-1640 containing 10% heat-inactivated foetal calf serum, HEPES 25 mM, L-glutamine 2 mM, non-essential amino acids 100 μM, 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin) and used in activation assays.
Activation of human T cells by PHA or ionomycin/PMA
Human T cells were added to a 96-well plate (125 μl total volume, 1.25×105 cells well−1) and incubated with Kv1.3 blockers for 15 min at room temperature. Following the addition of ionomycin (15.6 ng well−1) and PMA (0.625 ng well−1), or PHA (0.25 μg well−1), cells were incubated for 24 h at 37°C and then pulsed with 3H-thymidine (1 μCi ml−1; 25 μl well−1) for 18 h. The cells were then harvested using a TOMTEC Mach-2 harvester (TOMTEC International, Orange, CT, U.S.A.). Control wells contained T lymphocytes with and without stimulants and without test compound. Duplicate plates were set up to harvest the supernatants for interleukin-2 (IL-2) measurements. IL-2 plates were spun at 200×g for 10 min at room temperature and the supernatants were stored at −20°C until analysed with IL-2 ELISA kits (R & D Systems, Minneapolis, MN, U.S.A.).
IL-2 receptor (IL-2R) expression
Purified human T cells (106 cells ml−1) were stimulated with 125 ng ml−1 ionomycin and 5 ng ml−1 PMA at 37°C. At indicated times, cells were removed from culture and stained for IL-2R expression using anti-human CD25 (IL-2R a-chain) and analysed with a FACScan flow cytometer (Becton Dickinson, San Jose, CA, U.S.A.). Hydroxyurea (5 mg ml−1), a cell-cycle S phase blocker, was used as a control for cell-cycle studies.
86Rb efflux screen (Nguyen et al., 1996)
Purified human T cells (107 cells ml−1) in culture medium were incubated with 86RbCl (10 mCi ml−1) for 18 h at 37°C in 5% CO2, then washed in low K+ buffer (4.5 mM K+ in HBSS buffer, prepared according to the GIBCO manual) and resuspended at 5.5×106 cells ml−1 in low K+ buffer. Fifty μl of the cell suspension was incubated for 15 min at ambient temperature with 10 μl of the test compound (in low K+ buffer) in each well of a Millipore Multiscreen 96 well 0.65 micron filtration plate (cat. # MADV-N65-50, Millipore, Bedford, MA, U.S.A.). The mixture was then incubated for 20 min with 125 μl well−1 of high K+ buffer (140 mM K+ in HBSS, prepared by replacing NaCl with KCl) to initiate 86Rb efflux via membrane depolarization, and filtered with a TV-1 vacuum transfer manifold (Pall Trinity Micro Corp., Cortland, NY, U.S.A.) into standard 96-well microtiter plates (Costar, Cambridge, MA, U.S.A.); 100 μl of filtrate was removed from each well, mixed with 3 ml of scintillation cocktail, and counted in a Beckman LSC liquid scintillation counter for 1 min.
125I-ChTX binding assay (Nguyen et al., 1996)
Kv1.3 was heterologously expressed in HeLa cells using the vaccinia virus system (Spencer et al., 1997). These cells were incubated in normal ionic strength buffer (in mM): (NaCl 150, KCl 5, NaHEPES 10, glucose 6, pH 7.4) with 25 pM 125I-ChTX (custom iodinated by Amersham, Arlington Heights, IL, U.S.A.) and increasing concentrations of either cold ChTX (0–100 nM; Peptides International, Louisville, KY, U.S.A.) or the test compound, in a total volume of 800 μl. Test compounds were added 15 min before the radioiodinated toxin-probe and incubation was carried out on a rotary shaker for 1 h at ambient temperature. Binding was stopped with 4 ml of ice-cold quench buffer (NaCl 200 mM, HEPES 20 mM, pH 7.4). Cells were collected using a Hoefer filtration device (model FH224V, Hoefer Scientific Instruments, San Francisco, CA, U.S.A.) on GF/C filters (Whatman Inc., Fairfield, NJ, U.S.A.) that were pre-soaked in 0.3% polyethyleneimine in ultrapure water for 15 min. Filters were counted for 1 min with a Beckman 5500 gamma counter. Unlabelled ChTX inhibited binding of 125I-ChTX with an average IC50 value of 0.55±0.12 nM (n=13). The average c.p.m. of 125I-ChTX bound in the absence of cold ChTX was 4546±344 (s.e.mean, n=22) and the average c.p.m. in the presence of 100 nM cold ChTX was 848±25 (s.e.mean, n=22).
Electrophysiological analysis
Kv1.4, 1.6, Kv3.1, 3.2, 3.4, and 4.2, and all of the murine Kv1.3 mutants and the other channel constructs used in this study have been described previously (Grissmer et al., 1994; Nguyen et al., 1996; Rauer & Grissmer, 1996; Jäger et al., 1998). cRNA was transcribed in vitro (Stratagene, La Jolla, CA, U.S.A.), microinjected into RBL cells (Ikeda et al., 1992; Nguyen et al., 1996; Rauer & Grissmer, 1996; Jäger et al., 1998) and electrophysiologically characterized by patch-clamp recording in the whole-cell configuration as described previously. Patch-clamp recordings of purified human T lymphocytes or T-lymphoblasts (cells stimulated for 2–4 days with 2 μg ml−1 PHA) were performed in the whole-cell mode or in inside-out patch measurements as described previously (e.g. Nguyen et al., 1996; Rauer & Grissmer, 1996). Series-resistance compensation (80%) was employed if the current exceeded 2 nA. Capacitative and leak currents were subtracted using the P/8 procedure. The holding potential in all experiments was −80 mV, unless otherwise indicated, and the depolarizing potential was +40 mV. The depolarizing duration was 200 ms, unless otherwise indicated, and the interpulse interval was 30 s.
For inside-out patch experiments, the bath solution contained (in mM): K-aspartate 164.5, CaCl2 1, MgCl2 2, HEPES 10; EGTA 10; adjusted to pH=7.2 with KOH. The pipette solution was Normal Ringer.
3H-desmethoxyverapamil binding to Lewis rat cardiac calcium channels
Lewis rats (Charles River Labs, Portage, MI, U.S.A.) were sacrificed and cardiac tissues obtained. The hearts were homogenized in 20 volumes of 320 mM sucrose with a Polytron for ∼30 s, and the homogenate centrifuged at 3200 r.p.m. for 10 min. The membrane fraction, collected after an 18,000 r.p.m. spin, was resuspended in HEPES 50 mM, pH 7.4. and kept on ice. The membrane fraction was aliquoted into tubes (750ċ/tube) and incubated at room temperature for 60 min with 150ċ1 3H-desmethoxyverapamil (Amersham TRK834, Arlington Heights, IL, U.S.A.) (final concentration 0.25 nM) and either UK-78,282 or the buffer (100ċ1/tube). Incubation was rapidly terminated by rapid filtration under vacuum through Whatman GF/B filters (VWR, Boston, MA, U.S.A.) (pre-soaked in 1% polyethylenimine, Sigma Chemical Co., St. Louis, MO, U.S.A.) and rinsed three times with 4 ml of 50 mM NaCl. Non-specific binding was determined by the addition of 10 μM unlabelled verapamil (Sigma Chemical Co.). The filters were soaked in 7 ml Ready-Safe scintillation fluid (Beckman Instruments, Fullerton, CA, U.S.A.), vortexed and allowed to sit overnight to extract radioactivity. Quantification of radioactivity was determined by liquid scintillation counting. Data were expressed as IC50 values (the concentration of compound required to inhibit 50% of the specific binding).
