

Abstract
RSD 921 is a novel, structurally unique, class I Na+ channel blocking drug under development as a local anaesthetic agent and possibly for the treatment of cardiac arrhythmias. The effects of RSD 921 on wild-type heart, skeletal muscle, neuronal and non-inactivating IFMQ3 mutant neuronal Na+ channels expressed in Xenopus laevis oocytes were examined using a two-electrode voltage clamp.
RSD 921 produced similarly potent tonic block of all three wild-type channel isoforms, with EC50 values between 35 and 47 μM, whereas the EC50 for block of the IFMQ3 mutant channel was 110±5.5 μM.
Block of Na+ channels by RSD 921 was concentration and use-dependent, with marked frequency-dependent block of heart channels and mild frequency-dependent block of skeletal muscle, wild-type neuronal and IFMQ3 mutant channels.
RSD 921 produced a minimal hyperpolarizing shift in the steady-state voltage-dependence of inactivation of all three wild-type channel isoforms.
Open channel block of the IFMQ3 mutant channel was best fit with a first order blocking scheme with kon equal to 0.11±0.012×106 M−1 s−1 and koff equal to 12.5±2.5 s−1, resulting in KD of 117±31 μM. Recovery from open channel block occurred with a time constant of 14±2.7 s−1.
These results suggest that RSD 921 preferentially interacts with the open state of the Na+ channel, and that the drug may produce potent local anaesthetic or anti-arrhythmic action under conditions of shortened action potentials, such as during anoxia or ischaemia.
Keywords: Arylbenzacetamide, RSD 921, class I anti-arrhythmic, Xenopus oocytes, Na+ channel
Introduction
Voltage-gated Na+ channels are responsible for the initial rapid membrane depolarization during the early phase of an action potential in electrically excitable cells. The propagating action potential behaves as an electrical excitation wave that results in contraction of the heart, or proceeds down nerve axons and results in contraction of striated muscle or synaptic transmission. The diverse functions associated with Na+ channels within various tissues and the differences in pharmacological properties of Na+ currents have provided evidence for distinct channel subtypes. Molecular biological studies have demonstrated the existence of at least eight unique but homologous mammalian Na+ channel α subunits encoded by distinct genes (reviewed by Goldin, 1995). The α subunit is the primary, pore-forming subunit of the channel, and is composed of four homologous domains (I-IV), each of which is composed of six α-helical transmembrane segments (S1-S6). Although the α subunit forms the pore, functional properties of Na+ channels are modified by accessory β subunits when expressed in Xenopus oocytes (Isom et al., 1992; 1995; Smith & Goldin, 1998).
The Na+ channel can exist in resting (closed), open (active) and inactive (non-conducting) states, and the transitions between those states are both voltage and time-dependent (Hodgkin & Huxley, 1952). Local anaesthetic and anti-arrhythmic drugs have been shown to interact differentially with each of the states of the channel (reviewed by Strichartz et al., 1987). Hille (1977) proposed a modulated receptor hypothesis for the state-dependent interaction of local anaesthetics with the neuronal Na+ channel. Concurrently, Hondeghem & Katzung (1977) proposed a similar model for the interaction of anti-arrhythmic drugs with the cardiac Na+ channel. In general, both models suggest that most clinically useful local anaesthetic and anti-arrhythmic drugs have a low affinity for the resting state of the Na+ channel and that the affinity of the drug is greatest for the open and inactive states of the channel. This specificity is of fundamental importance for the drug treatment of cardiac arrhythmias.
Prior to the development of molecular biological techniques, it was difficult to analyse the relationship between drug block and the state-dependence of Na+ channel block. Agents that were used to remove the fast component of inactivation, such as toxins (e.g. veratridine) or enzymes (e.g., trypsin) were not specific in their actions and resulted in damage to the channel protein (Armstrong et al., 1973). The lack of specificity of action of these agents made it difficult to determine if drug block was dependent on channel inactivation. The identification by West et al. (1992) that glutamine (Q) replacement of the I1488, F1489 and M1490 amino acids in domain III-IV of the neuronal rBIIA Na+ channel (the IFMQ3 mutation) resulted in the loss of fast inactivation provided a specific means to examine the relationship between drug block and the state-dependence of channel block.
RSD 921 (+)-trans-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzo[b]thiophene-4-acetamide (Figure 1) is a novel class I antiarrhythmic drug currently under development as a potent Na+ channel blocking agent for use as a local anaesthetic agent (Beatch et al., 1997) and may possibly be used in the treatment of cardiac arrhythmias. RSD 921 is the (+)-enantiomer of a κ opioid receptor agonist compound and is an aryl derivative of benzacetamide (Clark et al., 1988a,1988b). Walker et al. (1996) recently developed a sensitive method to quantify the levels of RSD 921 in rat whole blood as well as cardiac and neuronal tissue. Their studies showed that tissues such as the heart rapidly absorb RSD 921. The use of RSD 921 as a potential therapeutic agent was recently examined in an open label, ascending dose, phase I clinical trial. This study was used to evaluate the safety of RSD 921 in healthy human volunteers. RSD 921 was shown to be well-tolerated in humans, had a half-life of approximately 10 h in man, and showed no evidence of drug-related adverse effects over the dose-range that was tested (Bain et al., 1997; G.N. Beatch, Nortran Pharmaceuticals Ltd., personal communication).
Figure 1.
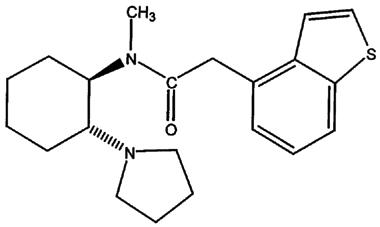
Diagram of the molecular structure of RSD 921.
In this study, we used the Xenopus oocyte expression system to characterize the electrophysiological actions of RSD 921 on wild-type rat heart (rH1), skeletal muscle (rSkM1), and neuronal (rBIIA) Na+ channels. RSD 921 blocked all three Na+ channel isoforms in a tonic and frequency-dependent manner. The effects of RSD 921 on the non-inactivating IFMQ3 mutant of the neuronal Na+ channel suggest that the drug preferentially interacts with the open state of the channel.
Methods
Transcription of RNA and expression of Xenopus oocytes
The plasmid pSkM2 contains the coding region for the rat heart (rH1) Na+ channel α subunit (Kallen et al., 1990), μ1 contains the coding region for the skeletal muscle (rSkM1) α subunit (Trimmer et al., 1989), and pVA2580 contains the coding region for the rat neuronal IIA (rBIIA) Na+ channel α subunit (Auld et al., 1990). The pSkM2 plasmid was generously provided by Dr Roland Kallen (University of Pennsylvania) and the μ1 plasmid was generously provided by Dr Gail Mandel (SUNY Stony Brook). The fast inactivation-deficient IFMQ3 mutant of the rBIIA Na+ channel contains substitutions of glutamine (Q) for I1488, F1489 and M1490 in the III-IV linker of the channel (West et al., 1992).
