

Abstract
The effects of several phosphodiesterase (PDE) inhibitors on the L-type Ca current (ICa) and intracellular cyclic AMP concentration ([cAMP]i) were examined in isolated rat ventricular myocytes. The presence of mRNA transcripts encoding for the different cardiac PDE subtypes was confirmed by RT–PCR.
IBMX (100 μM), a broad-spectrum PDE inhibitor, increased basal ICa by 120% and [cAMP]i by 70%, similarly to a saturating concentration of the β-adrenoceptor agonist isoprenaline (1 μM). However, MIMX (1 μM), a PDE1 inhibitor, EHNA (10 μM), a PDE2 inhibitor, cilostamide (0.1 μM), a PDE3 inhibitor, or Ro 20-1724 (0.1 μM), a PDE4 inhibitor, had no effect on basal ICa and little stimulatory effects on [cAMP]i (20–30%).
Each selective PDE inhibitor was then tested in the presence of another inhibitor to examine whether a concomitant inhibition of two PDE subtypes had any effect on ICa or [cAMP]i. While all combinations tested significantly increased [cAMP]i (40–50%), only cilostamide (0.1 μM)+Ro20-1724 (0.1 μM) produced a significant stimulation of ICa (50%). Addition of EHNA (10 μM) to this mix increased ICa to 110% and [cAMP]i to 70% above basal, i.e. to similar levels as obtained with IBMX (100 μM) or isoprenaline (1 μM).
When tested on top of a sub-maximal concentration of isoprenaline (1 nM), which increased ICa by (≈40% and had negligible effect on [cAMP]i, each selective PDE inhibitor induced a clear stimulation of [cAMP]i and an additional increase in ICa. Maximal effects on ICa were ≈8% for MIMX (3 μM), ≈20% for EHNA (1–3 μM), ≈30% for cilostamide (0.3–1 μM) and ≈50% for Ro20-1724 (0.1 μM).
Our results demonstrate that PDE1-4 subtypes regulate ICa in rat ventricular myocytes. While PDE3 and PDE4 are the dominant PDE subtypes involved in the regulation of basal ICa, all four PDE subtypes determine the response of ICa to a stimulus activating cyclic AMP production, with the rank order of potency PDE4>PDE3>PDE2>PDE1.
Keywords: Rat heart, L-type Ca2+ current, intracellular cyclic AMP, phosphodiesterase subtypes, phosphodiesterase inhibitors, MIMX (PDE1 inhibitor), EHNA (PDE2 inhibitor), cilostamide (PDE3 inhibitor), Ro20-1724 (PDE4 inhibitor), β-adrenoceptor agonist
Introduction
Phosphorylation of cardiac L-type Ca2+ channels by cyclic AMP-dependent protein kinase (PKA) plays a determinant role in the hormonal regulation of myocardial contraction. PKA increases the mean open probability of individual Ca2+ channels which results in an increase in the macroscopic L-type calcium current (ICa) (McDonald et al., 1994). Activation of PKA usually results from an increased production of cyclic AMP by activation of membrane receptors positively coupled to adenylyl cyclase via stimulatory G proteins (Gs). The best documented of such a regulation is the positive inotropic effect of sympathomimetic amines, such as isoprenaline (Hartzell et al., 1991; Hove-Madsen et al., 1996). However, cardiac myocytes, as most other cell types, also possess a negative feedback mechanism to adenylyl cyclase activation which is constituted of the cyclic nucleotide phosphodiesterases (PDEs), a family of enzymes that break down cyclic AMP into 5′-AMP (Beavo, 1995). Cyclic nucleotide PDE activity, at any given location within the cell, will counterbalance the synthesis of cyclic AMP and determine the extent of PKA activation and, hence, of protein phosphorylation. In particular, at the sarcolemmal membrane, this balance between adenylyl cyclase and PDE activities will control the degree of ICa stimulation upon hormonal activation (Fischmeister & Hartzell, 1991; Hove-Madsen et al., 1996). Other factors are involved, such as cyclic AMP compartmentation (Jurevicius & Fischmeister, 1996), PKA tethering to the membrane (Gao et al., 1997), or phosphatase activity (Wiechen et al., 1995).