Results
UK-78,282 is a potent use-dependent blocker of Kv1.3
Figure 1 shows the structure of UK-78,282 along with that of an analogue, CP-190,325, differing only in the type of alkoxymethyl substituent at position 4; CP-190,325 has a benzyl moiety in place of the benzhydryl group in UK-78,282. Drug potency against the Kv1.3 channel was measured using three independent assays: patch-clamp recording, inhibition of 125I-ChTX binding, and 86Rb efflux; we found correspondence between the IC50 values measured by these three methods (Table 1). Detailed biophysical and physiological studies were conducted on UK-78,282. Externally applied drug blocked Kv1.3 peak currents in human peripheral blood T lymphoblasts in a concentration-dependent manner (Figure 2A), with an IC50 value of 202±25 nM (mean±s.e.mean, n=14) (Table 1) and a drug:channel stoichiometry of 1 : 1 (Figure 2B). Similar IC50 values were obtained when the drug was tested on Kv1.3 channels expressed either in RBL (IC50=333±28 nM, n=6) or stably transfected L929 cells (IC50=280±40, n=3) (Table 2). Depending on the assay, CP-190,325 blocked Kv1.3 channels between 5–12 fold less potently than UK-78,282, suggesting that the benzhydryl moiety at position four might be important for blockade.
Figure 1.
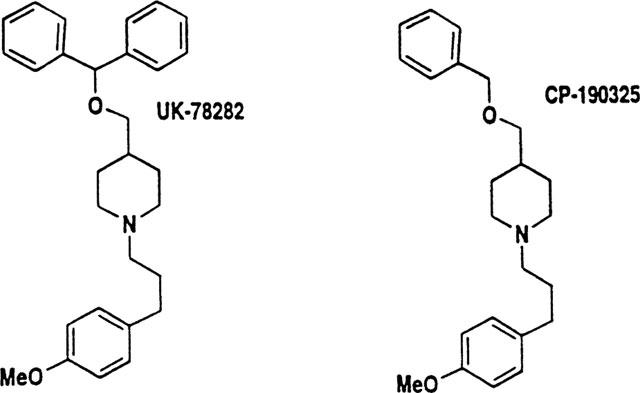
Chemical structures of UK-78,282 and CP-190,325.
Table 1.
Inhibition of Kv1.3 currents, 86Rb Efflux and 125I-ChTX binding

Figure 2.
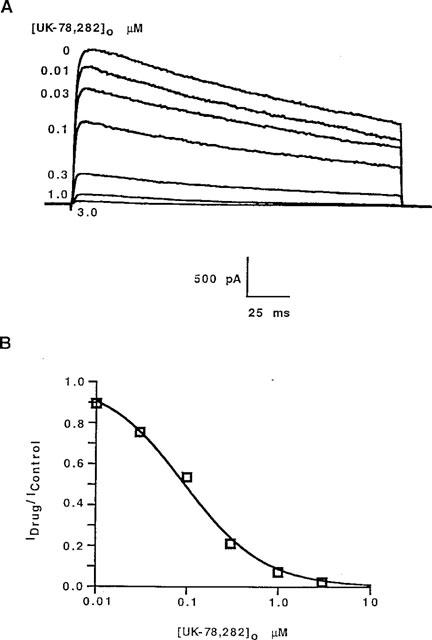
>(A) UK-78,282 blocks Kv1.3 from the outside. Typical Kv1.3 currents in human peripheral blood T lymphocytes studied in the whole cell configuration and blocked with drug applied to the external bath. (B) Hill plot of these data showing a stoichiometry of one drug per channel.
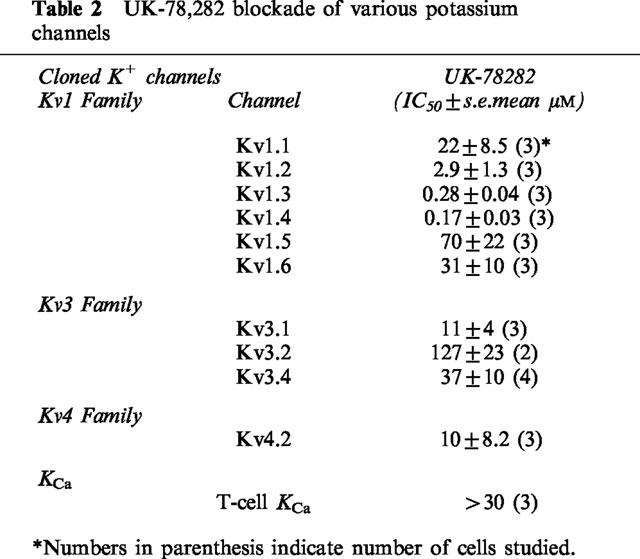
Table 2.
UK-78,282 blockade of various potassium channels
UK-78,282, at a concentration (1 μM) which blocks almost all the Kv1.3 current, required multiple openings of the channel to reach steady-state block, a phenomenon termed `use-dependent inhibition'. The time required to achieve steady-state block depended on the duration of the depolarizing pulse (Figure 3). When repeated depolarizing pulses of 200 ms duration (to +40 mV at 30 s intervals) were applied from a holding potential of −80 mV, steady-state block was reached in ∼200 s. Shortening the pulse duration to 20 ms significantly lengthened the time (∼600 s) to reach steady-state block (Figure 3). This result suggests that UK-78,282 blocks a post-activation state, possibly the open and/or inactivated conformations of the channel.
Figure 3.
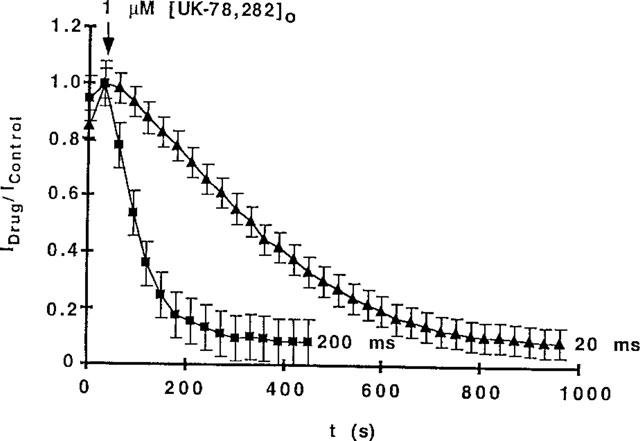
>(A) Effect of duration of depolarizing pulse on time to reach steady-state block. A human T lymphoblast was patch-clamped in the whole-cell configuration and 1 μM of UK-78,282 was applied to the external bath solution. Abscissa: time after drug addition. Ordinate: ratio of current at time t versus time 0. Holding potential=−80 mV; depolarizing potential=+40 mV; depolarizing durations=20 ms or 200 ms, interpulse interval=30 s. 200 ms: n=9 cells; 20 ms: n=4 cells.