Plasmid DNA was linearized by digestion with AseI (rH1) or NotI (rSkM1 and rBIIA), and RNA transcripts were synthesized using the message machine SP6 (rH1) or T7 (rSkM1 and rBIIA) RNA polymerase transcription kit (Ambion, Austin, TX, U.S.A.). Stage V oocytes were obtained from adult female Xenopus laevis frogs, defolliculated with collagenase (2 mg ml−1) and injected with 50 nl of in vitro transcribed RNA at a concentration empirically determined to obtain current amplitudes between 1–5 μA, as previously described (Goldin & Sumikawa, 1992). The RNAs for the rSkM1, rBIIA and fast-inactivation deficient IFMQ3 mutant channels were co-injected with RNA encoding the β1 subunit (Isom et al., 1992) transcribed in vitro from a plasmid encoding the rat β1 subunit (Smith & Goldin, 1998). The oocytes were incubated for 48 h at 20°C in ND96 (in mM): NaCl 96, KCl 2, CaCl2 1.8 and HEPES 5, pH 7.4 supplemented with gentamicin 0.1 mg ml−1, theophylline 0.5 mM and pyruvate 0.55 mg ml−1. All experiments were performed according to guidelines established by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Solutions and drugs
All oocyte experiments were performed at room temperature (20–22°C) in ND96 without supplements. RSD 921, (+)-trans - N -methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzo[b]thiophene-4-acetamide monohydrochloride (a gift from Dr M.J.A. Walker of Nortran Pharmaceuticals Ltd., Vancouver, BC, Canada), was solubilized in distilled water as a 10 mM stock solution prior to dilution to the final concentrations in the ND96 bath solution. RSD 921 was used at concentrations ranging from 1–1000 μM. A low volume (0.5 ml) plexiglass recording bath allowed for efficient exchange (15–30 s) between control and drug solutions from gravity-flow reservoirs. A suction device ensured continuous perfusion at a flow rate of 1–2 ml min−1 and maintained a constant fluid level.
Data recording and analysis
Recording electrodes were prepared from borosilicate glass using a two-stage P-87 puller (Sutter Instrument Co. Novato, CA, U.S.A.). Microelectrodes were filled with filtered 3 M KCl/0.5% low melting point agarose and had resistances between 0.5–4.0 MΩ. A grounded copper shield was inserted between the recording electrodes to minimize electrode coupling. Currents were recorded using a virtual ground circuit, and the data were filtered at 3 kHz on-line and digitized at a sampling frequency of 12.5 kHz. Currents were recorded and analysed using pCLAMP 6.0.4 software (Axon Instruments, Foster City, CA, U.S.A.). Capacitance transients and leak currents were corrected by P/4 subtraction, with the depolarizations for subtraction applied after each protocol. Non-linear curve fitting was performed using SigmaPlot® (version 4.0, Jandel Scientific, San Rafael, CA, U.S.A.). Data are shown as the mean±standard deviation for n experiments. Statistical analyses were performed using SigmaStat® statistical software (Jandel Scientific), with P less than 0.05 considered statistically significant.
Cumulative concentration-response curves for RSD 921 were determined by measuring peak inward current for cells depolarized from −100 mV (for rSkM1, rBIIA and IFMQ3) or −120 mV (for rH1) to −10 mV in the absence and presence of RSD 921 (1–1000 μM). The more negative holding potential (−120 mV) was necessary for recording from the rH1 channel to allow for complete recovery from slow inactivation (Pugsley & Goldin, 1998). Currents were allowed to recover from slow inactivation for 10 min before beginning any electrophysiological protocol. RSD 921 was then perfused for 5 min into the bath before recording current. The resulting fractional block of Na+ current by RSD 921 at each concentration examined was plotted against the log concentration of drug, and fitted with the Hill equation, INa=[1+([A]/EC50)nH]−1. This equation describes a first order blocking scheme for the drug-channel interaction, in which INa is the fractional block of Na+ current, [A] is the concentration of RSD 921, EC50 is concentration of RSD 921 that produced half-maximal block of the Na+ current and ‘nH' is the Hill coefficient describing the stoichiometry of drug binding to the channel.
The voltage-dependence of Na+ channel conductance (G) was calculated by measuring the peak current during 12 ms depolarizations ranging from −90–+50 mV in 10 mV increments every 6 s and dividing by (V-Vrev), in which V is the test potential and Vrev is the reversal potential for Na+. Reversal potentials were determined by individually fitting current-voltage data as described previously (Kontis & Goldin, 1993). Conductance values were obtained in the absence and presence of RSD 921 (100 μM), which was perfused onto the oocyte for 5 min before determining conductance. Peak conductance values were fit with a two-state Boltzmann equation of the form G=1*[1+exp(−0.03937*zapp*(V-V½½)], in which V is the potential of the voltage pulse, V½ is the half-maximal voltage for activation, and zapp is the apparent gating charge.
The frequency-dependent effects of RSD 921 were examined from a holding potential of −100 mV (−120 mV for rH1) with 35 depolarizations from −100 to −10 mV for 24.8 ms each. Trains of pulses were delivered at frequencies of 1, 5 and 30 Hz in the absence and presence of RSD 921 (10 and 100 μM). Current amplitude during each pulse was normalized to the peak maximal current (pulse number 1) and plotted as a function of pulse number.
The voltage-dependence of Na+ channel inactivation was determined using 500 ms conditioning pre-pulses at 15 s in-tervals from a holding potential of −100 mV (−120 mV for rH1) to +15 mV in 5 mV increments, followed by a test pulse to −5 mV for 22.5 ms. The peak current amplitude evoked during the test depolarization was normalized to the maximum current amplitude, and plotted as a function of the conditioning pre-pulse potential in the absence and presence of RSD 921 (100 μM). The data were fit with a two-state Boltzmann equation of the form I-Imax*[1+(exp(V–V½)/k)]−1, in which Imax is the maximal current evoked, V is the potential of the voltage pulse, V½ is the voltage at which 50% of the current is inactivated (the midpoint of the inactivation curve), and k is the slope factor.
The kinetics of RSD 921 block of IFMQ3, the fast-inactivation deficient mutant of the neuronal channel, were examined by clamping oocytes at −100 mV in ND-96 bath solution. A single 1000 ms depolarizing plus to +20 mV was applied and Na+ current recorded. RSD 921 (at 10, 30, 70 or 100 μM) was perfused into the bath for 5 min and a second, single depolarizing pulse from −100 to +20 mV was given. The data were individually fit to either a single (1-Aslow*exp(−t/τslow)) or double (1-[(Aslow*exp(−t/τslow)+Afast *exp(−t/τfast)]) exponential equation, in which Afast and Aslow represent the proportion of current decaying with time constants τfast and τslow, and t is the time interval. If we assume a first order blocking scheme to describe the dependence of the blocking rate on the concentration of the blocking drug, as with the Hill equation in Figure 2, then the apparent rate constants for binding (kon) and unbinding (koff) can be obtained by fitting the τfast values with the equation: 1/τfast=kon*[RSD 921]+koff (Lansman et al., 1986; Valenzuela et al., 1996). These values can then be used to calculate an apparent affinity constant, Kd, such that Kd=koff/kon (Valenzuela et al., 1996).
