

Abstract
Intracellularly recorded excitatory junction potentials (e.j.ps) were used to study the effects of adenosine receptor antagonists on neurotransmitter release from postganglionic sympathetic nerve terminals in the guinea-pig vas deferens in vitro.
The A1 adenosine receptor antagonists, 8-phenyltheophylline (10 μM) and 8-cyclopentyl-1,3-dipropylxanthine (0.1 μM), increased the amplitude of e.j.ps evoked during trains of 20 stimuli at 1 Hz in the presence, but not in the absence, of the α2-adrenoceptor antagonist, yohimbine (1 μM) or the non-selective α-adrenoceptor antagonist, phentolamine (1 μM).
Adenosine (100 μM) reduced the amplitude of e.j.ps, both in the presence and in the absence of phentolamine (1 μM). This inhibitory effect of adenosine is most likely caused by a reduction in transmitter release as there was no detectable change in spontaneous e.j.p. amplitudes.
In the presence of phentolamine, application of the adenosine uptake inhibitor, S-(p-nitrobenzyl)-6-thioinosine (0.1 μM), had no effect on e.j.p. amplitudes.
The phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine (100 μM), significantly increased the amplitudes of all e.j.ps evoked during trains of 20 stimuli at 1 Hz, both in the presence and in the absence of phentolamine (1 μM).
These results suggest that endogenous adenosine modulates neurotransmitter release by an action at prejunctional A1 adenosine receptors only when α2-adrenoceptors are blocked.
Keywords: adenosine receptors, α2-adrenoceptors, neurotransmitter release, electrophysiology, excitatory junction potentials, guinea-pig vas deferens, sympathetic nerves, purinergic transmission
Introduction
Adenosine is now widely viewed as a modulator of neurotransmitter release at a number of central and peripheral synapses (see Fredholm, 1995). At the sympathetic neuroeffector junction, exogenously applied adenosine has been shown to inhibit electrically evoked [3H]-noradrenaline release in several preparations including guinea-pig vas deferens (Hedqvist & Fredholm, 1976; Driessen et al., 1994). In addition, in guinea-pig vas deferens, exogenously applied adenosine or adenosine analogues have been demonstrated to inhibit neurally evoked contraction and to reduce both the overflow of endogenous adenosine 5′ triphosphate (ATP) and the amplitude of intracellularly recorded excitatory junction potentials (e.j.ps) evoked by electrical stimulation (Sneddon et al., 1984; Kirkpatrick & Burnstock, 1992; Driessen et al., 1994). These inhibitory effects of adenosine are prevented by the A1-adenosine receptor antagonist, 8-phenyltheophylline (8-PT, Sneddon et al., 1984; Kirkpatrick & Burnstock, 1992) and are believed to be mediated through a decrease in neurotransmitter release caused by activation of prejunctional A1 adenosine receptors.
It has been suggested that adenosine released from the cytosol of cells or generated from the extracellular breakdown of neurally released ATP may play an important role in the regulation of neurotransmitter release from the nerve terminals of many types of neurone (Fredholm, 1995). Although the inhibitory effects of exogenously applied adenosine or adenosine analogues can be reversed by 8-PT in the guinea-pig vas deferens, application of this agent alone has been reported to have no effect on electrically evoked contractions or e.j.ps in this tissue (Sneddon et al., 1984; Dalziel & Sneddon, 1988). Indeed, in a number of sympathetically innervated tissues in which adenosine has been shown to inhibit neurotransmitter release, application of A1-adenosine receptor antagonists alone does not detectably modify neurotransmitter release (e.g. mouse vas deferens, Blakeley et al., 1988; rat rail artery, Gonçalves & Queiroz, 1996; human atrial muscle, Munch et al., 1996). These findings suggest that in these tissues endogenous adenosine does not normally act prejunctionally to inhibit neurotransmitter release at the sympathetic neuroeffector junction.
In the previous studies investigating the effects of adenosine receptor antagonists on guinea-pig vas deferens (Sneddon et al., 1984; Dalziel & Sneddon, 1988), the action of released NA at prejunctional α2-adrenoceptors was not blocked. However, it has been reported that the inhibition of neurotransmitter release produced by activating prejunctional α2-adrenoceptors can occlude that produced by activating prejunctional A1 adenosine receptors (Limberger et al., 1988; Allgaier et al., 1991; Bucher et al., 1992). Thus blockade of prejunctional α2-adrenoceptors may reveal a presynaptic inhibitory effect of endogenous adenosine. In support of this suggestion, the A1 adenosine receptor selective antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), increased the stimulated overflow of [3H]-NA from rat vas deferens (Gonçalves & Queiroz, 1993; Kurz et al., 1993) and rat iris (Fuder et al., 1992) when prejunctional α2-adrenoceptors were blocked.
The aim of the present study was to examine the effects of the A1 adenosine receptor antagonists 8-PT and DPCPX on the amplitude of e.j.ps evoked by field stimulation of the guinea-pig vas deferens. In this tissue, intracellularly recorded e.j.ps are believed to measure, impulse-by-impulse, the release of the co-transmitter ATP (see Sneddon, 1992). In particular, this study has compared the effects of these A1 adenosine receptor antagonists in the presence and in the absence of α-adrenoceptor blockade.
