

Abstract
Pharmacological profiles of tritiated KMD-3213, a new antagonist of α1-adrenoceptor (AR), were examined in recombinant and native α1-AR, and compared with those of prazosin (PZ) and tamsulosin (YM-617).
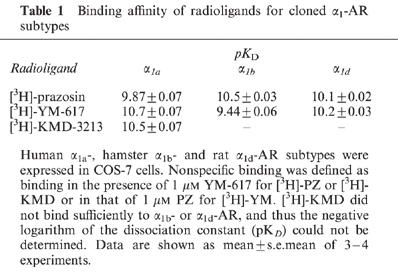
In saturation experiments, [3H]-KMD (10–2000 pM) showed high affinity for α1a-AR (pKD=10.5). However, no significant binding to α1b-AR and insufficient/unsaturated binding to α1d-AR were observed at concentrations up to 2000 pM. In contrast, [3H]-PZ and [3H]-YM bound to all subtypes with high affinity (pKD>9). In competition experiments, KMD-3213 also had higher affinity for α1a-AR than for other two subtypes; pKi=10.4, 8.1 and 8.6 for α1a-, α1b- and α1d-AR, respectively.
[3H]-KMD also bound to the native α1A-AR (rat submaxillary gland) with high affinity, but not to α1B-AR (rat liver). In rat kidney which expresses α1A- and α1B-AR, [3H]-KMD and [3H]-PZ bound to a single high-affinity site (pKD=10.8 and 10.1, respectively) with distinct amount of binding sites (Bmax=159 and 267 fmol mg−1 protein, respectively). [3H]-PZ binding sites consisted of low- and high-affinity sites for KMD-3213 (pKi=7.6 and 10.7, respectively), for WB4101 (pKi=8.1 and 10.0) and for YM-617 (pKi=8.7 and 10.8). The proportion of the high affinity site was approximately 60% in these drugs which was compatible to the ratio between Bmax of [3H]-KMD and [3H]-PZ. [3H]-KMD binding sites consisted of a single site for these drugs with affinities which were similar to those of the high affinity sites in [3H]-PZ binding.
In functional experiments, KMD-3213 antagonized the contractile responses to NS-49 or noradrenaline (NA) with higher affinity in functional α1A- (rat caudal artery, pA2=10.0 against NS-49) and α1L-AR (dog mesenteric artery, pA2=9.9 against NA) than in α1B- (dog carotid artery, pA2=7.7 against NA) and α1D-AR (rat thoracic aorta, pA2=8.3 against NA).
These results confirm the α1A-AR selectivity and high affinity of KMD-3213, and indicate that [3H]-KMD can label selectively α1A-AR.
Keywords: α1-Adrenoceptor subtypes, KMD-3213, prazosin, tamsulosin
Introduction
α1-Adrenoceptors (ARs) constitute a heterogeneous family of receptors (McGrath, 1982). The existence of two subtypes, α1A- and α1B-AR, was initially suggested in pharmacological studies (Han et al., 1987; Minneman, 1988; Morrow & Creese, 1986). Molecular cloning techniques have revealed the existence of at least three receptor subtypes (α1a-, α1b- and α1d-AR), and pharmacological studies indicated that these clones subtypes correspond to native α1A-, α1B- and α1D-AR subtypes, respectively (Bylund et al., 1995; Hieble et al., 1995). The fourth subtype, α1L-AR which is defined principally based on a distinct low affinity for prazosin (PZ), has been proposed mainly from functional studies (Flavahan & Vanhoutte, 1986; Muramatsu et al., 1990; 1991; Oshita et al., 1991; Ford et al., 1996). However, the molecular identity of α1L-AR still remains to be clarified (Bylund et al., 1998).
Various α1-AR ligands are used for characterizing the binding profiles and functions of receptor subtypes; however, few ligands showing subtype selectivity are available. Recently, KMD-3213 [(−)-(R)-1-(3-hydroxypropyl)-5-[2-[[2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl] amino]propyl]indoline - 7 - carboxamide] was reported as an α1-AR antagonist (Shibata et al., 1995). In the present study, we characterized the binding profiles of KMD-3213, and its tritiated form as a radioligand, by using recombinant α1-AR subtypes and native tissues, and compared α1-AR subtype selectivity of KMD-3213 with other α1-AR antagonists such as PZ and tamsulosin (YM-617). The characteristics of KMD-3213 were also compared with those of other drugs in functional experiments.
Methods
Male Wistar rats (250–350 g) and Beagle dogs (10–15 kg) were used for the experiments. Animals were sacrificed under anaesthesia with pentobarbital, and tissues were isolated immediately thereafter.
Membrane preparation
For the cloned ARs, COS-7 cells were transfected with the cDNA clones of human α1a-, hamster α1b- and rat α1d-AR. The harvested cells were suspended in ice-cold assay buffer (Tris-HCl 50 mM, EDTA 1 mM, pH 7.4), sonicated and centrifuged at 3000×g for 10 min. The supernatant was then centrifuged at 80,000×g for 30 min, and the resulting pellet was resuspended in assay buffer and used for binding experiments.
For preparation of native receptor subtypes, isolated rat tissues (submaxillary gland, liver and kidney) were homogenized in 20 vol. of ice-cold homogenization buffer (mM): Tris-HCl 50, NaCl 100, EDTA 2, pH 7.4) with a Polytron (setting 8, 15 s×3) and filtered through four layers of cheese cloth. The supernatants were centrifuged at 80,000×g for 30 min, and the resulting pellets were suspended in ice-cold assay buffer, and then again centrifuged at 80,000×g for 30 min. All centrifugation was done at 4°C. Final pellets were resuspended in assay buffer and used for the following experiments.