Figure 2.
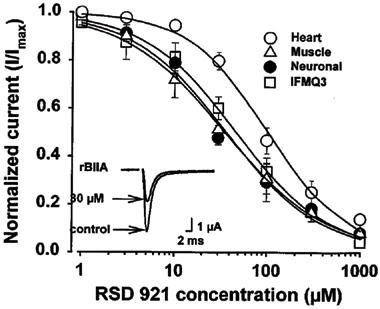
Concentration-response curves for the effects of RSD 921 on heart (○), skeletal muscle (▵), neuronal (•), and mutant IFMQ3 (□) Na+ channels. Oocytes were injected with Na+ channel RNA and currents were recorded in ND96 using two-electrode voltage clamp, as described in the Methods. Peak Na+ currents, evoked every 6 s, were measured at test potentials that elicited maximum inward current (−10 mV). Peak Na+ currents were measured again after 5 min perfusion of the cell at a flow rate of 1–2 ml min−1 of ND96 containing increasing concentrations of RSD 921. Data points represent the means of at least six individual oocytes, and error bars represent standard deviations. The curves described by the solid lines were fit by the Hill equation as described in the Methods. The inset shows a current trace for the neuronal Na+ channel during a depolarization to −10 mV in the absence and presence of 30 μM RSD 921.
Recovery from open channel block was measured from a holding potential of −100 mV with a 1000 ms depolarizing pre-pulse to −10 mV (P1) followed by a variable recovery period from 240 s to 200 ms, which in turn was followed by a 1000 ms test pulse to −10 mV (P2). When recovery was examined in control oocytes, the measured currents were similar but not exactly identical. Thus, to remove the confounding effect of incomplete channel recovery from inactivation in control oocytes in the absence of RSD 921, we subtracted out the current amplitude difference at each recovery time interval (Δt) in controls from that of recovery in the presence of 100 μM RSD 921. This analysis is based on the assumption that the extent of slow inactivation was comparable in the absence or presence of RSD 921. The resulting corrected fractional peak current recovery amplitude during the test pulse (P2) was normalized to P1 and plotted as a function of the duration of the recovery time interval (Δt). The data were individually fit to a single exponential equation (I=1-a*exp(−t/τ)), in which a represents the proportion of channels recovering from open channel block with a time constant τ, and t is the recovery time interval.
Results
RSD 921 effects on wild-type and mutant IFMQ3 Na+ currents
RSD 921 is a novel derivative of the arylacetamide compound U-50,488H, and it has previously been shown that U-50,488H reduces the incidence of cardiac arrhythmias by blockade of cardiac Na+ channels (Pugsley et al., 1992a,1992b). Therefore, it seemed likely that RSD 921 would also affect Na+ channel function. To determine if this were the case, we examined the effects of RSD 921 on the electrophysiological properties of wild-type rat heart, skeletal muscle and neuronal Na+ channels and a mutant of the neuronal channel (IFMQ3) in which fast inactivation had been removed. All of the channels were expressed in Xenopus oocytes and analysed using a two-electrode voltage-clamp. Oocytes were held at −100 mV for skeletal muscle, neuronal and IFMQ3 channels and −120 mV for heart channels, and currents were evoked by depolarizations to −10 mV every 6 s. This infrequent pulsing protocol may reveal drug interactions with the resting or open state of the Na+ channel and minimize the effects of frequency dependent block, therefore providing a reasonable estimation of the extent to which RSD 921 produces tonic block of Na+ current.
Figure 2 shows the concentration-response curves for RSD 921 on the wild-type and mutant Na+ currents. The smooth lines represent the best fits of the data using the Hill equation, with the parameters of the fits shown in Table 1. The stoichiometries of RSD 921 binding to the Na+ channels were not significantly different from 1 for any of the channels (nH in Table 1), suggesting that only one drug molecule is necessary to block the channel. Unlike many class I anti-arrhythmic drugs examined in the oocyte expression system, RSD 921 demonstrated potent block of all three wild-type Na+ channel isoforms. When EC50 values were compared, RSD 921 blocked heart, skeletal muscle and neuronal Na+ channels with similar potency (Table 1). However, the EC50 for block of the IFMQ3 channel was 2.7 fold less than for the neuronal channel (Table 1). RSD 921 block of sodium channels was reversible (data not shown). These data suggest that RSD 921 is a potent Na+ channel blocker and that the effect of RSD 921 on Na+ currents does not require fast inactivation of the channel, although the drug is less potent on the IFMQ3 mutant that lacks fast inactivation.
Table 1.
Inhibition of heart, skeletal muscle and neuronal Na+ channels by RSD 921

RSD 921 did not affect the voltage-dependence of Na+ channel activation
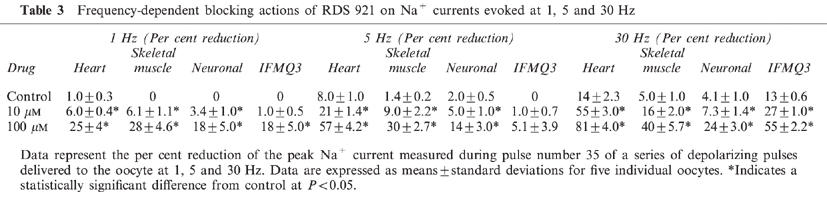
To determine if RSD 921 blocked Na+ channel conductance in a voltage-dependent manner, the effects of 100 μM RSD 921 were examined on the voltage-dependence of heart, skeletal muscle and neuronal Na+ channel activation (Figure 3). The concentration of RSD 921 used was chosen to block 75% of Na+ channel current (EC75), which would ensure that a sufficient fraction of channels would be blocked to reveal any effects on conductance. When the data for conductance were examined in the presence of drug and compared to the control data, no significant changes in either V½ or slope factor (zapp) were observed (Figure 3 and Table 2). Therefore, RSD 921 did not significantly alter the voltage-dependence of activation for any of the Na+ channel isoforms.
Figure 3.
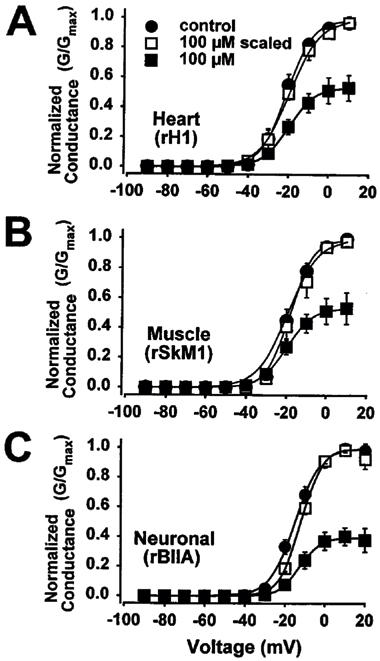
Effects of 100 μM RSD 921 on the voltage-dependence of conductance of heart, skeletal muscle and neuronal Na+ channels. Conductance was examined by 12 ms depolarizations from −100 mV (−120 mV for the heart channel) to potentials ranging from −90 to +50 mV in 10 mV increments every 6 s and dividing by (V-Vrev), in which V is the test potential and Vrev is the reversal potential for Na+. The reversal potentials were determined as described in the Methods. The data points in all panels were determined from at least five oocytes and error bars represent standard deviations. All curves were fit with a two-state Boltzmann function as described in the Methods, with the parameters of the fits in Table 2. Conductance curves are shown in the absence and presence of 100 μM RSD 921. The data for 100 μM RSD 921 are scaled to the maximum to demonstrate the shifts in the voltage-dependence of conductance.