Methods
Male guinea-pigs (200–300 g) were killed by an overdose of pentobarbitone sodium (100 mg kg−1, i.p.). Vasa deferentia were removed and pinned to the Sylgard (Dow Corning Corporation, Midland, MI, U.S.A.) coated base of a 1 ml recording chamber. The tissue was superfused continuously at 3–5 ml min−1 with physiological saline of the following ionic composition (mM): Na+ 151, K+ 4.7, Ca2+ 2, Mg2+ 1.2, Cl− 144.5, H2PO4− 1.3, HCO3− 16.3 and glucose 9.8. The physiological saline was gassed with a mixture of 95% O2 and 5% CO2 (to pH 7.4) and maintained at 35–36°C. For some experiments, the α2-adrenoceptor antagonist, yohimbine (1 μM), or the non selective α-adrenoceptor antagonist, phentolamine (1 μM), was added to the physiological saline in order to block α-adrenoceptor-mediated autoinhibition of transmitter release. Postganglionic sympathetic nerve fibres were excited by electrical stimulation through a pair of ring electrodes positioned around the prostatic end of the vas deferens.
Intracellular recording
Conventional glass micro-electrodes filled with 2 M KCl (resistances 80–120 MΩ) and connected to an Axoclamp bridge amplifier (Axon Instruments, Inc., Foster City, CA, U.S.A.) were used for intracellular recording of e.j.ps. In all experiments investigating the effects of drugs, the stimulation parameters (pulse width 0.5–1 ms, voltage 5–15 V) were adjusted under control conditions (i.e. with or without α-adrenoceptor blockers) to evoke e.j.ps of approximately 10 mV amplitude at full facilitation in order to reduce the effects of non-linear summation on e.j.p. amplitude. Once set, the stimulus parameters were not changed for the duration of the experiment. During impalements the tissues were stimulated at 2 min intervals with trains of 20 pulses at 1 Hz. In the majority of experiments the effects of the drugs were determined during a single impalement. In those experiments in which we failed to maintain impalements for the duration of the experiment, recordings from two or three separate impalements before and during drug treatment were analysed. In each tissue all recordings were made from a similar location. Drugs were applied by their addition, at the required concentration, to the superfusing solution and were left in contact with the tissue for a period of 20 min or until their effect on e.j.p. amplitude had stabilized. In the absence of drugs (n=7 tissues), no change in the amplitudes of e.j.ps evoked by trains of 20 stimuli at 1 Hz was detected during recording periods of 30 min. For each impalement, the resting membrane potential (r.m.p.) was determined upon withdrawal of the microelectrode. Only impalements in which the measured r.m.p was greater than −60 mV were used in this study.
The electrophysiological signals were digitized (sampling frequencies of 0.1–0.2 kHz) and stored using a Maclab recording system (AD Instruments, Castle Hill, N.S.W., Australia) attached to a Macintosh computer. The amplitude and time constant of decay of e.j.ps were determined using the program Igor Pro (Wavemetrics, Lake Oswego, OR, U.S.A.). Before analysis, e.j.ps evoked by three successive trains of stimuli under control and test conditions were averaged. In experiments where we failed to maintain the impalements during the drug treatment, e.j.ps evoked by three successive trains of stimuli in each cell in the presence and absence of the drug were averaged and analysed. In these tissues the e.j.p. amplitudes for the control cells and test cells were averaged before statistical comparisons.
Statistics
Data are presented as mean±s.e.mean. Statistical comparisons were made between the control and test e.j.p. amplitudes using repeated measures analysis of variance and paired t-tests. When multiple pairwise comparisons were made, the P value was corrected using the Dunn–Sidak procedure. The other statistical tests used are indicated in the text. P values <0.05 were considered significant. In all cases the number n refers to the number of tissues studied.
Drugs
Adenosine (Sigma Chemical Company, Castle Hill, N.S.W., Australia), phentolamine mesylate (Regitine: Ciba, Pendle Hill, N.S.W., Australia), yohimbine (Sigma), 8-phenyltheophylline (8-PT, Sigma), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, Sigma), S-(p-nitrobenzyl)-6-thioinosine (NBTI, Sigma) and 3-isobutyl-1-methylxanthine (IBMX; RBI, Natick, MA, U.S.A.) were used. 8-PT, DPCPX, NBTI and IBMX were prepared as 10 mM stock solutions in 80% v v−1 methanol in water containing 0.2 M NaOH. All remaining drugs were prepared as stock solutions in water.
Results
General observations
In α-adrenoceptor antagonist untreated vasa deferentia, stimulation with a train of 20 pulses at 1 Hz produced e.j.ps which facilitated in amplitude, reaching a plateau level after about the eighth stimulus in the train (Figure 1a and b). In tissues treated with the α2-adrenoceptor antagonist, yohimbine (1 μM, Figure 1c and d) or the non-selective α-adrenoceptor antagonist, phentolamine (1 μM Figure 1e and f) the period of facilitation was lengthened so that the amplitude of e.j.ps continued to increase until about the fifteenth stimulus in the train. Following adjustment of the stimulation strength (see Methods), the mean e.j.p. amplitude measured during the last 5 stimuli in the train was 9.1±0.4 mV (n=27) in the α-adrenoceptor antagonist untreated tissues, 11.6±0.5 (n=17) in yohimbine treated tissues and 10.9±0.4 mV (n=38) in phentolamine treated tissues.