Sensitivity of the Kv1.3 channel to block by UK-78,282 is dependent on its ability to undergo C-type inactivation
Voltage-gated K+ channels inactivate via two distinct mechanisms. N-type inactivation, involves a tethered ball in the amino terminus interacting with and blocking the inner mouth of the pore (see review by Chandy & Gutman, 1995). In contrast, C-type inactivation involves a conformational change resulting in the closure of the external mouth of the channel pore (Grissmer & Cahalan, 1989a,1989b; Busch et al., 1991; Lopez-Barneo et al., 1993; Panyi et al., 1995; Ogielska et al., 1995; Levy & Deutsch, 1996; Liu et al., 1996; Nguyen et al., 1996; Jäger et al., 1998). Kv1.3 exhibits only C-type inactivation (Cahalan et al., 1985; Grissmer & Cahalan, 1989a,1989b; Busch et al., 1991; Panyi et al., 1995; Levy & Deutsch, 1996; Nguyen et al., 1996; Jäger et al., 1998). If UK-78,282 binds to the C-type inactivated conformation, then removal or slowing of inactivation should reduce the sensitivity of Kv1.3 to this drug by decreasing the substrate for drug-binding (i.e. the fraction of channels in the inactivated conformation). On the other hand, if UK-78,282 binds primarily to the open conformation of the channel, changing C-type inactivation should have no effect on drug potency. We used three approaches to distinguish these two possibilities.
Holding at −50 mV increases sensitivity to UK-78,282
As a first test of this idea, we examined the effect of changing the holding potential from −80 to −50 mV on the sensitivity of the Kv1.3 channel to UK-78,282. The depolarized holding potential would significantly increase the proportion of channels in the inactivated conformation by slowing recovery from inactivation without changing the open duration (Cahalan et al., 1985; Grissmer & Cahalan, 1989a,1989b; Busch et al., 1991; Panyi et al., 1995; Levy & Deutsch, 1996; Nguyen et al., 1996; Jäger et al., 1998). As shown in Figure 4, the Kv1.3 channel was more sensitive to block by UK-78,282 at −50 mV compared to −80 mV, suggesting that the drug preferentially blocks the C-type inactivated state of the channel.
Figure 4.
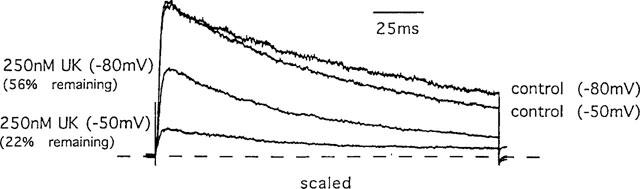
Effect of varying the holding potential (as indicated) on the sensitivity to block by UK-78,282. Superimposition of Kv1.3 currents measured at holding potentials of −80 mV or −50 mV in the presence or absence of 250 nM UK-78,282. Pulse duration=200 ms; interpulse interval=30 s.
[K+]0 slows C-type inactivation and decreases sensitivity to UK-78,282
As a second test of the hypothesis, we slowed C-type inactivation of Kv1.3 by increasing the extracellular potassium concentration (Cahalan et al., 1985; Grissmer & Cahalan, 1989a,1989b; Lopez-Barneo et al., 1993; Panyi et al., 1995; Levy & Deutsch, 1996; Nguyen et al., 1996; Jäger et al., 1998). Fewer Kv1.3 channels transition into the C-type inactivated conformation in the presence of 160 mM external K+ (e.g. Nguyen et al., 1996). If UK-78,282 does block the C-type inactivated conformation, as suggested by the holding potential experiment (Figure 4), exposure to high external K+ should decrease UK-78,282 potency by reducing the number of inactivated channels available for drug binding. Consistent with this prediction, 164.5 mM [K+]0 significantly slowed inactivation of native Kv1.3, and in parallel reduced the sensitivity of this channel to UK-78,282 (Figure 5).
Figure 5.
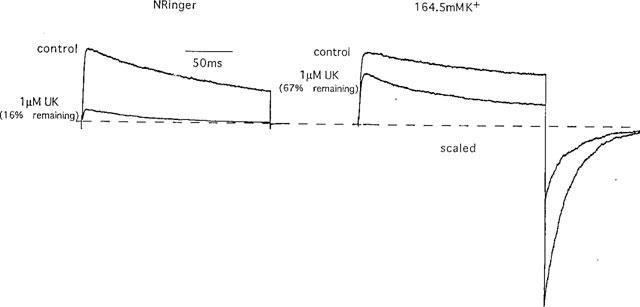
Effect of external K+ concentration ([K+]0) on inactivation and sensitivity to block by UK-78,282. Superimposition of currents from Kv1.3 in K-Ringer (164.5 mM external K+) or normal Ringer (4.5 mM external K+), in the presence or absence of 1 μM UK-78,282.
His404, a residue located at the entrance to the ion channel pore (Aiyar et al., 1995; 1996) is the K+-modulatory site responsible for the slowing of C-type inactivation by 160 mM external K+ (Nguyen et al., 1996). This residue, and the homologous residue in Shaker (Thr449), also play a critical role in the conformational change underlying C-type inactivation (Busch et al., 1991; Lopez-Barneo et al., 1993; Liu et al., 1996; Nguyen et al., 1996; Jäger et al., 1998). Replacement of His404 with either a hydrophobic valine (H404V) or a polar asparagine (H404N), abrogated both the 160 mM [K+]0-dependent slowing of C-type inactivation (Figure 6A) and the parallel decrease in UK-78,282 sensitivity (Figure 6B). These results provide further support for the notion that UK-78,282 binds to and blocks the inactivated state of the channel.
Figure 6.
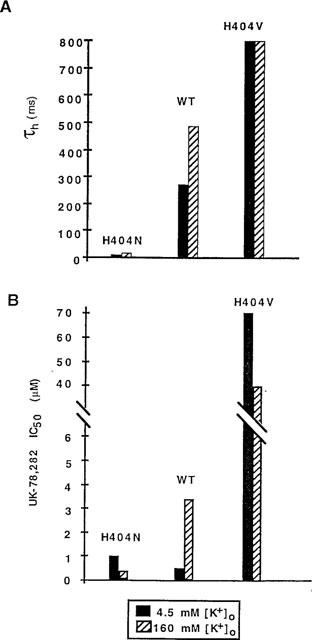
Effect of external K+ concentration ([K+]0) on τh (A) and UK-78,282 IC50 values (B) using wild type (WT) Kv1.3 and two His404 mutants.