Table 2.
Effect of RSD 921 on Na+ channel conductance

RSD 921 produced use-dependent block of wild-type and IFMQ3 Na+ channels
Differential block of Na+ channels in closed, open and inactive states of anti-arrhythmic drugs can result in use-dependent block, which is an important parameter to assess efficacy against arrhythmias characterized by rapidly firing action potentials. Therefore, we examined use-dependent block by RSD 921 of the three wild-type isoforms and IFMQ3 Na+ channels. The IFMQ3 mutation specifically removes fast inactivation (West et al., 1992), making it possible to determine if the inactive state is necessary for RSD 921 induced channel block. Steady-state tonic block of Na+ current was achieved using single depolarizing current pulses delivered every 6 s in the absence and presence of RSD 921. Two concentrations of RSD 921 were utilized to obtain minimal current block (EC10) and marked current block (EC75). Following steady-state drug block (tonic block), trains of depolarizing pulses were delivered to assess the extent of phasic block at different frequencies. The results are shown in Figure 4 for RSD 921 examined at a pulse frequency of 30 Hz. The per cent of current block during the 35th pulse is shown at frequencies of 1, 5 and 30 Hz in Table 3.
Figure 4.
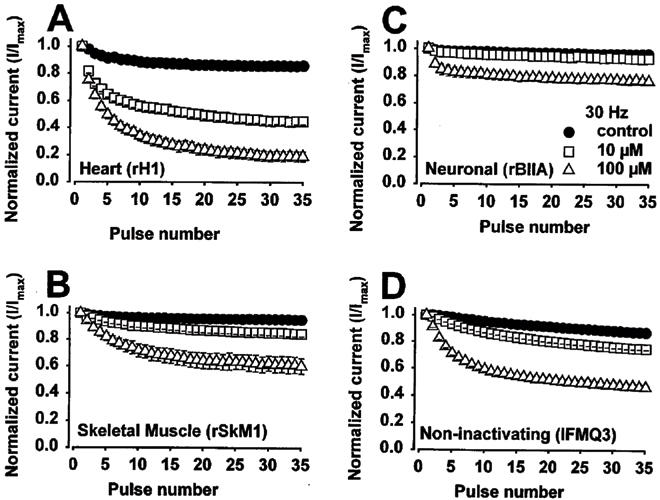
Effects of RSD 921 on the use-dependent block of heart, skeletal muscle, neuronal and IFMQ3 Na+ channels in the absence or presence of 10 or 100 μM RSD 921. A series of depolarizing pulses of 20 ms duration to −10 mV were applied from a holding potential of −100 mV at 30 Hz. The peak currents were normalized to the current during the first depolarization, and plotted as a function of pulse number. The peak currents measured in the presence of the drug were normalized to the current during the first depolarization in the presence of the drug. Those currents measured ∼80% for the low RSD 921 concentration and ∼30% for the high RSD 921 concentration compared to current amplitudes in the absence of RSD 921. The normalization emphasizes the amount of use-dependent block that developed independently of the tonic block by RSD 921.
Table 3.
Frequency-dependent blocking actions of RDS 921 on Na+ currents evoked at 1, 5 and 30 Hz
RSD 921 blocked the heart channel in a use-dependent manner, so that 81% of the current was blocked by 100 μM RSD 921 at 30 Hz (Figure 4A). RSD 921 moderately blocked (40%) the skeletal muscle and minimally blocked (24%) the neuronal channels in a use-dependent manner (Figure 4B and C). Figure 4D shows use-dependent block of the mutant IFMQ3 channel. During the 1 and 5 Hz series of depolarizing pulses, significant percentages of all three wild-type Na+ channel isoforms were blocked by RSD 921 at the low and high concentrations examined. However, the fast-inactivation deficient mutant IFMQ3 channel showed no significant level of use-dependent block at 1 and 5 Hz at the same drug concentrations (Table 3). Rather, only at a very high rate of channel stimulation (30 Hz) was a frequency-dependent component of block observed. Thus, under identical pulse conditions, the heart Na+ channel was more sensitive to block by RSD 921 than was the skeletal muscle or neuronal Na+ channel. The use-dependent block observed at 30 Hz with the IFMQ3 mutant suggests that the very short recovery interval (∼8 ms) associated with this high rate of channel stimulation did not provide a sufficient amount of time for RSD 921 to completely unbind from the channel, so that drug block accumulated with each successive pulse.
RSD 921 did not affect the voltage-dependence of inactivation
Many anti-arrhythmic drugs such as lidocaine produce use-dependent block from an interaction of the drug with both open and inactive states of the Na+ channel (Bean et al., 1983; Sanchez-Chapula et al., 1983; Bennett et al., 1995). Drugs that preferentially bind to the inactive state of the channel generally shift the voltage-dependence of inactivation in the negative direction (Bean et al., 1983). To determine which state of the Na+ channel (open or inactive) was blocked by RSD 921, we characterized the effects of the drug on the voltage-dependence of inactivation. Inactivation was examined using a two-pulse protocol with a 500 ms inactivation pre-pulse to ensure that all of the channels were inactivated. A concentration of RSD 921 (100 μM) was utilized to obtain marked current block. The effects of 100 μM RSD 921 on the heart (A), skeletal muscle (B) and neuronal (C) channels are shown in Figure 5. The smooth curves represent the best fits to the data using a two state Boltzmann function, with the parameters of the fits shown in Table 4.
Figure 5.
Effects of 100 μM RSD 921 on the voltage-dependence of inactivation of heart, skeletal muscle and neuronal Na+ channels. Inactivation was examined using a two-pulse protocol in which oocytes were held at −100 mV (−120 mV for the heart channel) and depolarized every 15 s from −90 to +15 mV for a duration of 500 ms, followed by a test-pulse to −5 mV for 22.5 ms to determine channel availability. The data points in all panels were determined from at least five oocytes and error bars represent standard deviations. All curves were fit with a two-state Boltzmann function as described in the Methods. Inactivation curves are shown in the absence and presence of 100 μM RSD 921. The data for 100 μM RSD 921 are scaled to the maximum to demonstrate the shifts in the voltage-dependence of inactivation.
Table 4.