Figure 1.
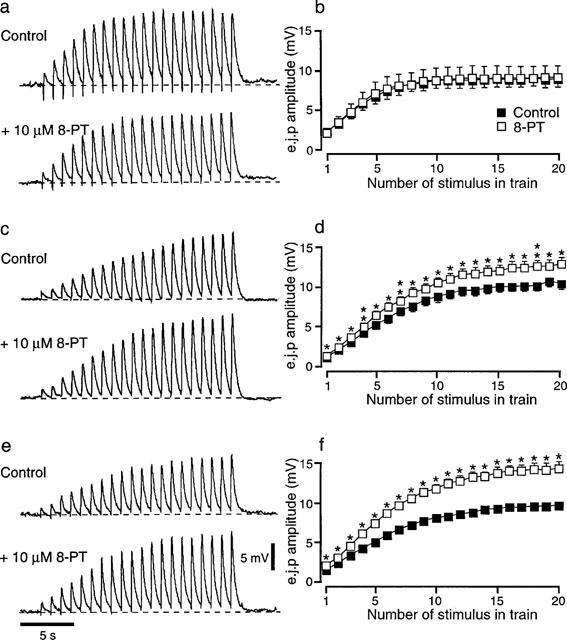
Effects of 8-PT (10 μM) on the amplitude of e.j.ps recorded during trains of 20 pulses at 1 Hz in the absence (a and b) and in the presence of (c and d) yohimbine (1 μM) or (e and f) phentolamine (1 μM). (a, c and e) Traces recorded before and during application of 8-PT in single impalements. (b, d and f) Graphs showing the mean effects of 8-PT on e.j.p. amplitude. In (b) n=6 tissues and in (d) and (f) n=7 tissues. *P<0.05, **P<0.01. The symbols in (b) also apply in (d and f).
Effects of 8-PT on e.j.p. amplitude
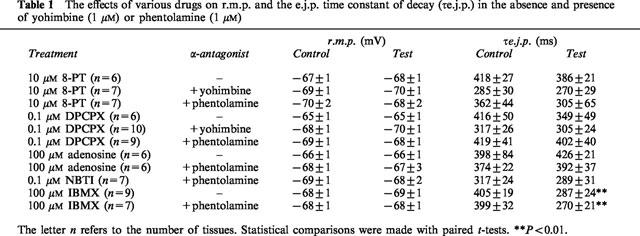
Figure 1 shows the effects of the adenosine antagonist, 8-PT (10 μM), on the amplitude of e.j.ps evoked during trains of 20 pulses at 1 Hz in the absence (Figure 1a and b) and in the presence of yohimbine (1 μM, Figure 1c and d) or phentolamine (1 μM, Figure 1e and f). In the absence of α-adrenoceptor blockade (n=6), application of 8-PT had no significant effect on the amplitude of e.j.ps evoked during the trains of stimuli (Figure 1b). In contrast, when yohimbine (n=7, Figure 1d) or phentolamine (n=7, Figure 1f) was present, 8-PT significantly increased the amplitudes of all e.j.ps evoked during the train of stimuli. In all sets of experiments, application of 8-PT had no significant effect on the r.m.p. or the e.j.p. time constant of decay (Table 1).
Table 1.
The effects of various drugs on r.m.p. and the e.j.p. time constant of decay (τe.j.p.) in the absence and presence of yohimbine (1 μM) or phentolamine (1 μM)
Comparison between the facilitatory effect of 8-PT on the mean amplitude of the last 5 e.j.ps evoked during the trains of stimuli in yohimbine (increased by 22±8%) and phentolamine (increased by 36±9%) treated tissues reveal no significant difference (unpaired t-test, P=0.19).
Effects of DPCPX on e.j.p. amplitude
Like 8-PT, the selective A1-receptor antagonist, DPCPX (0.1 μM), had no significant effect upon e.j.p. amplitude in the absence of α-adrenoceptor blockade (n=6, Figure 2a). In the presence of yohimbine (1 μM, n=10, Figure 2b) or phentolamine (1 μM, n=9, Figure 2c), DPCPX did not significantly change the amplitudes of e.j.ps evoked early in the train of stimuli, but those evoked during the second half of the train of stimuli were significantly increased in amplitude. DPCPX had no significant effect on the r.m.p. or the e.j.p. time constant of decay (Table 1).
Figure 2.
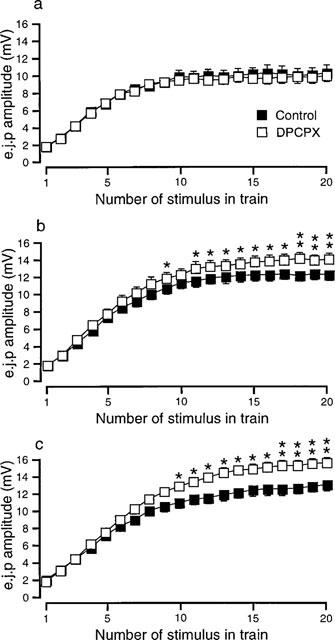
Graphs showing the effects of DPCPX (0.1 μM) on the mean amplitude of e.j.ps recorded during trains of 20 pulses at 1 Hz in the absence (a) and in the presence of (b) yohimbine (1 μM) and (c) phentolamine (1 μM). In (a) n=6 tissues, (b) n=10 tissues and (c) n=9 tissues. *P<0.05, **P<0.01. The symbols in (a) also apply in (b) and (c).