Cyclic nucleotide PDEs exist in multiple molecular forms (Stoclet et al., 1995; Loughney & Ferguson, 1996) and at least four different subtypes have been shown to coexist in the heart muscle (Shahid & Nicholson, 1990; Bode et al., 1991; Dubois et al., 1993; Taira et al., 1993; Engels et al., 1994; Kostic et al. 1997): (1) a Ca2+/calmodulin-activated PDE (PDE1) which hydrolyzes cyclic AMP and cyclic GMP; (2) a cyclic GMP-stimulated PDE (PDE2) which hydrolyzes cyclic AMP and cyclic GMP; (3) a cyclic GMP-inhibited PDE (PDE3) which hydrolyzes cyclic AMP with high affinity (low Km) and (4) and a cyclic GMP-independent PDE (PDE4) which hydrolyzes selectively cyclic AMP with high affinity. All these PDEs can be inhibited by xanthine derivatives, such as 3-isobutyl-1-metylxanthine (IBMX) or caffeine, which have been frequently used to evaluate the role of PDEs in the metabolism of cyclic AMP in cardiac preparations (see e.g. Katano & Endoh, 1993).
The use of selective or non selective PDE inhibitors has provided some information about the functional importance of PDEs in ICa regulation (Fischmeister & Hartzell, 1991). IBMX stimulates basal ICa in rat (Méry et al., 1991) and guinea-pig ventricular myocytes (Levi et al., 1989; Ono & Trautwein, 1991; Shiriyama & Pappano, 1996) and enhances the stimulatory effect of cyclic AMP in frog ventricular myocytes (Fischmeister & Hartzell, 1990; Méry et al., 1995). Erythro-9-[2-Hydroxy-3-nonyl]adenine (EHNA), a selective PDE2 inhibitor (Méry et al., 1995; Podzuweit et al., 1995), stimulates basal ICa in human atrial cells (Rivet-Bastide et al., 1997), and antagonizes the inhibitory effect of intracellular cyclic GMP or NO-donors on isoprenaline- or cyclic AMP-stimulated ICa in frog ventricular myocytes (Méry et al., 1995). Inhibition of PDE3 with milrinone results in an increase in basal ICa in human atrial myocytes (Kirstein et al., 1995). Similarly, application of intracellular cyclic GMP in guinea-pig ventricular (Shiriyama & Pappano, 1996; Ono & Trautwein, 1991) or human atrial myocytes (Rivet-Bastide et al., 1997) or activation of guanylyl cyclase by NO donors in human atrial myocytes (Kirstein et al., 1995) leads to an increase in ICa mediated by cyclic GMP inhibition of PDE3. Ro20-1724 or rolipram, two selective PDE4 inhibitors, enhance the response of ICa to cyclic AMP, isoprenaline or forskolin in frog cardiomyocytes (Fischmeister & Hartzell, 1990; Méry et al., 1995).
Despite these previous studies, to our knowledge the respective contribution of any given PDE subtype to the regulation of ICa or to the control of intracellular cyclic AMP concentration has not been examined at once in the same mammalian cardiac preparation. Thus, the aim of the present study was to get some insight into the respective roles of the four different PDE subtypes in the regulation of cardiac ICa and intracellular cyclic AMP levels in freshly isolated rat ventricular myocytes. After verifying the expression of PDE1-4 subtypes in rat heart by RT–PCR, we have examined how selective inhibition of one PDE subtype or simultaneous inhibition of two PDE subtypes modulate basal or isoprenaline-stimulated ICa and intracellular cyclic AMP. ICa was recorded using the whole-cell patch-clamp technique and intracellular cyclic AMP was measured using radioimmunoassay. Selective PDE inhibition was achieved using 8-methoxymethyl-3-isobutyl-1-methylxanthine (MIMX) for PDE1, EHNA for PDE2, cilostamide for PDE3, and Ro20-1724 for PDE4 (for reviews, see Beavo, 1995; Stoclet et al., 1995).