Vestibular mutations that slow τh also reduce sensitivity to UK-78,282
As a third test of the hypothesis, we evaluated the sensitivity of six Kv1.3 mutants with widely differing abilities to transition into the C-type inactivated state but with no differences in the open duration (Nguyen et al., 1996; Jäger et al., 1998). If our interpretation of the results presented in Figure 4 and 5 is correct, mutant channels that are able to rapidly enter the C-type inactivated conformation should be potently blocked by UK-78,282 by providing more substrate for drug binding. In contrast, mutants that exhibit little or no C-type inactivation should be less sensitive to the drug. Consistent with this prediction, the rapidly inactivating H404N and H404S mutants are more sensitive to block by UK-78,282 than slowly inactivating Kv1.3 mutants (Figure 7). A direct, although weak linear relationship (r=0.78) was noted between τh (a measure of the rate of inactivation) and the IC50 values (Figure 7). A similar relationship between τh and IC50 values was observed for the dihydroquinoline compound, CP-339,818 (r=0.9; dashed line in Figure 7), that selectively interacts with the C-type inactivated conformation of Kv1.3. Collectively, these results suggest that UK-78,282 preferentially binds to and blocks the C-type inactivated conformation of Kv1.3.
Figure 7.
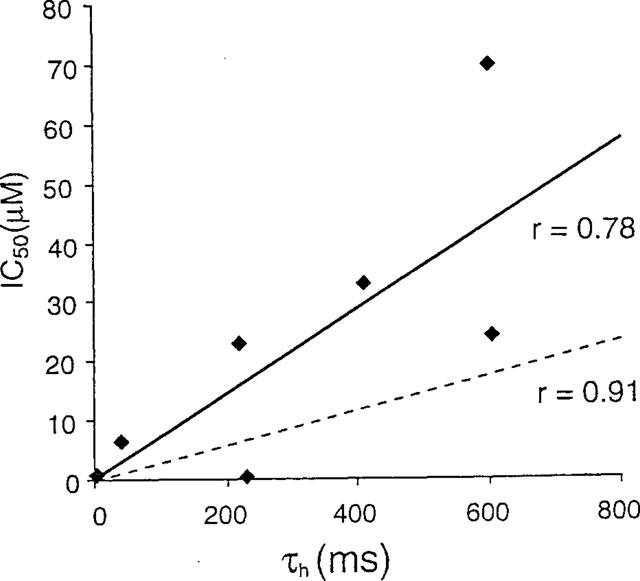
Direct relationship between τh for each Kv1.3 mutant and its sensitivity to block by UK-78,282 (IC50). All experiments were performed in the whole-cell mode with repeated 200-ms pulses to +40 mV at 30-s intervals. Solid regression line: all UK-78,282 data. Dashed regression line: all CP-339,818 data (Nguyen et al., 1996) included for comparison.
UK-78,282 does not bind to residues in the external vestibule?
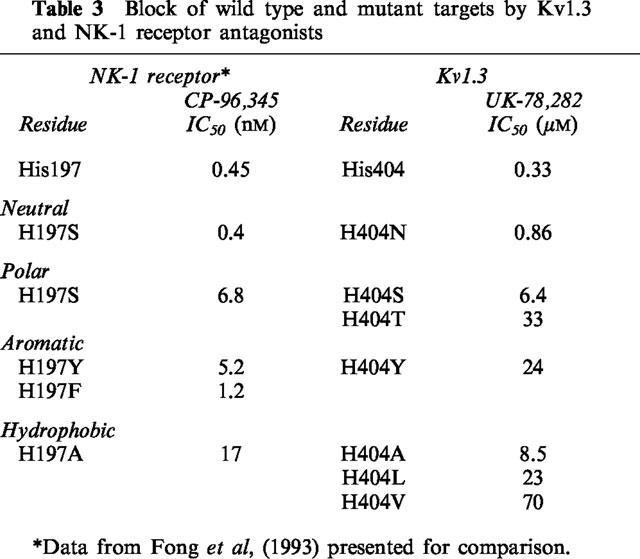
Because UK-78,282 competitively inhibits 125I-ChTX binding to its receptor in the outer vestibule of Kv1.3, this compound may interact with vestibular residues that become exposed to solvent in the C-type inactivated conformation. Cysteine-scanning mutagenesis studies on the Shaker channel show that the side chains of residues corresponding to Met403, His404 and Pro405 in Kv1.3 become more accessible to cysteine-modifying reagents in the C-type inactivated conformation compared with the closed or open states of the channel, suggesting that the conformational changes associated with C-type inactivation enhance the exposure of external vestibular residues to the solvent phase (Liu et al., 1996). If a similar conformational change occurs in Kv1.3, the increased exposure of vestibular residues in the C-type inactivated state might provide binding sites for UK-78,282. For example, UK-78,282 could bind to an exposed His404 via an aromatic-amino interaction, similar to the one reported between a histidine (His197) in the neurokinin-1 (NK-1) receptor and a benzhydryl in its antagonist, CP-96,345 (Fong et al., 1993). Interestingly, potency of UK-78,282 is substantially affected by substitutions at Kv1.3-position 404 in a manner analogous to the reported effects of His197 mutations in the NK-1 receptor on CP-96345 affinity (Table 3). In both cases, only neutral residues (Gln or Asn) maintain the amino-aromatic interaction (Fong et al., 1993; and Table 3). Replacement of this residue with Ser, Ala or Tyr produces a similar reduction in drug affinity in both Kv1.3 and the NK-1 receptor (Table 3). Replacing the benzhydryl moiety in CP-96345 with a benzyl substituent produces a derivative (L703766) that is unable to associate via an amino-aromatic interaction with His197 in the NK1 receptor (Fong et al., 1993). Similarly, CP-190,325, a UK-78,282 derivative that contains a benzyl moiety in place of the benzhydryl group, is a significantly less potent inhibitor of Kv1.3 (Table 1). These observations support the suggestion that the imidazole of His404 may be involved in an amino-aromatic interaction with the benzhydryl in UK-78,282. However, since His404 mutations also alter C-type inactivation, this interpretation must be taken with caution. Another reason for caution is that UK-78,282, a highly lipophilic compound, is added to cells at least 15 min before 125I-ChTX in the binding assay, giving it enough time to cross the membrane, interact with a site internally and allosterically hinder ChTX binding. Such a cross-pore interaction has been observed between ChTX and selective small molecule antagonists of the maxi KCa channel (Knaus et al., 1994).
Table 3.