The facilitatory effect of DPCPX on the mean amplitude of the last 5 e.j.ps evoked during the trains of stimuli in yohimbine treated tissues (increased by 13±2%) was significantly less (unpaired t-test, P<0.05) than that observed in phentolamine treated tissues (increased by 25±5%).
Effects of adenosine on e.j.p. amplitude
To determine whether the inhibitory effects on e.j.p. amplitude of activating A1-adenosine receptors (see Sneddon et al., 1984) were modified by blockade of α-adrenoceptors, the effects of adenosine were studied in the absence (n=6) and in the presence (n=6) of phentolamine. Under both conditions, adenosine (100 μM) reduced the amplitude of all e.j.ps evoked during the trains of 20 stimuli at 1 Hz (Figure 3a and b). The reduction in amplitude for the last five e.j.ps evoked during the train of stimuli was 40±16% in the absence of phentolamine and 44±6% in the presence of phentolamine. Statistical comparison revealed no significant difference between these inhibitory effects (unpaired t-test, P=0.52). Adenosine had no significant effect upon r.m.p. or e.j.p. time constant of decay (Table 1). 8-PT (10 μM) partially reversed the inhibitory effects of adenosine on e.j.p. amplitude in the absence (n=6) and in the presence (n=6) of phentolamine (results not shown).
Figure 3.
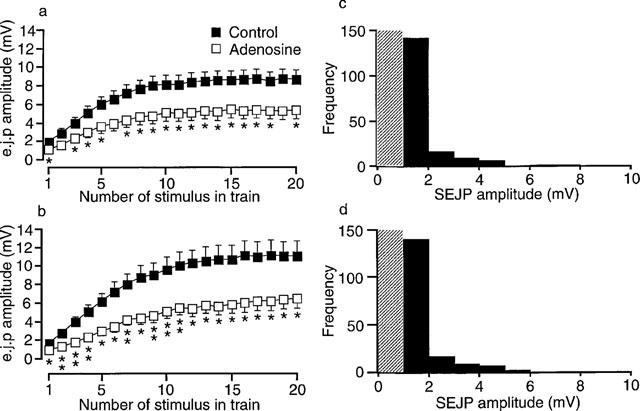
Effects of adenosine (100 μM) on the amplitudes of electrically evoked e.j.ps and spontaneous e.j.ps. (a and b) Graphs showing the effects of adenosine (100 μM) on the amplitude of e.j.ps recorded during trains of 20 stimuli at 1 Hz in the absence (a) and in the presence (b) of phentolamine (1 μM). In (a) and (b) n=6 tissues. (c and d) Amplitude frequency distributions for spontaneous e.j.ps recorded in phentolamine treated tissues before (c) and during application of 100 μM adenosine (d). Each distribution contains the amplitude of 35 sequentially recorded spontaneous e.j.ps from each of five tissues (175 in total) under each condition. The hatched area represents the detection level (1 mV) used to collect the spontaneous e.j.ps. *P<0.05, **P<0.01. The symbols in (a) also apply in (b).
As previously reported (Sneddon et al., 1984) the inhibitory effect of adenosine on e.j.p. amplitude was most likely due to a reduction in neurotransmitter release as adenosine had no effect (chi-squared test) on the amplitude frequency distribution of spontaneous e.j.ps (Figure 3c and d). These events are believed to monitor the sensitivity of the postjunctional membrane to spontaneously released quanta of ATP (Brock & Cunnane, 1993).
Effects of NBTI on e.j.p. amplitude
The adenosine uptake inhibitor, NBTI (0.1 μM), in tissues treated with phentolamine (n=7) had no effect on e.j.p. amplitudes evoked during trains of 20 stimuli at 1 Hz. In the absence and presence of NBTI the mean amplitude of the e.j.ps evoked by first stimulus in the train was 1.0±0.1 mV and 1.4±0.3 mV (paired t-test, P=0.24) respectively and by the last five stimuli was 12.2 mV and 12.4±1.0 mV respectively (paired t-test, P=0.5). NBTI also had no effect on r.m.p. or the e.j.p. time constant of decay (Table 1).
Effects of IBMX on e.j.p. amplitude
The effects of phosphodiesterase inhibitor, IBMX (100 μM), were investigated to assess the possibility that 8-PT may, in part, be acting through an inhibition of phosphodiesterase (Nicholson & Wilke, 1989). Both in the absence (Figure 4a, n=9) and presence (Figure 4b, n=7) of phentolamine, IBMX significantly increased the amplitudes of all e.j.ps evoked during the train of stimuli. IBMX had no significant effect upon r.m.p., but significantly reduced the e.j.p. time constant of decay (Table 1).
Figure 4.