A preliminary account of this work was presented at the XVIIIth European Section Meeting of the International Society for Heart Research, Versailles, France, July 1997 (Verde et al., 1997).
Methods
The investigation conforms with the European Community guiding principles in the care and use of animals (86/609/CEE, CE Off J noL358, 18 December, 1986) and the French decree no87/748 of October 19, 1987 (J Off République Française, 20 October, 1987, pp. 12245–12248). Authorizations to perform animal experiments according to this decree were obtained from the French Ministère de l'Agriculture et de la Forêt (no04226, April 12, 1991).
Preparation of rat ventricular myocytes
Rat ventricular myocytes were obtained by retrograde perfusion from hearts of male Wistar rats (200–250 g) as described (Méry et al., 1991), with slight modifications. Briefly, the rats were subjected to anaesthesia by intraperitoneal injection of penthotal and hearts were excised rapidly. The ionic composition of the Ca2+-free Ringer solution was as follows (in mM): NaCl 117, KCl 57, NaHCO3 4.4, KH2PO4 1.5, MgCl2 1.7, D-glucose 11.7, sodium phosphocreatine 10, taurine 20, and HEPES 21, adjusted to pH 7.1 with NaOH at room temperature. For enzymatic dissociation, 1 mg ml−1 collagenase A (Boehringer Mannhein, Mannhein, Germany) and 300 μM EGTA were added to the Ca2+-free Ringer solution, so that the free Ca2+ concentration was adjusted to 20 μM. The hearts were perfused retrogradedly at a constant flow of 6 ml min−1 and at 37°C by Ca2+-free solution during 5 min followed by 1 h of perfusion at 4 ml min−1 with the same solution containing collagenase. The ventricles were then separated from atria, chopped finely and agitated gently to dissociate individual cells. The resulting cell suspension was filtered on a gauze and the cells were allowed to settle down. The supernatant was discarded and cells resuspended four more times in Ca2+-free solution containing a progressively increasing calcium concentration. The cells were maintained at 37°C until use.
Measurement of rat PDE mRNA expression by reverse transcriptase polymerase chain reaction (RT–PCR)
Total RNA was prepared from rat isolated ventricular myocytes and rat ventricular tissue using the Trizol RNA purification system (Gibco BRL, Cergy-Pontoise, France). All RNAs were checked in 1% formaldehyde agarose gel. cDNA was prepared from mRNA with random hexanucleotide primers (20 pmol μg−1 RNA) using MMLV reverse transcriptase and the conditions recommended by the manufacturer (Clontech, Palo Alto, CA, U.S.A.). Fifty ng of cDNA were amplified in 50 μl PCR reaction mixture (200 μM dNTPs final concentrations) containing 2.5 U of Taq polymerase in the buffer supplied by the manufacturer (Bioprobe Sytems, Montreuil, France), and 1 μM primers. Oligonucleotide primers designed against the C-terminal region of each PDE sub-type were as follows:
PDE1C (Genbank accession number L41045): Forward primer 5′-TTTTCTCCTCTGTGTGACCG-3′, reverse primer 5′-GTGTTCCGTTGACTTGACCT-3′, fragment size 505 bp;
PDE2A2 (Genbank accession number U21101): Forward primer 5′-GAAGGACTATCAGCGAATGC-3′, reverse primer 5′-GGATGGTGAACTTGTGGGAC-3′, fragment size 461 bp;
PDE3B1 (Genbank accession number Z22867): Forward primer 5′-CACCCAGGAAGAACAAATGC-3′, reverse primer 5′-AAGCCAGCAGCATCATAGGA-3′, fragment size 551 bp;
PDE4A5 (Genbank accession number L27057): Forward primer 5′-TACAGTGGTGGAAGTGGCAG-3′, reverse primer 5′-GAGCAGAGATGATGGCAGAA-3′, fragment size 278 bp;
PDE4 B2 (Genbank accession number L27058): Forward primer 5′-GTTCTCCTCTCTACGCCAGCA-3′, reverse primer 5′-ACTTGGTAGGGTTGCTCAGGTC-3′, fragment size 454 bp;
PDE4 D3 (Genbank accession number U09457): Forward primer 5′-GCGTCCTCCTCCTTGATAACTATT-3′, reverse primer 5′-CTGACTCGCCATCTTCCTCTAA-3′, fragment size 447 bp.