Block of wild type and mutant targets by Kv1.3 and NK-1 receptor antagonists
As a further test of whether UK-78,282 binds in the external vestibule, we performed an acute competition experiment where UK-78,282 (0.2 μM) and unlabelled ChTX (10 nM) were applied to the external surface of Kv1.3, alone or in combination, and Kv1.3 currents were measured. If both these drugs interact with identical or overlapping sites, the normalized current reduction of one compound in the presence of the other should be predictably less than if these compounds interacted with the channel at independent sites. As shown in Figure 8A, 0.2 μM UK-78282 reduced the current to ∼50%, consistent with the results presented in Figure 2. In a separate cell, addition of 10 nM ChTX alone reduced current to 25% (Figure 8B, middle trace). Subsequent application of 10 nM ChTX and 0.2 μM UK-78,282 reduced the residual current by another 50% to a final value of 12.5% (Figure 8B, bottom trace), which is greater reduction than that predicted (final value of 20%) for the two compounds binding at overlapping sites. Since the degree of reduction of current by UK-78,282 in the absence (Figure 8A) and the presence (Figure 8B) of ChTX are almost identical, these results suggest that the two compounds act at independent sites.
Figure 8.
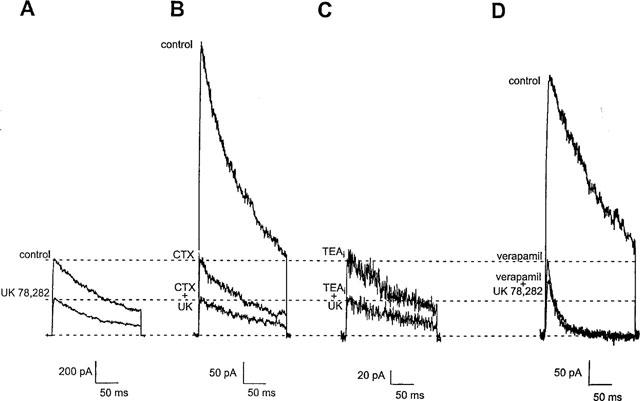
Competition of UK-78,282 with external ChTX, internal TEA or external verapamil. Membrane currents were elicited in the absence and presence of 0.2 μM UK 78,282 applied either alone (A), or in combination with either 10 nM ChTX (B), 1 mM intracellularly applied TEA, TEAi, (C), or 20 μM verapamil (D). The dotted lines show the degree of block with compounds added alone or in combination.
UK-78,282 binds to an internal site of the channel that overlaps the site of action of verapamil, but not internal TEA
Since UK-78282 does not appear to act in the external vestibule, we applied it to the cytoplasmic surface of the channel in inside-out patches and observed rapid inhibition of Kv1.3 currents with potency (Kd=0.39±0.05 μM; steady-state block reached after 183±43 s) comparable to whole-cell studies. This suggests that UK-78,282 might bind residues on the cytoplasmic surface of the channel. Internally-applied TEA might therefore be expected to competitively inhibit UK-78,282 from binding to its receptor; internal TEA has been reported to inhibit Kv1.3 currents with a Kd of ∼0.3 mM by interacting with residues in the inner pore (Aiyar et al., 1994). We performed competition experiments to determine whether the sites of action of UK-78,282 and internal TEA overlapped. Application of internal TEA (1 mM) via the pipette solution in the whole-cell mode produced block of the current (Figure 8C, top trace). After the current reached equilibrium we applied 0.2 μM UK-78,282 externally. As shown in Figure 8C (bottom trace), current reduction is almost identical to that in the absence of internal TEA (Figure 8A), suggesting that these two drugs act at independent sites.
As a final test, we performed competition experiments with verapamil and UK-78,282. Verapamil has been reported to interact with residues at the cytoplasmic surface of Kv1.3, at a site that overlaps that of internal TEA (Rauer & Grissmer, 1996). Addition of 20 μM verapamil alone reduced current to ∼28% (Figure 8D, middle trace). Subsequent application of 20 μM verapamil and 0.2 μM UK-78,282 reduced the residual current by only ∼25% to a final value of 21% (Figure 8D; bottom trace). This result is consistent with the two compounds competing for overlapping binding sites, since the residual current would be expected to be reduced by 0.2 μM UK-78,282 to a final value of 14%.
UK-78,282 displays selectivity for Kv1.3
We tested the potency of UK-78,282 against a panel of ten related cloned K+ channel targets, nine of which were 1–3 orders of magnitude less sensitive to block by this compound than Kv1.3 (Table 2). In addition, the ChTX-sensitive intermediate-conductance calcium-activated K+ channel (KCa) expressed in activated T cells was not blocked by UK-78,282 (Table 2). Since the binding site of UK-78,282 overlaps that for verapamil on the cytoplasmic surface of Kv1.3, we examined whether this compound could competitively inhibit radiolabelled verapamil binding to calcium channels in cardiac tissue. In this assay, UK-78,282 was found to be ∼20 fold more selective for Kv1.3 compared to cardiac calcium channels (IC50=4.4± 2.1 μM; s.e.mean, n=4). An exception to this pattern of Kv1.3-selectivity was the rapidly inactivating voltage-gated K+ channel in cardiac and neuronal tissues, Kv1.4, whose activity resembles the neuronal after hyperpolarizing current (Stühmer et al., 1989; Ruppersberg et al., 1991; Wymore et al., 1996). This channel was blocked by UK-78,282 with a potency (IC50=170 ±30 nM; mean±s.e.mean, n=3) comparable to the pandinotoxin peptide inhibitors (Rogowski et al., 1996) and CP-339,818 (Nguyen et al., 1996). Interestingly, the selectivity profile of CP-339,818 (Nguyen et al., 1996) is remarkably similar to that of UK-78,282, suggesting that both these drugs that block the C-type inactivated conformation might interact with identical or overlapping sites. However, since Kv1.4 is not expressed in T lymphocytes, UK-78,282 can serve as a pharmacological probe to study the role of Kv1.3 in T-lymphocyte activation.
UK-78,282 suppresses human peripheral blood T cell activation
Studies with the peptide inhibitors MgTX (Leonard et al., 1992; Lin et al., 1993; Koo et al., 1997) and ShK-Dap22 (Kalman et al., 1998), and the dihydroquinoline, CP-339,818 (Hill et al., 1995; Nguyen et al., 1996), indicate that blockade of Kv1.3 alone is sufficient to suppress T-lymphocyte mitogenesis. However, other experiments with the peptide inhibitors KTX and ChTX suggest that both Kv1.3 and the intermediate-conductance KCa channel may need to be blocked in order to inhibit T cell activation (Rader et al., 1996). To distinguish between these two possibilities, UK-78,282 was used as a specific pharmacological probe of Kv1.3. UK-78,282 inhibited 3H-thymidine incorporation and IL-2 production by human peripheral blood T cells stimulated by phytohaemagglutinin (PHA) with IC50 values of 2.0±0.2 μM (n=10) and 2.9±0.3 μM (n=8), respectively. As shown in Table 4, the compound was only effective at blocking IL-2 production when added together with stimulant and at blocking 3H-thymidine uptake when added during the first 20 h after stimulation. These findings are compatible with cell cycle studies which indicated that UK-78,282 blocks lymphocyte activation in the late G1/early S phase of the cell cycle (data not shown).
Table 4.