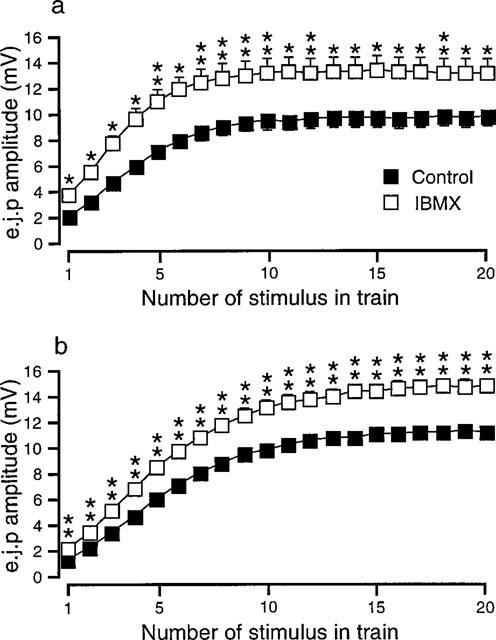
Graphs showing the effects of IBMX (100 μM) on the amplitude of e.j.ps recorded during trains of 20 stimuli at 1 Hz in the absence (a) and in the presence (b) of phentolamine (1 μM). In (a) n=9 tissues and (b) n=5 tissues. *P<0.05, **P<0.01. The symbols in (a) also apply in (b).
Discussion
The main finding of this study is that the A1 adenosine receptor antagonists, 8-PT and DPCPX, increased the amplitude of e.j.ps only when α-adrenoceptors were blocked. In the absence of α-adrenoceptor blockade, these agents had no detectable effect on e.j.p. amplitude. The latter finding is in agreement with previous reports that 8-PT, without prior blockade of α-adrenoceptors, had no detectable effect on neurally evoked contraction or e.j.p. amplitude in guinea-pig vas deferens (Sneddon et al., 1984; Dalziel & Sneddon, 1988). In this tissue, e.j.ps are believed to provide a measure of ATP released from the postganglionic sympathetic nerve terminals (see Sneddon, 1992). Previous studies have shown that exogenously applied adenosine or adenosine receptor agonists act at prejunctional A1 adenosine receptors to reduce both the neural release of NA and ATP (Kirkpatrick & Burnstock, 1992; Driessen et al., 1994) and the amplitude of e.j.ps (Sneddon et al., 1984; Dalziel & Sneddon, 1988). Therefore the findings of the present study suggest a role for endogenous adenosine in modulating neurotransmitter release in the guinea-pig vas deferens.
The facilitatory effect of the DPCPX was significantly greater in tissues treated with non-selective α-adrenoceptor antagonist, phentolamine, than in those treated with selective α2-adrenoceptor antagonist, yohimbine. While not significant, the mean facilitatory effect of 8-PT in phentolamine treated tissues was also greater than in yohimbine treated tissues. These findings can probably be explained by the observation that phentolamine has a significantly greater facilitatory effect on e.j.p. amplitude than yohimbine (Hardy & Brock, unpublished observations). It is unlikely that the effects of phentolamine on e.j.p. amplitude can be explained, in part, by blockade of pre- or postjunctional α1-adrenoceptors, as the potent α1-adrenoceptor antagonist, prazosin, at concentrations up to 0.1 μM has been reported to be without effect on e.j.p. amplitude in guinea-pig vas deferens (Blakeley et al., 1981). Perhaps the best explanation for our finding is that, as reported for sympathetic nerve terminals in guinea-pig ileum and heart (Funk et al., 1995; Trendelenberg et al., 1995), the prejunctional α2-adrenoceptors in guinea-pig vas deferens are of the α2D-subtype. This receptor subtype has been reported to have a higher affinity for phentolamine than for yohimbine (Lanier et al., 1991).
There have been a number of reports that activation of presynaptic α2-adrenoceptors can reduce the inhibitory effects on neurotransmitter release of activating presynaptic A1 adenosine receptors (e.g. Limberger et al., 1988; Allgaier et al., 1991; Bucher et al., 1992). To explain this finding it has been postulated that both receptors use the same second messenger pathway to inhibit neurotransmitter release or, alternatively, use different intracellular pathways which control transmitter release at a common site (e.g. N-type Ca2+ channels). Thus one possible explanation for why α-adrenoceptor blockade is needed to demonstrate the facilitatory effects of the adenosine receptor antagonists, is that activation of presynaptic α2-adrenoceptors by released NA occludes the effects of activating adenosine receptors. In support of this suggestion, Driessen et al. (1994) demonstrated in guinea-pig vas deferens that exogenous adenosine had a more pronounced inhibitory effect on NA and ATP release when the α-adrenoceptors were blocked. However, in the present study, application of adenosine (100 μM) had a similar inhibitory effect on e.j.p. amplitude in the presence and in the absence of phentolamine. The latter finding using bath applied adenosine, which presumably can access all the adenosine receptors in the tissue, does not exclude the possibility that endogenous NA may act at α2-adrenoceptors located close to its site of release and thereby occlude the effects of locally released/generated adenosine.
Another possible explanation for why α-adrenoceptor blockade is required to demonstrate the effects of the adenosine receptor antagonists is that normally both presynaptic α-adrenoceptors and adenosine receptors contribute functionally to the control of transmitter release. In this case, the increase in neurotransmitter release produced by adenosine receptor antagonists may be counteracted by an increase in the level of α-adrenoceptor-mediated autoinhibition leading to only a small (i.e. undetectable) change in neurotransmitter release (Fredholm, 1995).