Effects of RSD 921 on the voltage-dependence of inactivation

RSD 921 did not significantly affect the voltage-dependence of inactivation for any of the Na+ channel isoforms, even at high blocking concentrations (Table 4). At a concentration of RSD 921 (1000 μM) which resulted in block of >85% of Na+ channels, the shift in V½ was −6 mV for the skeletal muscle channel, −5 mV for the neuronal channel, and −3 mV for the heart channel (data not shown). In addition, RSD 921 did not significantly change the slope factor for any of the channel isoforms. These results suggest that RSD 921 does not preferentially bind to the inactive state of the channel.
Rate of development of open channel block by RSD 921
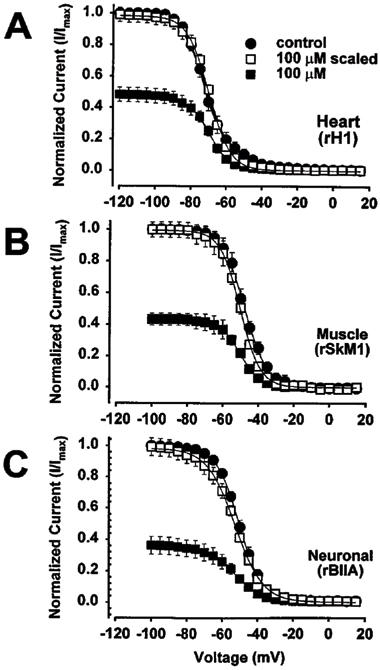
Since RSD 921 produced a minimum hyperpolarizing shift in the voltage-dependence of Na+ channel inactivation, we examined whether the observed block of Na+ channels resulted from interaction with the open state by using the IFMQ3 mutant Na+ channel. Figure 6A shows IFMQ3 block by 100 μM RSD 921. The control trace shows that the IFMQ3 Na+ current during a depolarization to ±20 mV decayed with a single, slow time constant of 428±8.9 ms (n=4). When currents were elicited after 5 min of perfusion with various concentrations of RSD 921, the currents decayed with slow time constants that were similar, with an average of 392±15 ms. However, there was an additional concentration-dependent fast exponential component (tfast) representing drug block (tB). The time constants of the fast components were 13.6±2.1, 15.8±2.9, 20.1±1.8 and 23.4±1.9 ms for 10, 30, 70 and 100 μM RSD 921, respectively. These fast components were ≈24.5 fold faster on average than the slow components of channel decay.
Figure 6.
Kinetics of open channel block induction of RSD 921. The rate of open channel block development was examined during a 1000 ms depolarizing pulse from −100 mV to +20 mV. (A) shows current traces elicited during the depolarization in the absence (control) and presence of 100 μM RSD 921. (B) shows the rate of drug block as a function of RSD 921 concentration. The time constant of the RSD 921-induced fast component of block (τfast) was obtained from a double exponential fit to the 100 μM current trace shown in A as described in the Methods. For a first order blocking scheme, a linear dependence of the rate of block development is expected for drug concentrations according to the equation: 1/tfast=kon*[RSD 921]+koff. The solid line in B represents the fit from which the apparent binding (kon) and unbinding (kon) rate constants were obtained. The data points were determined from five oocytes and error bars represent standard deviations.
The simplest model that can be used to describe the interaction between an open ion channel and an ion channel blocker states that blocking rates vary linearly with the concentration of the blocker. Therefore, a plot of the reciprocal of tB (1/tB) against drug concentration can be used to approximate the drug-channel interaction kinetics (Lansman et al., 1986; Valenzuela et al., 1996). Figure 6B shows the relationship between 1/tB and RSD 921 concentration, with the straight line representing the best fit to the equation 1/tB=kon*[RSD 921]+koff. The slope of the line is the apparent binding constant (kon), which equals 0.11±0.012×106 M−1 s−1, and the intercept is the unbinding constant (koff), which equals 12.5±2.2 s−1. The apparent affinity constant for RSD 921 (Kd) equals koff/kon, which was calculated as 117±31 μM. This value is very similar to the value determined for steady-state tonic block of the IFMQ3 channel shown in Table 1. This result, along with the facts that RSD 921 is a quaternary charged arylbenzacetamide compound at physiological pH and block development occurred in the absence of fast inactivation, suggests that RSD 921 interacts with the open state of the Na+ channel.
RSD 921 slows recovery from open channel block
Since the studies examining the interaction of RSD 921 with the IFMQ3 Na+ channel showed that drug block resulted from an interaction with the open state of the Na+ channel, we examined whether RSD 921 delayed recovery of the channel from open channel block. A slowing in the recovery of the channel from block would explain why the IFMQ3 channel exhibited a use-dependent component of block. Figure 7A shows the pulse protocol used to examine IFMQ3 recovery from RSD 921 open channel block, as described in the Methods. Figure 7B shows a pair of current traces recorded in the absence (control, left) and presence (right) of 100 μM RSD 921 with a recovery time interval (Δt) of 10 s between pulses. In the absence of RSD 921, peak IFMQ3 current recovered as a mono-exponential process with a time constant of 4.8±2.4 s. In the presence of 100 μM RSD 921, recovery was also mono-exponential but the time constant was increased to 14±2.7 s (Figure 7C). The slow recovery kinetics of the IFMQ3 channel from open channel block suggest that an interaction between RSD 921 and Na+ channels in the open state resulted in both tonic and phasic, or use-dependent, block.
Figure 7.
RSD 921 delays the recovery of IFMQ3 Na+ channels from open channel block. (A) shows the pulse protocol used to elicit pairs of currents from oocytes co-expressing IFMQ3 and the β1 subunit. The kinetics of channel recovery were examined by giving a 1000 ms conditioning pre-pulse from −100 to −10 mV (P1) followed by a similar test pulse (P2) after a variable recovery time interval (Δt) between 200 ms and 240 s at −100 mV. (B) shows a pair of 1000 ms current traces after a 10 s recovery time interval in the absence (control) and presence of 100 μM RSD 921. Channels are nearly fully recovered at this recovery interval in the absence of drug, so that the current amplitudes are almost identical. In the presence of 100 μM RSD 921, the test current measured during the second pulse (P2) has not recovered after a 10 s interval. (C) shows the complete recovery profile for IFMQ3 channels from RSD 921 open channel block. As described in the Methods, fractional recovery (P2/P1) is plotted on a log scale as a function of recovery time for 100 μM RSD 921. The data were fit with a single exponential equation as described in the Methods. For 100 μM RSD 921, the recovery time constant from open channel block was 14±2.7 s.
Discussion
The studies described in this paper represent the first pharmacological study of the effects of RSD 921, a novel class I anti-arrhythmic drug, on cloned heart, skeletal muscle and neuronal Na+ channels expressed in Xenopus oocytes. RSD 921 produced concentration-dependent tonic block that was similarly potent for all three Na+ channel isoforms. RSD 921 also produced frequency-dependent block of Na+ currents, with some selectivity for the heart channel. These results confirm previous in vitro ion channel blocking studies with chemically-related arylbenzacetamide compounds (Pugsley et al., 1993b; 1994), and conclusively show that RSD 921 displays state-, time- and voltage-dependent interactions with the Na+ channel.