Radioligand binding experiments
In saturation binding experiments, the membranes were incubated with various concentrations of [3H]-ligands for 45 min at 30°C. Total incubation volume for [3H]-PZ, [3H]-YM and [3H]-KMD was 1 ml, 3 ml and 2∼3 ml, respectively (5–30 μg protein/tube for cultured cells or 50–150 μg protein/tube for native tissues). Nonspecific binding was defined as binding in the presence of 1 μM YM-617 for [3H]-PZ or [3H]-KMD or in that of 1 μM PZ for [3H]-YM, excepting the binding to rat liver and kidney (0.3 μM YM-617 for [3H]-PZ and 0.3 μM PZ for [3H]-KMD). In competition binding experiments, membranes were incubated with about 200 pM [3H]-PZ or 70∼100 pM [3H]-KMD and unlabelled drugs for 45 min at 30°C. Specific binding of both radioligands (200 pM [3H]-PZ and 100 pM [3H]-KMD) was approximately 90% and 80% of the total binding for cloned cells and native tissues, respectively. Reactions were terminated by rapid filtration with a Brandel cell harvester onto Whatman GF/C filters presoaked in 0.3% polyethyleneimine for 15 min. The filters were then washed four times with 4 ml of ice-cold 50 mM Tris-HCl (pH 7.4) and dried. The filter-bound radioactivity was determined by liquid scintillation counting. Experiments were conducted in duplicate. Binding affinities of [3H]-ligands and unlabelled drugs were expressed as negative logarithm of the equilibrium dissociation constant (pKD and pKi, respectively). Protein concentrations were quantified by the method of Bradford using bovine serum albumin as standard (Bradford, 1976).
Functional experiments
Freshly isolated arteries were cleaned of adherent connective tissue and cut helically, and the endoethelium was removed by gentle rubbing. The strip was then mounted vertically in an organ bath containing 20 ml of Krebs-Henseleit solution (composition in mM): NaCl 112.0, KCl 5.9, CaCl2 2.0, MgCl2 1.2, NaH2PO4 1.2, NaHCO3 25.0 and glucose 11.5, gassed with 5% CO2 and O2 and maintained at 37°C. Resting tension applied was 0.5 g for rat caudal artery or 1 g for other tissues, and the responses were recorded isometrically through force-displacement transducers. All preparations were equilibrated for 90 min, and the following experiments were performed.
Concentration-response curves were obtained by adding the agonist (NS-49 for rat caudal artery or noradrenaline (NA) for other arteries) in cumulative fashion. The curves were drawn at least five times for the same strip, and the third curve was used as the control. After the control experiment, strips were incubated with α1-AR antagonists for 30 min before, and during, the construction of the curves. All the experiments were performed in the presence of 0.1 μM desipramine and 5 μM deoxycorticosterone acetate to block neural and extraneural uptake, respectively, of NA and 3 μM propranolol to block β-AR induced responses. In rat caudal artery, 0.1 μM rauwolscine was further applied in addition to those three chemicals to avoid α2-AR mediated responses.
Data analysis
Data were given as means±s.e.mean. Saturation and competition binding data were first fitted to a one- and then a two-site model, and the two-site model was accepted only if it resulted in a significant improvement of the fit as judged by an F-test comparison with a P<0.05. Analysis of radioligand binding data was performed with LIGAND (Munson & Rodbard, 1980), a nonlinear curve-fitting program.
In functional studies, antagonist affinity estimates were obtained by construction of Schild regressions and were constrained to a slope of unity (if not statistically different) according to the equation: pKB=log(r-1)−log[antagonist], where r is the concentration ratio between EC50 values (concentration for a half-maximal response) in the presence and absence of the antagonist. Schild plots were also constructed by plotting the log(r-1) against the log of antagonist concentration, and pA2 values were determined from the intercept (Arunlakshana & Schild, 1959).
Drugs
The drugs used and their sources were following: (−)-(R)-1-(3-hydroxypropyl) -5 - [ 2- [[2 -[2 - (2,2,2 - trifluoroethoxy) phenoxy] -ethyl]amino]propyl]indoline-7-carboxamide (KMD-3213), tamsulosin HCl (YM-617) and (R)-(−)-3′-(2-amino-1-hydroxyethyl)-4′-fluoromethane-sulphonanilide hydrochloride (NS-49), from Kissei Pharmaceutical Co. Ltd. (Matsumoto, Japan); prazosin HCl (PZ), (−)-noradrenaline bitartrate (NA) and desipramine hydrochloride, from Sigma (St. Louis, U.S.A.); WB4101, BMY7378 and rauwolscine hydrochloride, from Research Biochemicals Inc. (Natick, U.S.A.); (±)-propranolol hydrochloride and deoxycorticosterone acetate, from Nacalai (Kyoto, Japan); [3H]KMD-3213 (49∼52 Ci mmol−1), from Amersham (England); and [3H]prazosin (77.2 Ci mmol−1) and [3H]-YM-617 (32.5 Ci mmol−1), from NEN (Boston, U.S.A.).
KMD-3213 was dissolved in dimethylsulphoxide and diluted in assay buffer for binding studies or Hartmann's solution (composition in w v−1%): NaCl 0.60, KCl 0.03, CaCl2 0.02 and sodium lacate 0.31, for functional studies. [3H]-ligands were diluted in assay buffer. PZ was dissolved in 50% ethanol and diluted in distilled water. Other drugs were dissolved in and diluted with distilled water.