Effects of UK-78,282 on human T-cell and non-T-cell proliferation, and T-cell IL2 production and IL2-R expression
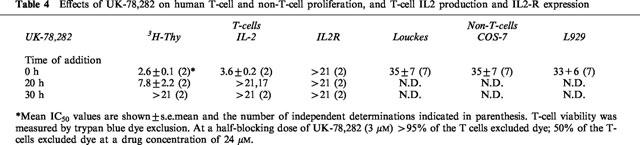
Suppression of T cell activation by UK-78,282 is not due to a toxic effect. First, at the concentrations used in our assays, the compound did not perceptibly alter cell viability of human T lymphocytes as judged by trypan blue exclusion (Table 4). Second, UK-78,282 was a significantly weaker (>10 fold) inhibitor of 3H-thymidine incorporation by non-T cells that do not express Kv1.3 (Table 3). Third, at concentrations required to effectively inhibit 3H-thymidine uptake and IL-2 production, UK-78,282 did not block IL-2 receptor expression (Table 3) or generally inhibit protein synthesis in non-T cells (data not shown). Collectively, these studies suggest that blockade of Kv1.3 alone by UK-78,282 is sufficient to suppress human T cell activation in vitro.
Discussion
In this report, we characterize a novel piperidine compound, UK-78,282, discovered through a high-throughput 86Rb-efflux screen. This compound potently blocks Kv1.3 (IC50 ∼200 nM) and suppresses T lymphocyte mitogenesis. Three independent lines of evidence suggest that UK-78,282 preferentially blocks the C-type inactivated state of the Kv1.3 channel. First, the sensitivity of the channel to UK-78,282 is enhanced as the fraction of inactivated channels increases by changing the holding potential from −80 to −50 mV (Figure 4). Second, substantial reduction in the proportion of inactivated channels via exposure to high external K+ significantly reduces drug potency (Figures 5 and 6). Third, studies with a panel of external vestibular Kv1.3 mutants with differing abilities to undergo C-type inactivation, reveal a direct relationship between the inactivation time constant and the sensitivity to block by UK-78,282 (Figure 7). Once bound, the drug might `lock' the channel in the inactivated conformation. This notion is generally consistent with effects of vestibular mutations on the rate of C-type inactivation and their sensitivity to block by UK-78,282. Mutants containing hydrophobic residues (e.g. valine) that might prefer to be buried, transition slowly into the C-type inactivated conformation and are insensitive to block by UK-78,282, possibly because the drug has lost essential binding sites on the channel.
Although our studies do not determine the precise binding site for UK-78,282, our competition experiments suggest that the site of action of this drug overlaps that of verapamil, but not internal TEA (Figure 8A, C and D). Interestingly, verapamil and internal TEA have overlapping sites of action. This suggests that the receptors for these three compounds are contiguous, with UK-78,282's receptor being at one end overlapping the receptor for verapamil, which in turn overlaps the receptor for internal TEA. If UK-78,282 binds to residues at the internal surface of Kv1.3, how does it hinder binding of radiolabeled ChTX to its receptor in the external vestibule? In the binding experiments, UK-78,282 is added at least 15–20 min prior to the toxin-probe, giving UK-78,282 enough time to cross the membrane, interact with its internal receptor, and allosterically interfere with 125I-ChTX binding to the external vestibule. Our `acute' competition experiments with unlabelled ChTX and UK-78,282 (Figure 8A and B) are compatible with this idea. Why do mutations in the external vestibule alter the channel's sensitivity to UK-78,282 if this compound binds to an internal receptor? Residues at this internal binding site may only be accessible in the C-type inactivated conformation, and mutations in the outer vestibule that remove inactivation might reduce the accessibility of these residues. In fact, mutations at the external His404 position in Kv1.3 interfere with verapamil binding to residues in the inner surface of the channel, at a site overlapping that of UK-78,282 (Rauer & Grissmer, 1996). In conclusion, our results are consistent with the drug binding to residues at the cytoplasmic surface of the channel. Our studies also suggest that high-throughput screening assays based on the competition of 125I-ChTX binding to channels are capable of discovering compounds that interact with distant sites in the channel, and not necessarily in the external vestibule.
Using UK-78,282 as a pharmacological probe of Kv1.3, we demonstrated that blockade of Kv1.3 alone is sufficient to inhibit both mitogen-induced IL-2 production and 3H-thymidine incorporation, without inhibiting expression of the IL-2 receptor. This confirms earlier studies with other peptide and non-peptide inhibitors of Kv1.3 (Chandy et al., 1984; Leonard et al., 1992; Lin et al., 1993; Koo et al., 1997, Kalman et al., 1998). T-cell activation requires multiple signal transduction pathways including the activation of calcineurin by a sustained or oscillatory rise in [Ca2+]i (reviewed in Lewis & Cahalan, 1995; Cahalan & Chandy, 1997). Ca2+ influx for a period of hours is required for accumulation of the NF-AT transcription factor in the nucleus leading to new gene transcription, including the IL-2 gene. The evidence is strong that K+ channel blockers indirectly inhibit the calcineurin/NF-AT pathway by depolarizing the membrane and thereby reducing the electrical driving force for Ca2+ entry (reviewed in Lewis & Cahalan, 1995; Cahalan & Chandy, 1997; Kath et al., 1997). Although maximal inhibition by Kv1.3 inhibitors of in vitro mitogen-stimulated 3H-thymidine incorporation by human T lymphocytes is never more than 60–80% (e.g. Kalman et al., 1998), these blockers display clear immunosuppressive activity in vivo (Koo et al., 1997).
What are the reasons for this discrepancy between the in vitro and in vivo efficacy of Kv1.3 blockers? One possibility is that the activation signals in in vitro mitogen assays (e.g. induced by phytohaemagglutin or by a combination of phorbol esters and ionomycin) are substantially more robust than that encountered physiologically during a normal immune response in vivo. Given that the opening of a few of the 400 or more Kv1.3 channels in human T lymphocytes is sufficient to maintain an adequate membrane potential and support Ca2+ influx (Cahalan et al., 1985; also reviewed in Lewis & Cahalan, 1995; Cahalan & Chandy, 1997), it is not surprising that half-blocking concentrations of Kv1.3 inhibitors required to suppress T-lymphocyte activation in vitro are higher than those required for channel blockade. The exact concentration of a Kv1.3 inhibitor necessary to suppress T lymphocyte activation depends upon the intensity of the activation stimulus, and on the number of Kv1.3 channels that have to be blocked to sufficiently depolarize the membrane. Studies with Kv1.3 inhibitors suggest that at least 80–90% of the Kv1.3 channels need to be blocked to achieve significant suppression of mitogenesis in vitro (Lewis & Cahalan, 1995; Cahalan & Chandy, 1997; Kath et al., 1997). Furthermore, small numbers of other Kv channels and KCa channels may help to buffer the membrane potential to some extent (reviewed in Cahalan & Chandy, 1997). In conclusion, UK-78,282, a reasonably potent and selective blocker of Kv1.3, might serve as an experimental tool to probe the conformational changes in the Kv1.3 channel leading to C-type inactivation and to further define the critical physiological role of Kv1.3 in T lymphocyte activation.