The tonic component of RSD 921 block of Na+ channels was examined using an infrequent pulsing protocol, which demonstrated block of all three Na+ channel isoforms with half-maximal blocking concentrations (EC50) between 35–47 μM. Using similar experimental conditions to compare the Na+ channel blocking actions of RSD 921 with those of lidocaine, a class Ib anti-arrhythmic drug, RSD 921 was 12 fold more potent than lidocaine against heart channels (EC50 equals 563±22 μM for lidocaine), 31 fold more potent against skeletal muscle channels (EC50 equals 1083±122 μM for lidocaine) and 25 fold more potent against neuronal Na+ channels (EC50 equals 935±100 μM for lidocaine) (Pugsley & Goldin, 1998). These marked differences in potency may result from the fact that RSD 921 is very different in structure compared to lidocaine, which might indicate that RSD 921 interacts with a different binding site on the Na+ channel α-subunit.
The frequency-dependent block of Na+ currents by RSD 921 can be interpreted in terms of the modulated receptor hypothesis as resulting from differences in the binding and unbinding ratio of the anti-arrhythmic agent to the binding site, with the resulting block ratio being dependent on higher binding affinity to the open or inactive state compared to the resting state of the channel (Hille, 1997; Hondeghem & Katzung, 1977). The decrease in Na+ current at high rates of stimulation results from an accumulation of drug-associated channels, since Na+ channels spend more time in the open and inactive states as the inter-pulse interval shortens. RSD 921 produced an enhanced inhibition of Na+ currents at high frequencies of stimulation for the heart channel, which is consistent with previous in vitro results for the structurally-related κ opioid receptor agonist spiradoline (Pugsley et al., 1998). This enhanced inhibition was also observed, although to a lesser extent, against skeletal muscle and neuronal Na+ channels (Figure 4). The differences in the isoform-specific frequency-dependent block by RSD 921 may result from differences in RSD 921 binding affinity for the open or inactive state in each of the three isoforms.
RSD 921 could preferentially interact with either the open or inactive state of the Na+ channel compared to the resting state. Differential block of open or inactive states is an important criterion for clinical efficacy, because Na+ channels spend more time in the open and inactive states in pathological conditions. An indication of higher affinity for the inactive state is a shift in the voltage-dependence of steady-state inactivation with increasing drug concentration, as is observed for lidocaine block of Na+ channels (Bean et al., 1983; Sanchez-Chapula et al., 1983; Bennett et al., 1995). However, RSD 921 did not shift the voltage-dependence of inactivation for Na+ channels in the hyperpolarizing direction (Figure 5), suggesting that RSD 921 interacts primarily with an active state of the Na+ channel. Similar results were observed with bisaramil, a charged tertiary amine class I anti-arrhythmic drug (Pugsley & Goldin, 1998). Pugsley & Saint (1995) showed that the block produced by bisaramil did not increase with a prolongation of the pre-pulse duration, which would be expected to occur if the drug had a greater affinity for the inactive state of the channel because channels are maintained in the inactive state for a longer period of time with longer depolarizations. Therefore, RSD 921 may interact preferentially with the open state of the Na+ channel because of the charged nature of the drug.
To directly assess the interaction of RSD 921 with the open state of the Na+ channel, we utilized the IFMQ3 mutant, which lacks fast inactivation (West et al., 1992). RSD 921 block of the IFMQ3 channel was approximately 3 fold less potent compared to that of the wild-type channel. This result suggests that either there is some component of drug block of the inactive state, or that the IFMQ3 mutation directly affects block by RSD 921. However, when the frequency-dependent actions of RSD 921 were examined on the IFMQ3 channel at the highest frequency tested (30 Hz), the additional use-dependent block that developed was greater than that observed for the wild-type neuronal channel. This result suggests that RSD 921 preferentially blocks the open state of the Na+ channel. Similar frequency-dependent open channel block was observed for disopyramide (Grant et al., 1996) and tetracaine (Cahalan, 1978).
The time course of decay of IFMQ3 Na+ currents in the presence of RSD 921 was bi-exponential, whereas the time course of IFMQ3 Na+ current decay in the absence of drug was mono-exponential (Figure 6). The slow time constant was similar in the presence and absence of RSD 921, and most likely represents slow inactivation. The fast time constant, which increased as drug concentration increased, most likely represents channel block by RSD 921 interacting with the open state of the channel. From these data, the apparent rates of RSD 921 binding (kon) and unbinding (koff) to the Na+ channel were determined to be 0.11±0.012×106 M−1 s−1 and 12.5±2.2 s−1, respectively (Figure 6). These values yielded an apparent affinity constant Kd (or EC50) of 117±31 μM. This value is similar to the 110 μM EC50 value that was determined for tonic block of the IFMQ3 channel (Table 1).
Previous studies by Ragsdale et al. (1994; 1996) indicate that many local anaesthetic and anti-arrhythmic drugs interact with the Na+ channel from the intracellular side of the membrane. Assuming that this is the case for RSD 921, then the drug must enter the cell to block the Na+ channel. RSD 921 can be considered a permanently charged molecule, because it has a tertiary nitrogen moiety with a pKa of 9.0±0.2, which is protonated under physiological conditions. The positive charge should limit the ability of RSD 921 to cross the cell membrane. One mechanism by which RSD 921 could enter the cell is through a hydrophilic pathway that develops as channels open, as was proposed for local anaesthetics by Hille (1977). This mechanism could be the reason why RSD 921 predominantly blocks open Na+ channels.
The use of selective arylbenzacetamide κ opioid receptor agonists has made it possible to establish the involvement of κ opioid receptors in sedation, analgaesia, diuresis, and most recently cardiac arrhythmogenesis (Martin, 1984; Pugsley et al., 1993a). Several arylbenzacetamide κ opioid receptor agonists such as U-50,488H and (−)Cl-977 have been shown to reduce the incidence and severity of ischaemic and electrical arrhythmias in rats (Pugsley et al., 1992a,1992b). Pugsley et al. (1992c) showed that these arylacetamide compounds were still anti-arrhythmic in the presence of specific κ opioid receptor blockade. Further studies showed that the observed anti-arrhythmic efficacy of these compounds was maintained when enantiomeric pairs such as (+) PD 129,290 (which lacks affinity for the κ receptor) and (−) PD 129,289 (which has high affinity for the κ receptor) were examined (Pugsley et al., 1993b). This study concluded that the anti-arrhythmic actions of these arylbenzacetamide compounds was inherent in their chemical structure and resulted from direct Na+ and K+ ion channel blockade in cardiac muscle, and was not related to κ opioid receptor agonism, as had been previously reported (Wong et al., 1990). A recent structure-activity relationship analysis of ten arylacetamide compounds provided additional evidence that the anti-arrhythmic activity of these compounds is not related to κ opioid receptor activity, but rather resides in the ability of these compounds to block cardiac Na+ (and K+) current (Yong et al., 1996). Our results with RSD 921 demonstrate that an arylacetamide drug can directly block Na+ channels.