Although 8-PT and DPCPX both increased e.j.p. amplitude, the two agents were not identical in their effects. 8-PT increased the amplitude of all e.j.ps evoked during the trains of 20 stimuli at 1 Hz, whereas the facilitatory effect of DPCPX was not observed until after the ninth stimulus in the train. The effect of DPCPX on e.j.p. amplitude is reminiscent of that produced by α2-adrenoceptor antagonists (Blakeley et al., 1984; Brock et al., 1990), although the facilitatory effect of these latter agents appeared earlier in the train of stimuli. This stimulus dependent effect of α2-adrenoceptor antagonists has given strong support for the idea that released NA acts at presynaptic α2-adrenoceptors to inhibit subsequent release (α-adrenoceptor mediated autoinhibition; see Brock, 1995). The findings with DPCPX also suggest that adenosine generated from released ATP acts presynaptically to inhibit transmitter release. The delayed onset of the facilitatory action of DPCPX on e.j.p. amplitude presumably reflects, at least in part, the time required to generate an effective concentration of adenosine at the receptors.
At present we cannot explain the differences between the effects of 8-PT and DPCPX on e.j.p. amplitude. Unlike DPCPX which is a relatively selective A1 adenosine receptor antagonist, 8-PT also antagonizes A2 adenosine receptors (see Fredholm et al., 1994). However, only increases in transmitter release from postganglionic sympathetic nerves have been reported following activation of presynaptic A2 adenosine receptors, whereas their blockade reduced transmitter release (e.g. Fuder et al., 1992; Gonçalves & Queiroz, 1996).
A problem often associated with adenosine antagonists is their ability to inhibit phosphodiesterase. It is normally assumed that 8-PT has little or no inhibitory action on phosphodiesterase activity in nerve terminals, although it has been reported to inhibit selectively the high affinity form of phosphodiesterase in some tissues (Nicholson & Wilke, 1989). For this reason we investigated the effects of the potent phosphodiesterase inhibitor, IBMX. Like 8-PT, IBMX increased the amplitude of all e.j.ps evoked during the train of stimuli. However, unlike 8-PT, this effect of IBMX was observed both in the presence and in the absence of phentolamine. As α2-adrenoceptor mediated inhibition of transmitter release from postganglionic sympathetic nerves is unlikely to involve regulation of cyclic AMP levels (Majewski & Barrington, 1995), disruption of α2-adrenoceptor mediated autoinhibition of transmitter release cannot explain the facilitatory action of IBMX in the absence of phentolamine. In addition to increasing e.j.p. amplitude, IBMX produced a significant decrease in the e.j.p. time constant of decay, which indicates that this agent acted postjunctionally to reduce the membrane time constant (see Cassell et al., 1988). This effect on e.j.p. time course was not observed with 8-PT. These findings indicate that phosphodiesterase inhibition is unlikely to account for the facilitatory action of 8-PT. A potential problem which complicates this conclusion is that IBMX is itself a weak A1-adenosine receptor antagonist (Kenakin & Beek, 1987). However, its actions on e.j.p. amplitude are different from those of the selective A1-adenosine receptor antagonist, DPCPX, which has no known inhibitory action on phosphodiesterase.
The stimulus dependent effect of DPCPX suggests that the main source of extracellular adenosine acting at presynaptic A1-adenosine receptors is the breakdown of neurally released ATP by ectonucleotidases. Adenosine is also released under basal conditions from most cell types through bi-directional membrane transporters (see Fredholm, 1995), its concentration in the extracellular space being in equilibrium with that in the cytosol of the surrounding cells. This leakage cannot be the main source of adenosine activating the presynaptic A1 adenosine receptors in the present study, as its effect at these receptors should be stimulus independent. Another potential stimulus-dependent source of adenosine is the smooth muscle, which releases ATP when contracted by neurotransmitters (von Kugelgen & Starke, 1991; Vizi et al., 1992). This ATP would also be broken down to adenosine by ectonucleotidases. However, this source of adenosine is unlikely to be important in the present study, as the stimulation parameters used did not cause a detectable contraction of the vas deferens.
Application of the adenosine uptake inhibitor, NBTI, which acts at the same bi-directional adenosine transporter as dipyramidole (see Thorn & Jarvis, 1996), had no significant effects on e.j.p. amplitude in the presence of phentolamine. This finding is in agreement with Sneddon et al. (1984) who reported that dipyramidole had no significant effect on e.j.p. amplitudes in guinea-pig vas deferens. In contrast, Kirkpatrick & Burnstock (1992) reported that the electrically evoked overflow of ATP from the guinea-pig vas deferens was inhibited by dipyramidole, a finding consistent with an increased presynaptic inhibitory action of extracellular adenosine. This difference can be explained if blockade of adenosine uptake has a more pronounced effect during the long trains of relatively high frequency stimuli (8 Hz for 1 min) used in the study of Kirkpatrick & Burnstock (1992).
In conclusion, this study has examined the effects of adenosine antagonists on neurotransmitter release from postganglionic sympathetic nerves in the guinea-pig vas deferens. The results suggest that, during short trains of stimuli, endogenous adenosine generated from neurally released ATP can inhibit neurotransmitter release through activation of A1 adenosine receptors when α2-adrenoceptors are blocked. The need to block α2-adrenoceptors to reveal the inhibitory effects of endogenous adenosine questions its physiological role in the regulation of neurotransmitter release in guinea-pig vas deferens. However, it remains possible that during periods of intense nerve activity that adenosine generated from neurally released ATP and/or leakage from the smooth muscle reaches extracellular concentrations which act like exogenously applied adenosine.