Acknowledgments
We thank Mr Brent Dethlefs and Dr Luette Forrest for technical assistance. Some of these studies were presented at the Keystone Meeting on Ion Channels as Therapeutic Targets, Tamarron, CO, U.S.A. Feb. 4–10, 1996. This work was supported by grants from the NIH (AI24783, K.G. Chandy; N.S 14609, M.D. Cahalan) and Pfizer Inc. (K.G. Chandy, M.D. Cahalan, G.A. Gatman, S. Grissmer).
References
- AIYAR J., NGUYEN A., CHANDY K.G., GRISSMER S. The P-region and S6 segment of Kv3.1 contribute to the formation of the potassium channel pore. Biophys. J. 1994;67:2261–2264. doi: 10.1016/S0006-3495(94)80710-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AIYAR J., RIZZI J.P., GUTMAN G.A., CHANDY K.G. The signature sequence of voltage-gated potassium channels projects into the external vestibule. J. Biol. Chem. 1996;271:31013–31016. doi: 10.1074/jbc.271.49.31013. [DOI] [PubMed] [Google Scholar]
- AIYAR J., WITHKA J.M., RIZZI J.P., SINGLETON D.H., ANDREWS G.C., LIN W., BOYD J., HANSON D.C., SIMON M., DETHLEFS B., LEE C-L., HALL J.E., GUTMAN G.A., CHANDY K.G. Topology of the pore-region of a K+ channel revealed by the NMR-derived structures of scorpion toxins. Neuron. 1995;15:1169–1181. doi: 10.1016/0896-6273(95)90104-3. [DOI] [PubMed] [Google Scholar]
- BURGESS L.E., KOCH K., COOPER K., BIGGERS M.S., RAMCHANDANI M., SMITROVICH J.H., GILBERT E.J., BRUNS M.J., MATHER R.J., DONOVAN C.B., HANSON D.C. The SAR of UK-78,282: a novel blocker of human T cell Kv1.3 potassium channels. Biorganic Med. Chem. Lett. 1997;7:1047–1052. [Google Scholar]
- BUSCH A.E., HURST R.S., NORTH R.A., ADELMAN J.P., KAVANAUGH M.P. Current inactivation involves a histidine residue in the pore of the rat lymphocyte potassium channel RGK5. Biochem. Biophys. Res. Commun. 1991;179:1384–1390. doi: 10.1016/0006-291x(91)91726-s. [DOI] [PubMed] [Google Scholar]
- CAHALAN M.D., CHANDY K.G. Ion channels in the immune system as targets for immunesuppression. Curr. Opin. Biotechnol. 1997;8:749–756. doi: 10.1016/s0958-1669(97)80130-9. [DOI] [PubMed] [Google Scholar]
- CAHALAN M.D., CHANDY K.G., DECOURSEY T.E., GUPTA S. A voltage-gated potassium channel in human T lymphocytes. J. Physiol. 1985;358:197–237. doi: 10.1113/jphysiol.1985.sp015548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDY K.G., DECOURSEY T.E., CAHALAN M.D., MCLAUGHLIN C., GUPTA S. Voltage-gated potassium channels are required for human T lymphocyte activation. J. Exp. Med. 1984;160:369–385. doi: 10.1084/jem.160.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANDY K.G., GUTMAN G.A.Voltage-gated potassium channel genes Handbook of Receptors and Channels: Ligand- and Voltage-gated Ion Channels 1995Boca Raton, FL: CRC Press, Inc; 1–71.ed North, R.A. pp [Google Scholar]
- DECOURSEY T.E., CHANDY K.G., GUPTA S., CAHALAN M.D. Voltage-gated potassium channels in human T lymphocytes: a role in mitogenesis. Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- DEUTSCH C., PRICE M., LEE S., KING V.F., GARCIA M.L. Characterization of high affinity binding sites for charybdotoxin in human T-lymphocytes. Evidence for association with the voltage-gated K+ channel. J. Biol. Chem. 1991;266:3668–3674. [PubMed] [Google Scholar]
- FONG T.M., CASCIERI M.A., YU H., BANSAL A., SWAIN C., STRADER C.D. Amino-aromatic interaction between histidine 197 of the neurokinin-1 receptor and CP-96345. Nature. 1993;362:350–353. doi: 10.1038/362350a0. [DOI] [PubMed] [Google Scholar]
- FREEDMAN B.D., PRICE M.A., DEUTSCH C.J. Evidence for voltage modulation of IL-2 production in mitogen-stimulated hu-man peripheral blood lymphocytes. J. Immunol. 1992;149:3784–3794. [PubMed] [Google Scholar]
- GRISSMER S., CAHALAN M.D. TEA prevents inactivation while blocking open K+ channels in human T-lymphocytes. Biophys. J. 1989a;55:203–206. doi: 10.1016/S0006-3495(89)82793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISSMER S., CAHALAN M.D. Divalent ion trapping inside potassium channels of human T lymphocytes. J. Gen. Physiol. 1989b;93:609–630. doi: 10.1085/jgp.93.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISSMER S., DETHLEFS B., WASMUTH J.J., GOLDIN A.L., GUTMAN G.A., CAHALAN M.D., CHANDY K.G. Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc. Natl. Acad. Sci. U.S.A. 1990;87:9411–9415. doi: 10.1073/pnas.87.23.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRISSMER S., NGUYEN A.N., AIYAR J., HANSON D.C., MATHER R.J., GUTMAN G.A., KARMILOWICZ M.J., AUPERIN D.D., CHANDY K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- HILL R.J., GRANT A.M., VOLBERG W., RAPP L., FALTYNEK C., MILLER D., PAGANI K., BAIZMAN E., WANG S., GUILES J.W., KRAFTE D.S. WIN 17317-3: novel nonpeptide antagonist of voltage-activated K+ channels in human T lymphocytes. Mol. Pharm. 1995;48:98–104. [PubMed] [Google Scholar]
- IKEDA S.R., SOLER F., ZUHLKE R.D., LEWIS D.L. Heterologous expression of the human potassium channel, Kv2.1, in clonal mammalian cells by direct cell injection. Pflueg. Arch. Eur. J. Physiol. 1992;422:201–203. doi: 10.1007/BF00370422. [DOI] [PubMed] [Google Scholar]
- JÄGER H., RAUER H., NGUYEN A.N., AIYAR J., CHANDY K.G., GRISSMER S. Regulation of mammalian Shaker-related potassium channels by extracellular potassium and pH: evidence for non-conducting closed and non-conducting inactivated states. J. Physiol. 1998;506:291–301. doi: 10.1111/j.1469-7793.1998.291bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KALMAN K., PENNINGTON M.W., LANIGAN M.D., NGUYEN A., RAUER H., MAHNIR V., PASCHETTO K., KEM W.R., GRISSMER S., GUTMAN G.A., CHRISTIAN E.P., CAHALAN M.D., NORTON R.S., CHANDY K.G. ShK-Dap22: A potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998;273:5851–5857. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- KATH J.C., HANSON D.C., CHANDY K.G. T lymphocyte potassium channel blockers. Ann. Reports in Med. Chem. 1997;32:181–190. [Google Scholar]