The PCR products were separated on 1.7% agarose gel containing 0.01% ethidium bromide and photographed under UV irradiation at 320 nm. To assess relative quantities of cDNA from each sample, a second PCR amplification was conducted with primers directed to the rat housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as previously described (Grimaldi et al., 1998). All PCR procedures were performed as follows: 40 cycles and 35 cycles for isolated myocytes and ventricular tissue respectively (45 s at 94°C, 50 s at 50°C and 1 min at 72°C) and a final elongation (7 min at 72°C).
Cyclic AMP radioimmunoassay
Freshly isolated ventricular myocytes were prepared as described above from hearts of adult male Wistar rats weighting 200–250 g. Rod-shaped myocytes were counted using a Mallassez cell and resuspended in an adequate volume of the Ca2+-free Ringer solution supplemented with 1 mM Ca2+, in order to obtain a density of 105 cell ml−1. Each incubation tube contained 500 μl of this cell suspension to which was added successively 5 μl of a 200× stock solution of the appropriate drug and 500 μl of Ringer solution. Each condition including control (i.e. in absence of any drug) was tested in triplicate. Incubation was carried out at room temperature (20–24°C) and lasted 15 min, allowing the cells to settle down. After this period, the supernatant was discarded and replaced by 500 μl of cold (4°C) ethanol to stop the reaction. Ethanol was evaporated (1 h at 37°C and low pressure) and the pellet was homogenized in 200 μl of buffer for cyclic AMP assay (cyclic AMP radioimmunoassay kit, Immunotech, Marseille, France). After centrifugation at 4500 r.p.m. for 5 min, the supernatants were diluted ten times in the same buffer and cyclic AMP was assayed according to the instructions provided by the manufacturer.
Electrophysiological experiments
The whole cell configuration of the patch-clamp technique was used to record the high-threshold L-type calcium current (ICa) on Ca2+-tolerant rat ventricular myocytes. The cell was routinely depolarized every 8 s from −50 to 0 mV for 400 ms. The use of a holding potential of −50 mV allowed the elimination of fast sodium currents. Potassium currents were blocked by replacing all K+ ions with intracellular and extracellular Cs+. For the determination of current-voltage relationships for ICa (see Figure 4A) and ICa inactivation curve (see Figure 4B), a double pulse voltage clamp protocol was used (Kirstein et al., 1995). Briefly, every 4 s, the membrane potential of the cell, which was normally maintained at its holding value of −50 mV, experienced the following sequence of events: −50 mV for 10 ms, different potentials values ranging from −100 to +100 mV for 200 ms, −50 mV for 3 ms, and 0 mV for 200 ms (see inset in Figure 4B). Voltage-clamp protocols were generated by a challenger/09-VM programmable function generator (Kinetic Software, Atlanta, GA, U.S.A.). The cells were voltage-clamped using a patch-clamp amplifier (model RK-400; Bio-Logic, Claix, France). Currents were analogue-filtered at 3 KHz and digitally sampled at a frequency of 10 kHz using a 12-bit analogue-to-digital converter (DT2827; Data translation, Marlboro, MA, U.S.A.) connected to a PC compatible (386/33 Systempro; Compaq Computer Corp., Houston, TX, U.S.A.). All experiments were done at room temperature (21–25°C).
Figure 4.