Results
Characteristics of tritiated KMD-3213
Chemical structures of tritiated KMD-3213, (−)-(R)-1-(3-hydroxypropyl) - 5 - [2 -[[2-[2-(2,2,2-trifluoroethoxy)[4,6(n)-3H]phenoxy]ethyl]amino]propyl]indoline-7-carboxamide, is shown in Figure 1a. This tritiated form of KMD-3213 ([3H]-KMD) with high specific radioactivity (49∼52 Ci mmol−1) effectively bound to α1a/α1A-AR at subnanomolar concentrations. A representative saturation curve of [3H]-KMD at human α1a-AR is shown in Figure 1b. The binding was equilibrated by incubation at 30°C for more than 40 min (data not shown). Non-specific binding was less than 10% at 100 pM [3H]-KMD. Since [3H]-KMD had high affinity for α1a/α1A-AR, 2∼3 ml of incubation volume was employed in binding experiments in order to avoid a significant reduction of free [3H]-KMD concentration during incubation.
Figure 1.
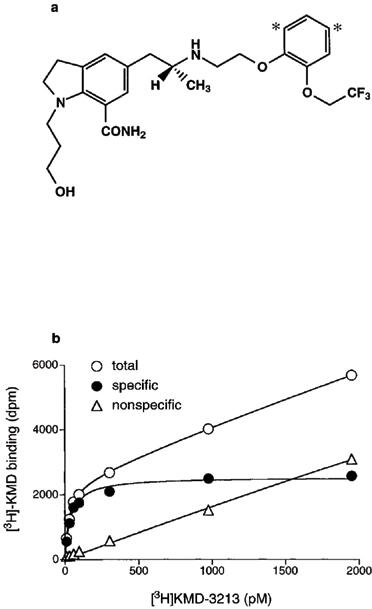
Characteristics of [3H]KMD-3213. (a) Chemical structure of [3H]KMD-3213. Asterisks show the tritiated sites. (b) Saturation of [3H]KMD3213 binding to human α1a-AR subtypes expressed in COS-7 cells. Each point represents the means of duplicate determinations from a single experiment. The experiments were replicated three times with similar results.
Radioligand binding experiments for cloned α1-AR subtypes
In saturation experiments, specific binding of the three [3H]-ligands ([3H]-PZ, [3H]-YM and [3H]-KMD) was determined by use of the same membranes in each experiment, and representative patterns are shown in Figure 2. All of the specific binding curves were saturable, except the curves for [3H]-KMD binding to α1b- and α1d-AR. In contrast to [3H]-PZ and [3H]-YM which showed high affinity for the three cloned α1-AR subtypes (pKD>9), [3H]-KMD clearly showed different sensitivity to them; high affinity to α1a-AR (pKD; 10.5±0.07), no significant binding to α1b-AR and insufficient/unsaturated binding to α1d-AR at concentrations up to 2000 pM (Table 1) were observed.
Figure 2.
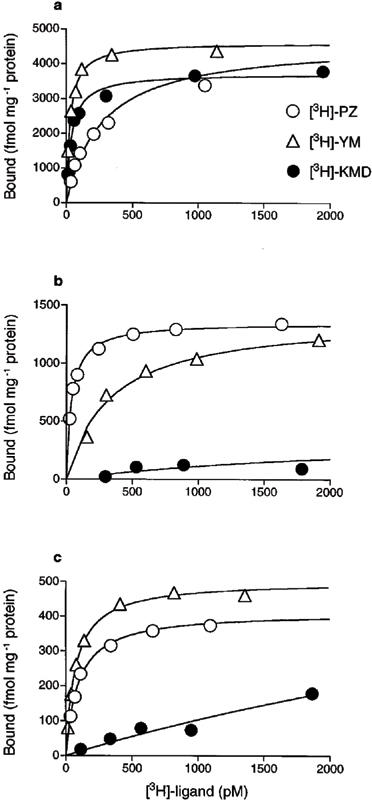
Saturation binding experiments of [3H]-PZ, [3H]-YM and [3H]-KMD to recombinant α1-AR subtypes. (a) Human α1a-, (b) hamster α1b- and (c) rat α1d-AR subtypes were expressed in COS-7 cells. Specific binding, drawn in Figures, was determined as the difference in radioligand binding in the presence or absence of 1 μM YM-617 for [3H]-PZ and [3H]-KMD or 1 μM PZ for [3H]-YM. All three radioligands were incubated with the same membrane in each experiment. A representative result for each subtype is shown as mean of duplicate in a single experiment (see Table 1).
Table 1.
Binding affinity of radioligands for cloned α1-AR subtypes
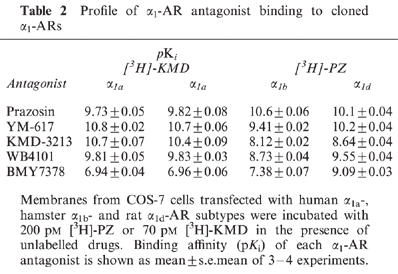
Several α1-AR antagonists were tested in competition experiments against [3H]-PZ or [3H]-KMD binding to cloned α1-AR subtypes. The competition experiment with [3H]-KMD binding was done only with α1a-AR, because [3H]-KMD at concentrations used in this experiment did not show sufficient binding to α1b- and α1d-AR (Figure 2). The affinities of the α1-AR antagonists to cloned α1-AR subtypes are shown in Table 2. The characteristics of each antagonist were basically similar between [3H]-PZ and [3H]-KMD binding. Regarding the selectivity for α1-AR subtypes, several patterns were observed. PZ showed no selectivity for the three α1-AR subtypes. YM-617 and WB4101 showed slightly lower affinity for α1b-AR than for other subtypes, and BMY7378 showed higher affinity for α1d-AR than for other two subtypes. KMD-3213 had prominent selectivity, showing more than 200 and 50 times higher affinity for α1a-AR than for α1b- and α1d-AR, respectively.