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council. We thank Elspeth McLachlan for her comments on the manuscript.
Abbreviations
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- e.j.p.
excitatory junction potential
- IBMX
3-isobutyl-1-methylxanthine
- NBTI
S-(p-nitrobenzyl)-6-thioinosine
- 8-PT
8-phenyltheophylline
- r.m.p.
resting membrane potential
References
- ALLGAIER C., GREBER R., HERTTING G. Studies on the interaction between pre-synaptic α2-adrenoceptors and adenosine A1 receptors located on noradrenergic nerve terminals. N.-S. Arch. Pharmacol. 1991;344:187–192. doi: 10.1007/BF00167217. [DOI] [PubMed] [Google Scholar]
- BLAKELEY A.G., CUNNANE T.C., MASKELL T., MATHIE A., PETERSEN S.A. Alpha-adrenoceptors and facilitation at a sympathetic neuroeffector. J. Auton. Pharmacol. 1984;4:53–58. doi: 10.1111/j.1474-8673.1984.tb00433.x. [DOI] [PubMed] [Google Scholar]
- BLAKELEY A.G., CUNNANE T.C., PETERSEN S.A. An electropharmacological analysis of the effects of some drugs on neuromuscular transmission in the vas deferens of the guinea-pig. J. Auton. Pharmacol. 1981;1:367–375. doi: 10.1111/j.1474-8673.1981.tb00075.x. [DOI] [PubMed] [Google Scholar]
- BLAKELEY A.G., DUNN P.M., PETERSEN S.A. A study of the actions of P1-purinoceptor agonists and antagonists in the mouse vas deferens in vitro. Br. J. Pharmacol. 1988;94:37–46. doi: 10.1111/j.1476-5381.1988.tb11497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROCK J.A.Modulation of neurotransmitter release by autoreceptors Neurotransmitter release and its modulations 1995Cambridge University Press: Cambridge; 81–103.(ed.) Powis, D.A. & Bunn, S.J. pp [Google Scholar]
- BROCK J.A., CUNNANE T.C. Neurotransmitter release mechanisms at the sympathetic neuroeffector junction. Exp. Physiol. 1993;78:591–614. doi: 10.1113/expphysiol.1993.sp003709. [DOI] [PubMed] [Google Scholar]
- BROCK J.A., CUNNANE T.C., STARKE K., WARDELL C.F. α2-Adrenoceptor-mediated autoinhibition of sympathetic transmitter release in guinea-pig vas deferens studied by intracellular and focal extracellular recording of junction potentials and currents. N.-S. Arch. Pharmacol. 1990;342:45–52. doi: 10.1007/BF00178971. [DOI] [PubMed] [Google Scholar]
- BUCHER B., CORRIU C., STOCLET J.-C. Pre-junctional opioid μ-receptors and adenosine A1-receptors on the sympathetic nerve endings of the rat tail artery interact with the α2-adrenoceptors. N.-S. Arch. Pharmacol. 1992;345:37–43. doi: 10.1007/BF00175467. [DOI] [PubMed] [Google Scholar]
- CASSELL J.F., MCLACHLAN E.M., SITTIRACHA T. The effect of temperature on neuromuscular transmission in the main caudal artery of the rat. J. Physiol. 1988;397:31–49. doi: 10.1113/jphysiol.1988.sp016986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALZIEL H.H., SNEDDON P. The mechanism of action of AMP-induced inhibition of sympathetic neurotransmission in the isolated vas deferens of the rat and guinea-pig. Br. J. Pharmacol. 1988;94:961–967. doi: 10.1111/j.1476-5381.1988.tb11610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRIESSEN B., VON KUGELGEN I., STARKE K. P1-purinoceptor-mediated modulation of neural noradrenaline and ATP release in guinea-pig vas deferens. N.-S. Arch. Pharmacol. 1994;350:42–48. doi: 10.1007/BF00180009. [DOI] [PubMed] [Google Scholar]
- FREDHOLM B.B.Modulation of neurotransmitter release by heteroceptors Neurotransmitter release and its modulation 1995Cambridge: Cambridge University Press; 104–121.(ed) Powis, D.A. & Bunn, S.J. pp [Google Scholar]
- FREDHOLM B.B., ABBRACCHIO M.P., BURNSTOCK G., DALY J.W., HARDEN T.K., JACOBSON K.A., LEFF P., WILLIAMS M. Nomenclature and classification of purinoceptors. Pharmacol. Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- FUDER H., BRINK A., MEINCKE M., TAUBER U. Purinoceptor-mediated modulation by endogenous and exogenous agonists of stimulation-evoked [3H]noradrenaline release in rat iris. N.-S. Arch. Pharmacol. 1992;345:417–423. doi: 10.1007/BF00176619. [DOI] [PubMed] [Google Scholar]
- FUNK L., TRENDELENBERG A.-U., LIMBERGER N., STARKE K. Subclassification of presynaptic α2-adrenoceptors: α2D-autoreceptors and α2D-adrenoceptors modulating release of acetylcholine in guinea-pig ileum. N.-S. Arch. Pharmacol. 1995;352:58–66. doi: 10.1007/BF00169190. [DOI] [PubMed] [Google Scholar]