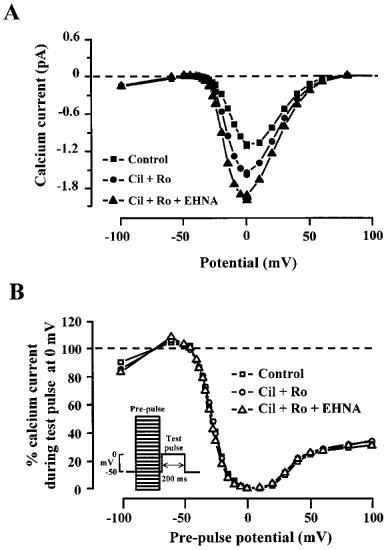
Effects of cilostamide, Ro20-1724 and EHNA on the current-voltage and inactivation relationships of ICa in a rat ventricular myocyte. (A) current-voltage relationships and (B) inactivation curves for ICa obtained in the experiment shown in Figure 3 in control (squares), in the presence of 0.1 μM cilostamide and 0.1 μM Ro20-1724 (Cil+Ro, circles), and in the presence of 0.1 μM cilostamide, 0.1 μM Ro20-1724 and 10 μM EHNA (Cil+Ro+EHNA, triangles). Inactivation curves were obtained using a double-pulse protocol indicated in the inset and described in Methods.
Solutions and drugs
Control external Cs+ Ringer solution contained (in mM): NaCl 107.1, CsCl 20, NaHCO3 4, NaH2PO4 0.8, MgCl2 1.8, CaCl2 1.8, D-glucose 5, sodium pyruvate 5 and HEPES 10, adjusted to pH 7.4 with NaOH. Control or drug-containing solutions were applied to the exterior of the cell by placing the cell at the opening of a 250 μm inner diameter capillary tubing from which the external solution was flowing at a rate of 10 μl min (Méry et al., 1991). Patch electrodes (0.5–1 MΩ) were filled with control internal solution containing (in mM): CsCl 119.8, EGTA 5, MgCl2 4, sodium phosphocreatine 5, Na2ATP 3.1, Na2GTP 0.42, CaCl2 (pCa 8.5) 0.062 and HEPES 10, adjusted to pH 7.3 with CsOH.
Isoprenaline, IBMX, MIMX and EHNA were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Cilostamide was purchased from Calbiochem (Meudon, France), and Ro20-1724 was gently provided by Hoffman-La-Roche (Switzerland).
Data analysis
The maximal amplitude of ICa was measured as the difference between the peak inward current and the end-pulse current (I200 or I400), which was the current amplitude at the end of the 200 or 400 ms duration pulse (Kirstein et al., 1995). Currents were not compensated for capacitive and leak currents. Cell membrane capacitance and series resistance were measured by exponential analysis of current responses to 1 mV step changes in membrane potential. Membrane capacitance was 130.2±5.0 pF and series resistance 4.0±0.2 MΩ (n=137 different cells). The on-line analysis was made possible by programming a PC compatible computer in Pascal language to determine for each depolarization, peak, and steady state current value. The results are expressed as mean±s.e.mean. In each experimental condition the effects of the drugs tested on ICa or cyclic AMP levels are expressed as percent change with respect to the values of these parameters under basal conditions, i.e., in the absence of any hormonal stimulation. The variations in ICa or cyclic AMP induced by the PDE inhibitors were tested for statistical significance by Student's t-test.
Results
Expression of PDE (1–4) in rat heart
Semi-quantitative RT–PCR analysis with primers specific for rat PDE genes was performed on rat ventricular tissue as well as on isolated rat ventricular myocytes in order to check for the expression of PDE1C, PDE2A, PDE3B and PDE4 (A, B and D) subtypes (Figure 1). Earlier studies have provided direct or indirect evidence for the presence of these PDE gene products in rat cardiac tissues (Bode et al., 1991; Taira et al., 1993; Engels et al., 1994; Kostic et al., 1997). Figure 1 shows that rat ventricles (Figure 1A) as well as isolated rat ventricular myocytes (Figure 1B) express significant amounts of transcripts for each PDE subtype, suggesting the presence of these enzymes in this tissue. In addition, we found that PDE2 and PDE4 transcripts were more abundant than those coding for PDE1 and PDE3.