Table 2.
Profile of α1-AR antagonist binding to cloned α1-ARs
Binding characteristics for native α1-AR subtypes
The binding of [3H]-KMD was further characterized in rat submaxillary gland, liver and kidney, which are pharmacologically defined as tissues predominantly containing α1A-AR, α1B-AR and a mixture of α1A- and α1B-ARs, respectively (Michel et al., 1989; Feng et al., 1991). As shown in Table 3, [3H]-PZ showed high affinity for all tissues, whereas [3H]-KMD bound with high affinity to the submaxillary gland and kidney but showed no saturable binding to liver membranes at concentrations up to 2000 pM (data not shown). Bmax values of these two tritiated compounds were similar in submaxillary gland, but in kidney Bmax of [3H]-KMD was only 60% of that of [3H]-PZ.
Table 3.
Characteristics of radioligand binding to native tissues

The binding characteristics of series of antagonists for the native α1-AR subtypes were examined in competition experiments as shown in Table 4. In submaxillary gland (α1A-AR) and liver (α1B-AR), the binding of [3H]-PZ and [3H]-KMD was displaced monophasically by the competitors used with affinities which were compatible with those shown in Table 2. In rat kidney, however, YM-617, KMD-3213 and WB4101 displaced the binding of [3H]-PZ with two distinct affinity sites (Figure 3). The affinities for the high and low affinity sites corresponded well to those for α1A- and α1B-AR subtype, respectively. The proportion of high affinity sites in the total binding of [3H]-PZ were 68, 58 and 66% for YM-617, KMD-3213 and WB4101, respectively, which matched well with the ratio (60%) between Bmax values of [3H]-KMD and [3H]-PZ seen in Tables 3 and 4. The binding of [3H]-KMD was uniformly displaced by a series of drugs with affinities corresponding to those for high affinity sites in the displacement of [3H]-PZ binding (Figure 3 and Table 4). These results suggest that [3H]-KMD recognized a single population of binding sites (α1A-AR) from the mixed populations (α1A- and α1B-AR) of binding sites in rat kidney because of its high affinity for α1A-AR.
Table 4.
Binding affinity of α1-AR antagonists to native tissues
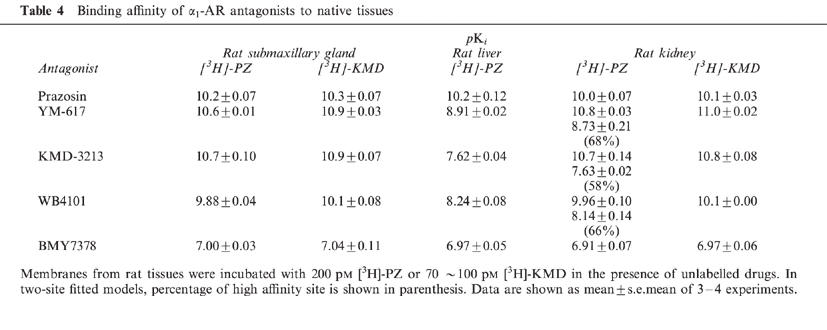
Figure 3.
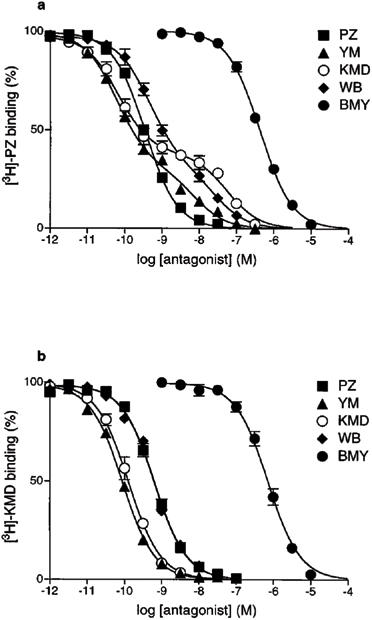
Competition experiments of specific [3H]-PZ and [3H]-KMD binding by various α1-AR antagonists in rat kidney. (a) 200 pM [3H]-PZ or (b) 100 pM [3H]-KMD was incubated with rat kidney membranes in the presence of unlabelled drugs. Competition curves of YM-617, KMD-3213 and WB4101 against [3H]-PZ binding were significantly fitted to two-site model (P<0.05), and other curves were fitted to one-site model. Data are shown as mean±s.e.mean of three independent experiments.