The block of active channels by class I anti-arrhythmic drugs may be of benefit in tissues in which the action potential is brief, or under conditions that shorten the ventricular action potential. In atrial tissue, the propagating action potential is inherently shorter than that found in ventricular tissue (Bailey, 1981). Therefore, Na+ channels spend much less time in the inactive state in the atrium compared to the ventricle, so that block of open channels by RSD 921 might play a significant role in terminating atrial tachyarrhythmias, potentially preventing the embolic risks associated with these arrhythmias. A shortening of the cardiac action potential may also result from pathological conditions associated with ischaemia (Cascio et al., 1995). In the setting of myocardial ischaemia, a number of significant changes occur in the intracellular and extracellular milieu, which results in modification of the cardiac action potential (Kleber et al., 1987). These action potential changes include a shortening of the duration and reduction in amplitude of the action potential (Barrett et al., 1997). Since RSD 921 produces a time-dependent, rapid block of Na+ channels, it may be highly effective at reducing conduction in ischaemic ventricular issue
In summary, we have demonstrated that RSD 921, an arylbenzacetamide anti-arrhythmic drug, blocks the open state of cardiac, skeletal muscle and neuronal Na+ channels expressed in Xenopus oocytes. Our studies suggest that RSD 921 may be a promising new drug for the treatment of cardiac arrhythmias associated with myocardial ischaemia, and may have increased anticonvulsant or local anaesthetic efficacy in neuronal tissue when compared to other Na+ channel blockers.
Acknowledgments
We thank Dr Gail Mandel (SUNY, Stony Brook) for generously providing the rat skeletal muscle Na+ channel clone, Dr Roland Kallen (University of Pennsylvania) for generously supplying the rat heart Na+ channel clone, Dr Michael Walker (Nortran Pharmaceuticals Ltd., Vancouver, BC, Canada) for generously providing RSD 921, Ted Shih, Marianne Smith and Daniel Allen for helpful discussions, and Miriam Reyes for excellent technical assistance. Supported by grants to A.L. Goldin from NIH (NS26729), the American Heart Association (9750642N) and the National Alliance for Research on Schizophrenia and Depression (23334). M.K.Pugsley is a MRC Post-Doctoral Research Fellow of Canada. A.L.Goldin is an Established Investigator of the American Heart Association.
Abbreviations
- rBIIA
rat brain IIA Na+ channel
- rH1
rat heart 1 Na+ channel
- rSkM1
rat skeletal muscle 1 Na+ channel
References
- ARMSTRONG C.M., BENZANILLA F., ROJAS E. Destruction of sodium conductance inactivation of squid axons perfused with pronase. J. Gen. Physiol. 1973;62:375–391. doi: 10.1085/jgp.62.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AULD V.J., GOLDIN A.L., KRAFTE D.S., CATTERALL W.A., LESTER H.A., DAVIDSON N., DUNN R.J. A neutral amino acid change in segment IIS4 dramatically alters the gating properties of the voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY J.C. The electrophysiological basis for cardiac electrical activity: normal and abnormal. Heart Lung. 1981;10:455–464. [PubMed] [Google Scholar]
- BAIN A.I., BARRETT T.D., BEATCH G.N., FEDIDA D., HAYES E.S., PLOUVIER B., PUGSLEY M.K., WALKER M.J.A., WALKER M.L., WALL R.A., YONG S.L., ZOLOTOY A. Better antiarrhythmics? Development of antiarrhythmic drugs selective for ischaemia-dependent arrhythmias. Drug Dev. Res. 1997;42:198–210. [Google Scholar]
- BARRETT T.D., MACLEOD B.A., WALKER M.J.A. A model of myocardial ischaemia for the simultaneous assessment of electrophysiological changes and arrhythmias in intact rabbits. J. Pharmacol. Tox. Meth. 1997;37:27–36. doi: 10.1016/s1056-8719(96)00145-1. [DOI] [PubMed] [Google Scholar]
- BEAN B.P., COHEN C.J., TSIEN R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEATCH G.N., MACLEOD B.A., PUGSLEY M.K., WALKER M.J.A. RSD 921, a potent new local anesthetic. ASEAN Cong. Anesthesiol. 1997. p. 60.
- BENNETT P.B., VALENZUELA C., CHEN L.-Q., KALLEN R.G. On the molecular nature of the lidocaine receptor of cardiac Na+ channel. Modification of block by alterations in the α-subunit III-IV interdomain. Circ. Res. 1995;77:584–592. doi: 10.1161/01.res.77.3.584. [DOI] [PubMed] [Google Scholar]
- CAHALAN M.D. Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophys. J. 1978;23:285–311. doi: 10.1016/S0006-3495(78)85449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CASCIO W.E., JOHNSON T.A., GETTES L.S. Electrophysiologic changes in ischemic ventricular myocardium: I. Influence of ionic, metabolic, and energetic changes. J. Cardiovasc. Electrophysiol. 1995;6:1039–1062. doi: 10.1111/j.1540-8167.1995.tb00381.x. [DOI] [PubMed] [Google Scholar]
- CLARK C.R., BIRCHMORE B., SHARIF N.A., HUNTER J.C., HILL R.G., HUGHES J. DD117302: A selective agonist for the κ-opioid receptor. Br. J. Pharmacol. 1988a;93:618–626. doi: 10.1111/j.1476-5381.1988.tb10319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLARK C.R., HALFPENNY P.R., HILL R.G., HORWELL D.C., HUGHES J., HARVIS T.C., REES D.C., SCHOFIELD D. Highly selective κ opioid analgesics. Synthesis and structure-agonist relationships of novel N-[(2-aminocyclohexyl)aryl]acetamide and N-[C2-aminocyclohexyl)aryloxy]acetamide derivatives. J. Med. Chem. 1988b;31:831–836. doi: 10.1021/jm00399a025. [DOI] [PubMed] [Google Scholar]
- GOLDIN A.L.Voltage-gated sodium channels Ligand- and Voltage-Gated Ion channels 1995Boca Raton, FL, CRC Press; 73–122.ed. North, R.A. pp [Google Scholar]
- GOLDIN A.L., SUMIKAWA K. Preparation of RNA for injection into Xenopus oocytes. Methods Enzymol. 1992;207:279–297. doi: 10.1016/0076-6879(92)07018-j. [DOI] [PubMed] [Google Scholar]
- GRANT A.O., JOHN J.E., NESTERENKO V.V., STARMER C.F., MOORMAN J.R. The role of inactivation in open-channel block of the sodium channel: studies with inactivation-deficient mutant channels. Mol. Pharmacol. 1996;50:1643–1650. [PubMed] [Google Scholar]