Figure 1.
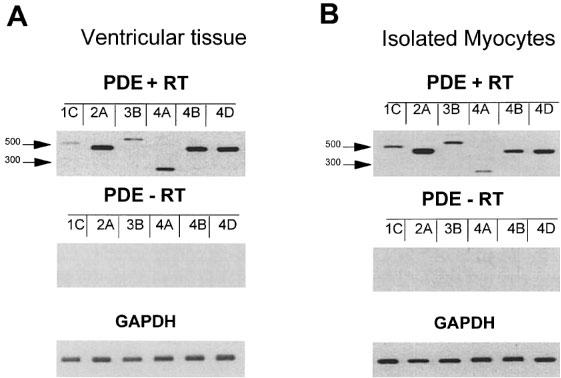
Expression analysis of PDE1, PDE2, PDE3 and PDE4 transcripts in rat ventricle. RT–PCR analysis was performed on mRNA extracted from whole ventricular tissue (A) and isolated ventricular myocytes (B) and in the presence (+RT) or in the absence (−RT) (as a negative control) of the reverse transcriptase. The PCR products were analysed on a 1.7% agarose gel and photographs of the ethidium bromide stained gels are shown. The PCR primers used for this analysis and expected length of the PCR products are described in Methods. Positive controls were performed in a second PCR using rat glyceraldehyde 3-phosphate dehydrogenase primers (GAPDH). Positions of two molecular weight markers are indicated in bp. Note that the PCR procedure was 40 cycles and 35 cycles for isolated myocytes and ventricular tissue respectively. This figure is representative of three separate determinations of the PDE mRNA expressions obtained by RT–PCR.
Effect of PDE inhibitors on basal ICa
ICa was measured in isolated rat ventricular myocytes using the whole-cell patch-clamp technique (Hamill et al., 1981). The effects of PDE inhibitors were first examined on basal ICa, i.e. in the absence of a stimulated cyclic AMP production. Basal ICa amplitude was on average 827.3±36.8 pA at 0 mV membrane potential, and mean ICa density, which represents the ratio of ICa amplitude to membrane capacitance, was 6.9±0.3 pA/pF (n=137). Figure 2 shows a typical experiment in which the effect of an extracellular application of Ro20-1724 (0.3 μM), a PDE4 inhibitor, was compared with the effect of 100 μM IBMX, a broad-spectrum PDE inhibitor. As shown, application of Ro20-1724 produced no effect on basal ICa. Table 1 shows that similar results were obtained with MIMX (1 μM), a PDE1 inhibitor, EHNA (10 μM), a PDE2 inhibitor, and cilostamide (0.1 μM), a PDE3 inhibitor, when the compounds were applied under basal conditions.
Figure 2.
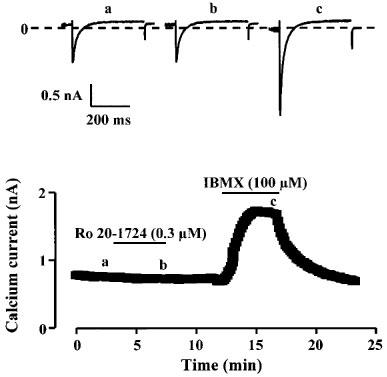
Effects of Ro20-1724 and IBMX on ICa in a rat ventricular myocyte. Each symbol corresponds to a measure of ICa at 0 mV obtained every 8 s. The cell was first superfused with control Cs+ Ringer solution and then exposed to the drugs during the periods indicated by the solid lines. 0.3 μM Ro20-1724 had no effect on ICa, while application of 100 μM IBMX produced a strong and reversible stimulation of ICa The individual current traces shown on the upper part were obtained at the times indicated by the corresponding letters in the bottom graph. The dotted line indicates the zero current level.