Functional experiments in isolated native tissues
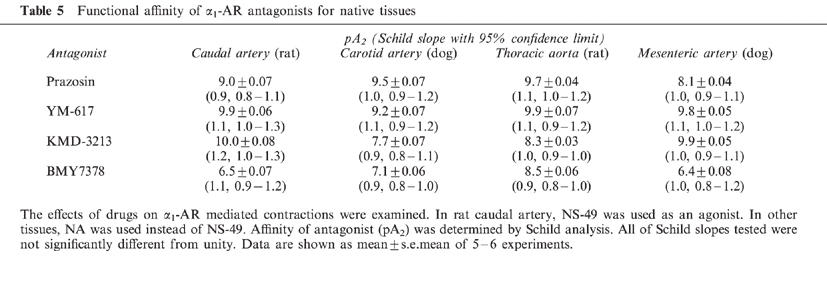
To further characterize the selectivity of KMD-3213, we performed functional experiments using isolated rat caudal arteries, dog carotid arteries, rat thoracic aortae and dog mesenteric arteries in which agonist-induced contraction is mediated by α1A-, α1B-, α1D- and α1L-AR subtype, respectively (Lachnit et al., 1997; Muramatsu et al., 1990; 1991; Kenny et al., 1995). In rat caudal artery, the contractile response to NA was mediated by at least two α1-AR subtypes; one is α1A-AR and the other remains to be defined (Lachnit et al., 1997). We employed NS-49, an α1A-AR selective agonist (Obika et al., 1995), to draw the response mediated by α1A-AR in rat caudal artery. In other tissues NA was employed as an agonist. The representative antagonism of KMD-3213 are shown in Figures 4 and 5, and the results are summarized in Table 5. All of the antagonists shifted the agonist-induced concentration-response curves to the right in a parallel fashion, and Schild analysis revealed that slopes of all the antagonists in any tissues were close to unity (Figures 4 and 5, Table 5). We, thus, concluded that the antagonist was fully equilibrated under these conditions. PZ displayed high affinity for α1A-, α1B- and α1D-AR (pA2 >9), but low affinity for α1L-AR. In contrast to PZ, KMD-3213 showed higher affinity for functional α1A- and α1L-AR than for α1B- and α1D-AR displaying pA2 values of 10.0, 9.9, 7.7 and 8.3, respectively. The ratios of affinity of KMD-3213 for α1-AR subtypes are approximately 200 for α1B/(α1A or α1L) and 50 for α1D/(α1A or α1L), which are similar values with those obtained in binding experiments. YM-617 showed high affinity for all subtypes (pA2 >9), and BMY7378 displayed α1D-AR selectivity.
Figure 4.
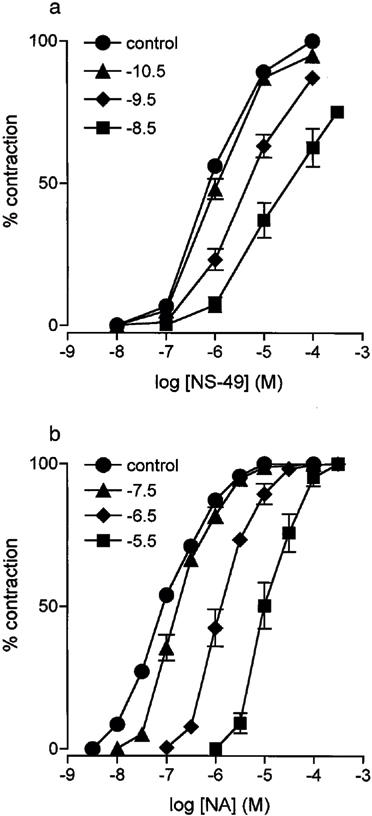
Antagonism of contractions to agonists by KMD-3213. (a) Concentration response curves to NS-49 in rat caudal artery are shown. •, Control; ▴, KMD-3213 0.03 nM; ⧫, 0.3 nM; ▪, 3 nM. (b) Concentration response curves to NA in dog carotid artery are shown. •, Control; ▴, KMD-3213 0.03 μM; ⧫, 0.3 μM; ▪, 3 μM. Data are shown as mean±s.e.mean of 5–6 independent experiments.
Figure 5.
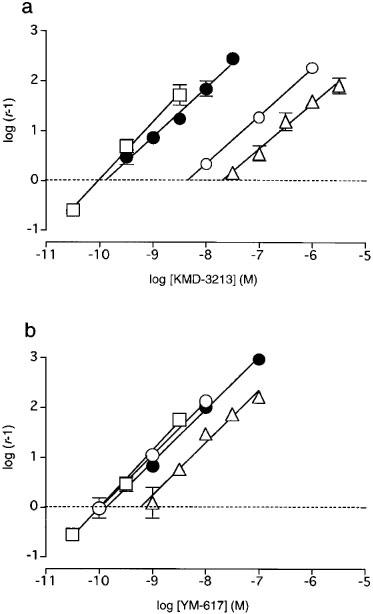
Comparison of Schild analyses for KMD-3213 and YM-617. Schild plots constructed with data from this Figure and Table 5 for KMD-3213 (a) or YM-617 (b) against contractile responses to NS-49 in rat caudal artery □, or to NA in dog carotid artery ▵, in rat thoracic aorta ○, and in dog mesenteric artery •. Data are shown as mean±s.e.mean of 5–6 independent experiments
Table 5.
Functional affinity of α1-AR antagonists for native tissues
Discussion
KMD-3213, a recently synthesized α1A-AR antagonist, displays antagonism with apparent tissue selectivity (Moriyama et al., 1997; Yamagishi et al., 1996). In the present study, we characterized the pharmacological profiles of KMD-3213 by use of its tritiated form as a radioligand and compared it with those of other α1-AR ligands, [3H]-PZ and [3H]-YM.
At first, we compared three α1-AR specific radioligands, [3H]-PZ, [3H]-YM and [3H]-KMD, in saturation binding experiments with recombinant α1a-, α1b- and α1d-AR subtypes. [3H]-PZ showed high affinity for all α1-AR subtypes without apparent subtype selectivity. [3H]-YM binding had slightly less affinity at α1b-AR than at α1a- and α1d-ARs, however, relatively high affinity was observed for all subtypes as has been described previously (Han et al., 1995). In contrast, [3H]-KMD showed high affinity for the α1a-AR subtype only; [3H]-KMD binding to α1b- and α1d-AR subtypes was insufficient and not saturated at concentrations up to 2000 pM (Table 1 and Figure 2). This high selectivity of KMD-3213 to α1a-AR was also confirmed in competition binding experiments. Although YM-617 and WB4101 also showed higher affinity for the α1a-AR subtype, the degree of selectivity was less than that of KMD-3213 (Table 2). These results were in good agreement with results of a previous report (Shibata et al., 1995), in which three recombinant subtypes of human were investigated with unlabelled KMD-3213. This suggests that the selectivity of KMD-3213 is not dependent on species and indicates that [3H]-KMD could distinguish the α1a-AR subtype from the other two recombinant subtypes.