- HILLE B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HODGKIN A.L., HUXLEY A.F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol., London. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONDEGHEM L.M., KATZUNG B.G. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- ISOM L.L., DEJONGH K.S., PATTON D.E., REBER B.F.X., OFFORD J., CHARBONNEAU H., WALSH K., GOLDIN A.L., CATTERALL W.A. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- ISOM L.L., RAGSDALE D.S., DE JONGH K.S., WESTENBROEK R.E., REBER B.F.X., SCHEUER T., CATTERALL W.A. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- KALLEN R.G., SHENG Z.-H., YANG J., CHEN L., ROGART R.B., BARCHI R.L. Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron. 1990;4:233–242. doi: 10.1016/0896-6273(90)90098-z. [DOI] [PubMed] [Google Scholar]
- KLEBER A.G., RIEGER C.B., JANSE M.J. Extracellular K+ and H+ shifts in early ischaemia: mechanisms and relation to changes in impulse propagation. J. Mol. Cell. Cardiol. 1987;19:34–44. doi: 10.1016/s0022-2828(87)80608-9. [DOI] [PubMed] [Google Scholar]
- KONTIS K.J., GOLDIN A.L. Site-directed mutagenesis of the putative pore region of the rat IIA sodium channel. Mol. Pharmacol. 1993;43:635–644. [PubMed] [Google Scholar]
- LANSMAN J.B., HESS P., TSIEN R.W. Blockade of current through single calcium channels by Cd2+, Mg2+ and Ca2+: voltage and concentration dependence of calcium entry into the pore. J. Gen. Physiol. 1986;88:321–347. doi: 10.1085/jgp.88.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN W.R. Pharmacology of opioids. Pharmacol. Rev. 1984;35:285–323. [PubMed] [Google Scholar]
- PUGSLEY M.K., GOLDIN A.L. Effects of bisaramil, a novel class I antiarrhythmic agent, on heart, skeletal muscle and brain Na channels. Eur. J. Pharmacol. 1998;342:93–104. doi: 10.1016/s0014-2999(97)01420-9. [DOI] [PubMed] [Google Scholar]
- PUGSLEY M.K., PENZ W.P., WALKER M.J.A. The cardiovascular actions of the kappa-opioid receptor agonist (−)Cl-977. Proc. Can. Fed. Biol. Sci. 1992b;35:220. [Google Scholar]
- PUGSLEY M.K., PENZ W.P., WALKER M.J.A. Cardiovascular actions of U50,488H and related kappa agonists. Cardiovasc. Drug Rev. 1993a;11:151–164. [Google Scholar]
- PUGSLEY M.K., PENZ W.P., WALKER M.J.A., WONG T.M. Antiarrhythmic effects of U50,488H in rats subject to coronary artery occlusion. Eur. J. Pharmacol. 1992a;212:15–19. doi: 10.1016/0014-2999(92)90066-d. [DOI] [PubMed] [Google Scholar]
- PUGSLEY M.K., PENZ W.P., WALKER M.J.A., WONG T.M. Cardiovascular actions of the κ-agonist, U50,488H, in the absence and presence of opioid receptor blockade. Br. J. Pharmacol. 1992c;105:521–526. doi: 10.1111/j.1476-5381.1992.tb09012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUGSLEY M.K., SAINT D.A. Tonic and use-dependent block of sodium currents in isolated cardiac myocytes by bisaramil. Br. J. Pharmacol. 1995;114:377–382. doi: 10.1111/j.1476-5381.1995.tb13237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUGSLEY M.K., SAINT D.A., HAYES E.S., KRAMER D., WALKER M.J.A.Spiradoline - a kappa-receptor agonist with non-opioid antiarrhythmic and electrophysiological properties J. Cardiovasc. Pharmacol 1998. in press [DOI] [PubMed]
- PUGSLEY M.K., SAINT D.A., PENZ W.P., WALKER M.J.A. Electrophysiological and antiarrhythmic actions of the κ agonist PD129290 and its R,R,(+)-enantiomer, PD129289. Br. J. Pharmacol. 1993b;110:1579–1585. doi: 10.1111/j.1476-5381.1993.tb14004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUGSLEY M.K., SAINT D.A., WALKER M.J.A. An electrophysiological basis for the antiarrhythmic actions of the κ-opioid receptor agonist U50,488H. Eur. J. Pharmacol. 1994;261:303–309. doi: 10.1016/0014-2999(94)90121-x. [DOI] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- RAGSDALE D.S., MCPHEE J.C., SCHEUER T., CATTERALL W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANCHEZ-CHAPULA J., TSUDA Y., JOSEPHSON I.R. Voltage- and use-dependent effects of lidocaine on sodium current in rat single ventricular cells. Circ. Res. 1983;52:557–565. doi: 10.1161/01.res.52.5.557. [DOI] [PubMed] [Google Scholar]
- SMITH R.D., GOLDIN A.L. Functional analysis of the rat I sodium channel in Xenopus oocytes. J. Neurosci. 1998;18:811–820. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRICHARTZ G., RANDO T., WANG G.K. An integrated view of the molecular toxinology of sodium channel gating in excitable cells. Ann. Rev. Neurosci. 1987;10:237–267. doi: 10.1146/annurev.ne.10.030187.001321. [DOI] [PubMed] [Google Scholar]
- TRIMMER J.S., COOPERMAN S.S., TOMIKO S.A., ZHOU J., CREAN S.M., BOYLE M.B., KALLEN R.G., SHENG Z., BARCHI R.L., SIGWORTH F.J., GOODMAN R.H., AGNEW W.S., MANDEL G. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- VALENZUELA C., DELPON E., FRANQUEZA L., GAY P., PEREZ O., TAMARGO J., SNYDERS D.J. Class III antiarrhythmic effects of zatebradine. Time-, state-, use- and voltage-dependent block of hKv1.5 channels. Circ. 1996;94:562–570. doi: 10.1161/01.cir.94.3.562. [DOI] [PubMed] [Google Scholar]
- WALKER M.L., WALL R.A., WALKER M.J.A. Determination of an arylacetamide antiarrhythmic in rat blood and tissues using reverse-phase high-performance liquid chromatography. J. Chromatography. 1996;675:257–263. doi: 10.1016/0378-4347(95)00360-6. [DOI] [PubMed] [Google Scholar]
- WEST J.W., PATTON D.E., SCHEUER T., WANG Y., GOLDIN A.L., CATTERALL W.A. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONG T.M., LEE A.Y.S., TAI K.-K. Effect of drugs interacting with opioid receptors during normal perfusion or ischaemia and reperfusion in the isolated rat heart - an attempt to identify cardiac opioid receptor subtypes(s) involved in arrhythmogenesis. J. Mol. Cell. Cardiol. 1990;22:1167–1175. doi: 10.1016/0022-2828(90)90080-l. [DOI] [PubMed] [Google Scholar]
- YONG S.L., ABRAHAM S., PUGSLEY M.K., HAYES E.S., ZOLOTOY A.B., WALKER M.J.A. SAR evidence that antiarrhythmic activity is unrelated to opioid kappa agonist activity. Br. J. Pharmacol. 1996;119:P35. [Google Scholar]