Table 1.
Effect of PDE inhibitors and isoprenaline on basal ICa

The lack of effect of selective inhibitors of PDE1-4 on ICa may indicate that none of the four PDEs is active under basal conditions. Alternatively, it may indicate that inhibition of a single subtype of PDE does not lead to a sufficient increase in cyclic AMP to stimulate basal ICa. To discriminate between these two hypotheses, we examined the effect of IBMX, a broad-spectrum PDE inhibitor, on basal ICa. The individual experiment of Figure 2 and the summary data of Table 1 show that application of 100 μM IBMX, a concentration which inhibits all PDE subtypes, produced a >2 fold and reversible stimulation of ICa. This result indicates that, while inhibition of any single PDE subtype is insufficient to enhance basal ICa, complete inhibition of all PDEs produces a sufficient accumulation of cyclic AMP to activate basal ICa.
We next wanted to examine which PDE subtype had a dominant activity under basal condition. To do this, we tested different combinations of selective PDE inhibitors for their effect on basal ICa. In the experiment shown in Figure 3, the myocyte was initially exposed to 0.1 μM cilostamide which had no effect on basal ICa (see also Table 1). However, when Ro20-1724 (0.1 μM) was applied to the cell in combination with cilostamide, ICa increased by ≈40%. Addition of EHNA (10 μM) on top of the two other PDE inhibitors induced a further increase in ICa. All the stimulatory effects were completely reversible upon washout of the drugs. Figure 4 shows the current-voltage (Figure 4A) and the inactivation relationships (Figure 4B) of ICa obtained in the experiment of Figure 3, under control condition, in the presence of cilostamide+Ro20-1724 or cilostamide+Ro20-1724+ EHNA. As shown, the combination of cilostamide and Ro20-1724 and the addition of EHNA increased ICa essentially by the same amount at every membrane potential (Figure 4A) which indicates that the stimulatory effects of the drugs were not dependent on membrane potential. The inactivation curve of ICa (Figure 4B) was also unchanged by the presence of the drugs which indicates that neither the voltage-dependent nor the Ca-dependent inactivation process (McDonald et al., 1994) was modified.
Figure 3.
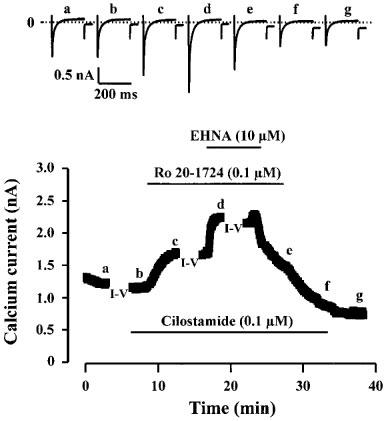
Effects of cilostamide, Ro20-1724 and EHNA on ICa in a rat ventricular myocyte. The cell was superfused for several minutes with a control solution and then challenged with different drugs during the periods indicated by the solid lines. While application of 0.1 μM cilostamide had no effect on basal ICa, application of 0.1 μM Ro20-1724 on top of cilostamide or of EHNA (10 μM) on top of cilostamide and Ro20-1724 induced a net increase in ICa. Test pulses were 0 mV except during the recording of current-voltage (I–V) relationships. The individual current traces shown on the upper part were obtained at the times indicated by the corresponding letters in the bottom graph. The dotted line indicates the zero current level.