At native α1-AR subtypes, [3H]-KMD showed similar subtype selectivity, i.e. high affinity for α1A-AR of rat submaxillary gland and no significant binding to α1B-AR of rat liver (Table 3). This selectivity was also applied to rat kidney which contains both α1A- and α1B-AR (Feng et al., 1991). Both [3H]-PZ and [3H]-KMD binding revealed single populations of binding sites with high affinity but showed different densities; Bmax of [3H]-KMD was 60% of that of [3H]-PZ. In addition, the binding of [3H]-PZ consisted of two components with high- (α1A-AR) and low- (α1B-AR) affinity sites discriminated by YM-617, KMD-3213 and WB4101 (Figure 3 and Table 4). The proportion of high affinity sites in the total binding sites of [3H]-PZ were 68, 58 and 66% for YM-617, KMD-3213 and WB4101, respectively, which matched well with the ratio (60%) between the Bmax values of the two [3H]-ligands (Table 4). In contrast, the binding of [3H]-KMD was displaced by these drugs monophasically with affinity estimates which correspond well with those for α1A-AR. These results demonstrated that [3H]-KMD exclusively detected an α1A-AR population from the mixture labelled by 3H-PZ. Since it is impossible to investigate the binding characters of α1D-AR in native tissues (Yang et al., 1997), we conducted further functional experiments.
Rat caudal artery, dog carotid artery, rat thoracic aorta and dog mesenteric artery were employed in functional experiment as tissues in which agonist-induced contraction is mediated by α1A-, α1B-, α1D- and α1L-AR subtype, respectively (Lachnit et al., 1997; Muramatsu et al., 1990; 1991; Kenny et al., 1995). The α1L-AR, the fourth subtype (Bylund et al., 1998), has been proposed based on its distinct low affinity for PZ mainly in functional experiments (Flavahan & Vanhoutte, 1986; Muramatsu et al., 1990; Ohmura et al., 1992). This α1L-AR has not yet been identified separately as a distinct gene product and is therefore not fully characterized. Recently, Ford et al. (1997) suggested that the cloned α1a-AR subtype, which showed typical characteristics of the α1a-AR in binding experiments, exhibited pharmacological characteristics as α1L-AR in the functional experiments. In fact, several investigators have indicated that there is more similarity of pharmacological characteristics between α1L- and α1A-ARs than between α1L- and α1B-/α1D-ARs (Williams et al., 1996; Daniels et al., 1996). In addition, α1A-AR has recently been further subdivided into four isoforms based on their structural bases derived from alternative splicing of the single gene transcript (Hirasawa et al., 1995; Chang et al., 1998). Although the pharmacological profiles of these isoforms do not differ much, as so far reported (Hirasawa et al., 1995; Chang et al., 1998), one of the isoforms may possibly represent the α1L-AR. In spite of this controversy, the existence of a distinct α1-AR with low affinity for PZ has been widely accepted especially in functional studies (Bylund et al., 1994; Graham et al., 1996; Muramatsu et al., 1995; Kava et al., 1998). Therefore, we examined the affinity of KMD-3213 for native α1-AR subtypes including α1L-AR in functional experiments. KMD-3213 showed a prominent subtype selectivity maintaining high affinity for α1A- and α1L-ARs. The values of pA2 were 10.0, 9.9, 7.7 and 8.3 for α1A-, α1L-, α1B- and α1D-ARs, respectively (Table 5). In rat caudal artery, KMD-3213 sometimes gave apparently insurmountable contractile responses even at 100 μM NS-49 especially at higher concentration of the antagonist (Figure 4a). However, we thought that this is due to its high affinity to the receptor in that tissue and that the antagonism by KMD-3213 is basically competitive in nature. This α1A (and/or α1L)-AR selectivity was not seen in both PZ and YM-617. Recently, however, several chemicals have been also developed as α1A-AR selective antagonists, such as SNAP5089 (Lachnit et al., 1997), RS17053 (Ford et al., 1996), Rec15/2739 (Testa et al., 1996), and L762459 (O'Malley et al., 1998). These including KMD-3213 are targeted to medical therapeutics for diseases such as benign prostatic hypertrophy.
In conclusion, KMD-3213 is an α1-AR antagonist that shows high affinity and selectivity for α1A-AR and functional α1L-AR subtypes compared with PZ or YM-617. Because of its high selectivity, KMD-3213 and its radioligand can be useful tools for analysing α1-AR pharmacology in native tissues or recombinant receptors.
Acknowledgments
The authors would like to thank Ms N. Aoki for secretarial assistance and Ms N. Saito for technical assistance. This work is supported in part by grant from the Smoking Research Foundation of Japan and by Grant-in-Aid for Scientific Research (09273105, 09470023 and 10670135) from the Ministry of Education, Science, Sports and Culture of Japan.
Abbreviations
- AR
adrenoceptor
- KMD-3213
(−)-(R)-1-(3-hydroxypropyl)-5-[2-[[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl] amino]propyl]indoline-7-carboxamide
- NA
noradrenaline
- PZ
prazosin
- YM-617
tamsulosin
References
- ARUNLAKSHANA O., SCHILD H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BYLUND D.B., BOND R.A., CLARKE D.E., EIKENBUR D.C., HIEBLE J.P., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., MOLINOF P.B., RUFFOLO R.R., STROSBERG A.D., TRENDELENBURG U.G. ADRENOCEPTORS. The IUPHAR Compendium of Receptor Characterization and Classification. 1998. pp. 58–74.