- KNAUS H.G., MCMANUS O.B., LEE S.H., SCHMALHOFER W.A., GARCIA-CALVO M., HELMS L.M., SANCHEZ M., GIANGIACOMO K., REUBEN J.P., SMITH A.B., III, KACZOROWSKI G., GARCIA M. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 1994;33:5819–5828. doi: 10.1021/bi00185a021. [DOI] [PubMed] [Google Scholar]
- KOO G.C., BLAKE J.T., TALENTO A., NGUYEN M., LIN S., SIROTINA A., SHAH K., MULVANY K., HORA D. , JR, CUNNINGHAM P., WUNDERLER D.L., MCMANUS O., SLAUGHTER R., BUGIANESI R., FELIX J., GARCIA M., WILLIAMSON J., KACZOROWSKI G., SIGAL N.H., SPRINGER M.S., FEENEY W. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J. Immunol. 1997;158:5120–5128. [PubMed] [Google Scholar]
- KOSCHAK A., BUGIANESI R.M., MITTERDORFER J., KACZOROWSKI G.J., GARCIA M.L., KNAUS H.G. Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom. J. Biol. Chem. 1998;273:2639–2644. doi: 10.1074/jbc.273.5.2639. [DOI] [PubMed] [Google Scholar]
- LEONARD R.J., GARCIA M.L., SLAUGHTER R.S., REUBEN J.P. Selective blockers of voltage-gated K+ channels depolarize human T lymphocytes: mechanism of the antiproliferative effect of charybdotoxin. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10094–10098. doi: 10.1073/pnas.89.21.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVY D.I., DEUTSCH C.J. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys. J. 1996;70:798–805. doi: 10.1016/S0006-3495(96)79619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIS R.S., CAHALAN M.D. Potassium and calcium channels in lymphocytes. Annu. Rev. Immunol. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- LIN C.S., BOLTZ R.C., BLAKE J.T., NGUYEN M., TALENTO A., FISCHER P.A., SPRINGER M.S., SIGAL N.H., SLAUGHTER R.S., GARCIA M.L., KACZOROWSKI G.J., KOO G.C. Voltage-gated potassium channels regulate calcium-dependent pathways involved in human T lymphocyte activation. J. Exp. Med. 1993;177:637–645. doi: 10.1084/jem.177.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU T., JURMAN M.E., YELLEN G. Dynamic arrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- LOPEZ-BARNEO J., HOSHI T., HEINEMANN S.H., ALDRICH R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Recept. Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- NGUYEN A., KATH J.C., HANSON D.C., BIGGERS M.S., CANNIFF P.C., DONOVAN C.B., MATHER R.J., BRUNS M.J., RAUER H., AIYAR J., LEPPLE-WIENHUES A., GUTMAN G.A., GRISSMER S., CAHALAN M.D., CHANDY K.G. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv1.3 and suppress T cell activation. Mol. Pharm. 1996;50:1672–1679. [PubMed] [Google Scholar]
- OGIELSKA E.M., ZAGOTTA W.N., HOSHI T., HEINEMANN S.H., ALDRICH R.W. Cooperative subunit interaction in C-type inactivation of K+ channels. Biophys. J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANYI G., SHENG Z., DEUTSCH C.J. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys. J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRICE M., LEE S.C., DEUTSCH C.J. Charybdotoxin inhibits proliferation and interleukin 2 production in human peripheral blood lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 1989;86:10171–10175. doi: 10.1073/pnas.86.24.10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADER R.K., KAHN L.E., ANDERSON G.D., MARTIN C.L., CHINN K.S., GREGORY S.A. T cell activation is regulated by voltage-dependent and calcium-activated potassium channels. J. Immunol. 1996;156:1425–1430. [PubMed] [Google Scholar]
- RAUER H., GRISSMER S. Evidence for an internal phenylalkylamine action on the voltage-gated potassium channel Kv1.3. Mol. Pharm. 1996;50:1625–1634. [PubMed] [Google Scholar]
- ROGOWSKI R.S., COLLINS J.H., O'NEILL T.J., GUSTAFSON T.A., WERKMAN T.R., ROGAWSKI M.A., TENENHOLZ T.C., WEBER D.J., BLAUSTEIN M.P. Three new toxins from the scorpion Pandinus imperator selectively block certain voltage-gated K+ channels. Mol. Pharm. 1996;50:1167–1177. [PubMed] [Google Scholar]
- RUPPERSBERG J.P., FRANK R., PONGS O., STOCKER M. Cloned neuronal Ik(A) channels reopen during recovery from inactivation. Nature (Lond). 1991;17:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- SABATH D.E., MONOS D.S., LEE S.C., DEUTSCH C., PRYSTOWSKY M.B. Cloned T-cell proliferation and synthesis of specific proteins are inhibited by quinine. Proc. Natl. Acad. Sci. U.S.A. 1986;83:4739–4743. doi: 10.1073/pnas.83.13.4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANDS S.B., LEWIS R.S., CAHALAN M.D. Charybdotoxin blocks voltage-gated K+ channels in human and murine T-lymphocytes. J. Gen. Physiol. 1989;93:1061–1074. doi: 10.1085/jgp.93.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAMOTIENKO O.G., PARCEJ D.N., DOLLY J.O. Subunit combinations defined for K+ channel Kv1 subtypes in synaptic membranes from bovine brain. Biochemistry. 1997;36:8195–8201. doi: 10.1021/bi970237g. [DOI] [PubMed] [Google Scholar]
- SPENCER R.H., SOKOLOV Y., LI H., TAKENAKA B., MILICI A.J., AIYAR J., NGUYEN A., PARK H., JAP B.K., HALL J.E., GUTMAN G.A., CHANDY K.G. Purification, visualization, and biophysical characterization of Kv1.3 tetramers. J. Biol. Chem. 1997;272:2389–2395. doi: 10.1074/jbc.272.4.2389. [DOI] [PubMed] [Google Scholar]
- STÜHMER W., RUPPERSBERG J.P., SCHROTER K.H., SAKMANN B., STOCKER M., GIESE K.P., PERSCHKE A., BAUMANN A., PONGS O. Molecular basis of functional diversity of voltage-gated potassium channels in mammalian brain. EMBO J. 1989;8:3235–3244. doi: 10.1002/j.1460-2075.1989.tb08483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WYMORE R., LAIDLAW D., KINOSHITA K., KALMAN K., GUTMAN G.A., CHANDY K.G. Characterization of the transcription unit of a mammalian K+ channel gene, Kv1.4. J. Biol. Chem. 1996;271:15629–15634. doi: 10.1074/jbc.271.26.15629. [DOI] [PubMed] [Google Scholar]