- GONÇALVES J., QUEIROZ G. Facilitatory and inhibitory modulation by endogenous adenosine of noradrenaline release in the epididymal portion of rat vas deferens. N.-S. Arch. Pharmacol. 1993;348:367–371. doi: 10.1007/BF00171335. [DOI] [PubMed] [Google Scholar]
- GONÇALVES J., QUEIROZ G. Purinoceptor modulation of noradrenaline release in rat tail artery: tonic modulation mediated by inhibitory P2Y- and facilitatory A2A-purinoceptors. Br. J. Pharmacol. 1996;117:156–160. doi: 10.1111/j.1476-5381.1996.tb15168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEDQVIST P., FREDHOLM B.B. Effects of adenosine on adrenergic neurotransmission: prejunctional inhibition and postjunctional enhancement. N.-S. Arch. Pharmacol. 1976;293:217–223. doi: 10.1007/BF00507344. [DOI] [PubMed] [Google Scholar]
- KENAKIN T.P., BEEK D. Measurement of antagonist affinity for purine receptors of drugs producing concomitant phosphodiesterase blockade: the use of pharmacologic resultant analysis. J. Pharmacol. Exp. Ther. 1987;243:482–486. [PubMed] [Google Scholar]
- KIRKPATRICK K.A., BURNSTOCK G. Evidence that the inhibition of ATP release from sympathetic nerves by adenosine is a physiological mechanism. Gen. Pharmacol. 1992;23:1045–1050. doi: 10.1016/0306-3623(92)90284-q. [DOI] [PubMed] [Google Scholar]
- KURZ K., VON KUGELGEN I., STARKE K. Prejunctional modulation of noradrenaline release in mouse and rat vas deferens: contribution of P1- and P2-purinoceptors. Br. J. Pharmacol. 1993;110:1465–1472. doi: 10.1111/j.1476-5381.1993.tb13986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANIER S.M., DOWNING S., DUZIC E., HOMEY C.J. Isolation of rat genomic clones encoding subtypes of the α2-adrenergic receptors. J. Biol. Chem. 1991;266:10470–10478. [PubMed] [Google Scholar]
- LIMBERGER N., SPÄTH L., STARKE K. Pre-synaptic α2-adrenoceptor, opioid k-receptor and adenosine A1-receptor interactions on noradrenaline release in rabbit brain cortex. N.-S. Arch. Pharmacol. 1988;338:53–61. doi: 10.1007/BF00168812. [DOI] [PubMed] [Google Scholar]
- MAJEWSKI H., BARRINGTON M.Second messenger pathways in the modulation of neurotransmitter release Neurotransmitter release and its modulation 1995Cambridge: Cambridge University Press; 163–181.(ed) Powis, D.A. & Bunn, S.J. pp [DOI] [PubMed] [Google Scholar]
- MUNCH G., KURZ T., URLBAUER T., SEYFARTH M., RICHARDT G. Differential presynaptic modulation of noradrenaline release in human atrial tissue in normoxia and anoxia. Br. J. Pharmacol. 1996;118:1855–1861. doi: 10.1111/j.1476-5381.1996.tb15614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLSON C.D., WILKE R. 8-Phenyltheophylline as an inhibitor of cyclic AMP hydrolysis by cyclic nucleotide phosphodiesterase. J. Auton. Pharmacol. 1989;9:159–165. doi: 10.1111/j.1474-8673.1989.tb00207.x. [DOI] [PubMed] [Google Scholar]
- SNEDDON P. Suramin inhibits excitatory junction potentials in guinea-pig vas deferens. Br. J. Pharmacol. 1992;222:171–174. doi: 10.1111/j.1476-5381.1992.tb14469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SNEDDON P., MELDRUM L.A., BURNSTOCK G. Control of transmitter release in guinea-pig vas deferens by pre-junctional P1-purinoceptors. Eur. J. Pharmacol. 1984;105:293–299. doi: 10.1016/0014-2999(84)90621-6. [DOI] [PubMed] [Google Scholar]
- THORN J.A., JARVIS S.M. Adenosine transporters. Gen. Pharmacol. 1996;27:613–620. doi: 10.1016/0306-3623(95)02053-5. [DOI] [PubMed] [Google Scholar]
- TRENDELENBURG A.-U., LIMBERGER N., STARKE K. Subclassification of presynaptic α1-adrenoceptors: α2D-autoreceptors in guinea pig atria and brain. N.-S. Arch. Pharmacol. 1995;352:49–57. doi: 10.1007/BF00169189. [DOI] [PubMed] [Google Scholar]
- VIZI E.S., SPERLAGH B., BARANYI M. Evidence that ATP, released from the postsynaptic site by noradrenaline, is involved in mechanical responses of guinea-pig vas deferens: cascade transmission. Neurosci. 1992;50:455–465. doi: 10.1016/0306-4522(92)90437-7. [DOI] [PubMed] [Google Scholar]
- VON KUGELGEN I., STARKE K. Noradrenaline-ATP co-transmission in the sympathetic nervous system. T.I.P.S. 1991;12:319–324. doi: 10.1016/0165-6147(91)90587-i. [DOI] [PubMed] [Google Scholar]