- BYLUND D.B., EIKENBERG D.C., HIEBLE J.P., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., MOLINOFF P.B., RUFFOLO R.R., TRENDELENBURG U.G. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- BYLUND D.B., REGAN J.W., FABER J.E., HIEBLE J.P., TRIGGLE C.R., RUFFOLO R.J. Vascular alpha-adrenoceptors: from the gene to the human. Can. J. Physiol. Pharmacol. 1995;73:533–543. doi: 10.1139/y95-068. [DOI] [PubMed] [Google Scholar]
- CHANG D.J., CHANG T.K., YAMANISHI S.S., SALAZAR F.H., KOSAKA A.H., KHARE R., BHAKTA S., JASPER J.R., SHIEH I.S., LESNICK J.D., FORD A.P., DANIELS D.V., EGLEN R.M., CLARKE D.E., BACH C., CHAN H.W. Molecular cloning, genomic characterization and expression of novel human alpha 1A-adrenoceptor isoforms. FEBS Lett. 1998;422:279–283. doi: 10.1016/s0014-5793(98)00024-6. [DOI] [PubMed] [Google Scholar]
- DANIELS D.V., GEVER J.R., MELOY T.D., CHANG D.J., KOSAKA A.H., CLARKE D.E., FORD A.P. Functional pharmacological characteristics of human, rat and rabbit cloned alpha 1A-adrenoceptors expressed in Chinese hamster ovary (CHO-K1) cells. Br. J. Pharmacol. 1996;119:360P. [Google Scholar]
- FENG F., PETTINGER W.A., ABEL P.W., JEFFRIES W.B. Regional distribution of alpha 1-adrenoceptor subtypes in rat kidney. J. Pharmacol. Exp. Ther. 1991;258:263–268. [PubMed] [Google Scholar]
- FLAVAHAN N.A., VANHOUTTE P.M. Alpha-adrenoceptor subclassification in vascular smooth muscle. Trends Pharmacol. Sci. 1986;7:347–349. [Google Scholar]
- FORD A.P., ARREDONDO N.F., BLUE D.J., BONHAUS D.W., JASPER J., KAVA M.S., LESNICK J., PFISTER J.R., SHIEH I.A., VIMONT R.L., WILLIAMS T.J., MCNEAL J.E., STAMEY T.A., CLARKE D.E. RS-17053 (N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-alpha, alpha-dimethyl-1H-indole-3-ethanamine hydrochloride), a selective alpha 1A-adrenoceptor antagonist, displays low affinity for functional alpha 1-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol. Pharmacol. 1996;49:209–215. [PubMed] [Google Scholar]
- FORD A.P.D.W., DANIELS D.V., CHANG D.J., GEVER J.R., JASPER J.R., LESNICK J.D., CLARKE D.E. Pharmacological pleiotropism of the human recombinant alpha A-adrenoceptor: implications for alpha 1-adrenoceptor classification. Br. J. Pharmacol. 1997;121:1127–1135. doi: 10.1038/sj.bjp.0701207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAHAM R.M., PEREZ D.M., HWA J., PIASCIK M.T. Alpha 1-adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ. Res. 1996;78:737–749. doi: 10.1161/01.res.78.5.737. [DOI] [PubMed] [Google Scholar]
- HAN C., ABEL P.W., MINNEMAN K.P. Alpha 1-adrenoceptor subtypes linked to different mechanisms for increasing Ca2+ in smooth muscle. Nature. 1987;329:333–335. doi: 10.1038/329333a0. [DOI] [PubMed] [Google Scholar]
- HAN C., HOLLINGER S., THEROUX T.L., ESBENSHADE T.A., MINNEMAN K.P. 3H-Tamsulosin binding to clonal alpha 1-adrenergic receptor subtypes expressed in human embryonic kidney 293 cells: antagonist potencies and sensitivity to alkylating agents. Pharmacol. Commun. 1995;5:117–126. [Google Scholar]
- HIEBLE J.P., BYLUND D.B., CLARKE D.E., EIKENBURG D.C., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., RUFFOLO R.J. International Union of Pharmacology. X. Recommendation for nomenclature of alpha 1-adrenoceptors: consensus update. Pharmacol. Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- HIRASAWA A., SHIBATA K., HORIE K., TAKEI Y., OBIKA K., TANAKA T., MURAMOTO N., TAKAGAKI K., YANO J., TSUJIMOTO G. Cloning, functional expression and tissue distribution of human alpha 1c-adrenoceptor splice variants. FEBS Lett. 1995;363:256–260. doi: 10.1016/0014-5793(95)00330-c. [DOI] [PubMed] [Google Scholar]
- KAVA M.S., BLUE D.J., VIMONT R.L., CLARKE D.E., FORD A.P. Alpha 1L-adrenoceptor mediation of smooth muscle contraction in rabbit bladder neck: a model for lower urinary tract tissues of man. Br. J. Pharmacol. 1998;123:1359–1366. doi: 10.1038/sj.bjp.0701748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KENNY B.A., CHALMERS D.H., PHILPOTT P.C., NAYLOR A.M. Characterization of an alpha 1D-adrenoceptor mediating the contractile response of rat aorta to noradrenaline. Br. J. Pharmacol. 1995;115:981–986. doi: 10.1111/j.1476-5381.1995.tb15907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LACHNIT W.G., TRAN A.M., CLARKE D.E., FORD A.P. Pharmacological characterization of an alpha 1A-adrenoceptor mediating contractile responses to noradrenaline in isolated caudal artery of rat. Br. J. Pharmacol. 1997;120:819–826. doi: 10.1038/sj.bjp.0700983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGRATH J.C. Evidence for more than one type of post-junctional alpha-adrenoceptor. Biochem. Pharmacol. 1982;31:467–484. doi: 10.1016/0006-2952(82)90147-2. [DOI] [PubMed] [Google Scholar]
- MICHEL A.D., LOURY D.N., WHITING R.L. Identification of a single alpha 1-adrenoceptor corresponding to the alpha 1A-subtype in rat submaxillary gland. Br. J. Pharmacol. 1989;98:883–889. doi: 10.1111/j.1476-5381.1989.tb14617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINNEMAN K.P. Alpha 1-adrenergic receptor subtypes, inositol phosphates, and sources of cell calcium. Pharmacol. Rev. 1988;40:87–119. [PubMed] [Google Scholar]
- MORIYAMA N., AKIYAMA K., MURATA S., TANIGUCHI J., ISHIDA N., YAMAZAKI S., KAWABE K. KMD-3213, a novel alpha 1A-adrenoceptor antagonist, potently inhibits the functional alpha 1-adrenoceptor in human prostate. Eur. J. Pharmacol. 1997;331:39–42. doi: 10.1016/s0014-2999(97)01009-1. [DOI] [PubMed] [Google Scholar]
- MORROW A.L., CREESE I. Characterization of alpha 1-adrenergic receptor subtypes in rat brain: a reevaluation of [3H]WB4101 and [3H]prazosin binding. Mol. Pharmacol. 1986;29:321–330. [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- MURAMATSU I., KIGOSHI S., OHMURA T. Subtypes of alpha 1-adrenoceptors involved in noradrenaline-induced contractions of rat thoracic aorta and dog carotid artery. Jpn. J. Pharmacol. 1991;57:535–544. doi: 10.1254/jjp.57.535. [DOI] [PubMed] [Google Scholar]
- MURAMATSU I., OHMURA T., HASHIMOTO S., OSHITA M. Functional subclassification of vascular alpha 1-adrenoceptors. Pharmacol. Commun. 1995;6:23–28. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAMATSU I., OHMURA T., KIGOSHI S., HASHIMOTO S., OSHITA M. Pharmacological subclassification of alpha 1-adrenoceptors in vascular smooth muscle. Br. J. Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OBIKA K., SHIBATA K., HORIE K., FOGLAR R., KIMURA K., TSUJIMOTO G. NS-49, a novel alpha 1a-adrenoceptor-selective agonist characterization using recombinant human alpha 1-adrenoceptors. Eur. J. Pharmacol. 1995;291:327–334. doi: 10.1016/0922-4106(95)90073-x. [DOI] [PubMed] [Google Scholar]
- OHMURA T., OSHITA M., KIGOSHI S., MURAMATSU I. Identification of alpha 1-adrenoceptor subtypes in the rat vas deferens: binding and functional studies. Br. J. Pharmacol. 1992;107:697–704. doi: 10.1111/j.1476-5381.1992.tb14509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'MALLEY S.S., CHEN T.B., FRANCIS B.E., GIBSON R.E., BURNS H.D., DISALVO J., BAYNE M.L., WETZEL J.M., NAGARATHNAM D., MARZABADI M., GLUCHOWSKI C., CHANG R.S. Characterization of specific binding of [125I]L-762,459, a selective alpha 1A-adrenoceptor radioligand to rat and human tissues. Eur. J. Pharmacol. 1998;348:287–295. doi: 10.1016/s0014-2999(98)00149-6. [DOI] [PubMed] [Google Scholar]
- OSHITA M., KIGOSHI S., MURAMATSU I. Three distinct binding sites for [3H]-prazosin in the rat cerebral cortex. Br. J. Pharmacol. 1991;104:961–965. doi: 10.1111/j.1476-5381.1991.tb12533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIBATA K., FOGLAR R., HORIE K., OBIKA K., SAKAMOTO A., OGAWA S., TSUJIMOTO G. KMD-3213, a novel, potent, alpha 1a-adrenoceptor-selective antagonist: characterization using recombinant human alpha 1-adrenoceptors and native tissues. Mol. Pharmacol. 1995;48:250–258. [PubMed] [Google Scholar]
- TESTA R., GUARNERI L., TADDEI C., POGGESI E., ANGELICO P., SARTANI A., LEONARDI A., GOFRIT O.N., MERETYK S., CAINE M. Functional antagonistic activity of Rec 15/2739, a novel alpha-1 antagonist selective for the lower urinary tract, on noradrenaline-induced contraction of human prostate and mesenteric artery. J. Pharmacol. Exp. Ther. 1996;277:1237–1246. [PubMed] [Google Scholar]
- WILLIAMS T.J., CLARKE D.E., FORD A.P. Whole-cell radioligand binding assay reveals alpha 1L-adrenoceptor (AR) antagonist profile for the human cloned alpha 1A-AR in chinese hamster ovary (CHO-K1) cells. Br. J. Pharmacol. 1996;119:359P. [Google Scholar]
- YAMAGISHI R., AKIYAMA K., NAKAMURA S., HORA M., MASUDA N., MATSUZAWA A., MURATA S., UJIIE A., KURASHINA Y., IIZUKA K., KITAZAWA M. Effect of KMD-3213, an alpha 1a-adrenoceptor-selective antagonist, on the contractions of rabbit prostate and rabbit and rat aorta. Eur. J. Pharmacol. 1996;315:73–79. doi: 10.1016/s0014-2999(96)00589-4. [DOI] [PubMed] [Google Scholar]
- YANG M., VERFURTH F., BUSHCER R., MICHEL M.C. Is alpha 1D-adrenoceptor protein detectable in rat tissues. Naunyn-Schmiedeberg's Arch Pharmacol. 1997;355:438–446. doi: 10.1007/pl00004966. [DOI] [PubMed] [Google Scholar]