Each of the selective PDE inhibitors was tested in the presence of another inhibitor. This led to six different experimental conditions, and the results of these experiments are summarized in Table 1. Of all the combinations tested, only Ro20-1724 and cilostamide had a significant stimulatory effect on basal ICa. The dual inhibition of PDE3 and PDE4 resulted in a ≈50% increase in basal ICa, an effect which corresponded to ≈40% of the stimulatory effect observed in the presence of 100 μM IBMX (Table 1). At this concentration, IBMX increased maximally ICa, since its effect was not statistically different from the stimulation induced by a saturating concentration (1 μM) of isoprenaline (Table 1). PDE1 inhibition (with 1 μM MIMX) had no effect on basal ICa under any condition tested (data not shown). PDE2 inhibition (with 10 μM EHNA) had no effect unless PDE3 and PDE4 were simultaneously blocked (data not shown). As shown in Table 1, the concomitant inhibition of PDE2, PDE3 and PDE4 with a combination of EHNA, cilostamide and Ro20-1724 led to a similar increase in ICa as that produced by IBMX or isoprenaline.
Effect of PDE inhibitors on isoprenaline-stimulated ICa
We next examined the respective contribution of each PDE subtype in the regulation of phosphorylated Ca2+ channels. To do this, we examined the effects of each selective PDE inhibitor on ICa which had been stimulated by isoprenaline. However, it was important to use a non-saturating concentration of the β-adrenoceptor agonist in order to allow ICa to be further increased by the drugs. A concentration of 1 nM isoprenaline was chosen, which increased ICa by 38.1±4.1% (n=82, Figure 5), i.e. which produced ≈30% of the maximal stimulatory effect observed with 1 μM isoprenaline or 100 μM IBMX (Table 1).
Figure 5.
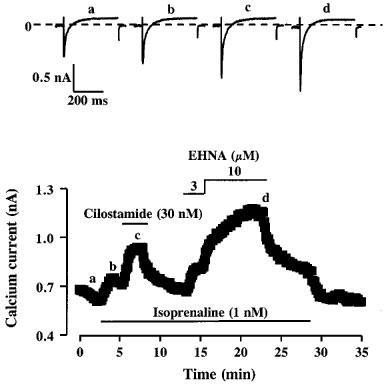
Effects of cilostamide and EHNA on ICa pre-stimulated by isoprenaline. The cell was superfused for few minutes with a control solution and then challenged with different drugs during the periods indicated by the solid lines. Application of 1 nM isoprenaline slightly increased ICa. Addition of 30 nM cilostamide or of 3 or 10 μM EHNA in the continuing presence of isoprenaline induced an additional and reversible increase in ICa. The individual current traces shown on the upper part were obtained at the times indicated by the corresponding letters in the bottom graph. The dotted line indicates the zero current level.
Figure 5 shows a typical experiment in which the effects of PDE2 and PDE3 inhibition with EHNA and cilostamide, respectively, were successively tested on a rat ventricular myocyte exposed to 1 nM isoprenaline. While, as shown above, 100 nM cilostamide or 10 μM EHNA had no effect on basal ICa, as low as 30 nM cilostamide or 3 μM EHNA induced a net and reversible stimulation of ICa on top of that obtained with isoprenaline alone. A similar finding was obtained with PDE4 inhibition by Ro20-1724. Indeed, Figure 6 shows that application of 30 nM Ro20-1724 induced a net further increase in ICa in the presence of 1 nM isoprenaline, while this drug had no effect on basal ICa when used at a 3 fold higher concentration (Table 1).
Figure 6.
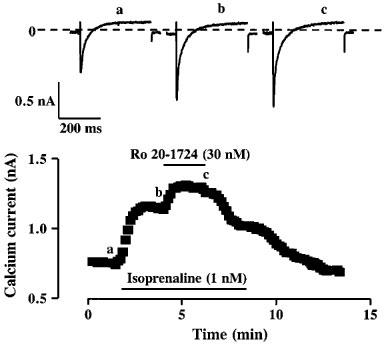
Effects of R020-1724 on ICa pre-stimulated by isoprenaline. The cell was superfused for few minutes with a control solution and then challenged with different drugs during the periods indicated by the solid lines. Application of 1 nM isoprenaline increased ICa. Addition of 30 nM Ro 20-1724 in the continuing presence of isoprenaline induced an additional and reversible increase in ICa. The individual current traces shown on the upper part were obtained at the times indicated by the corresponding letters in the bottom graph. The dotted line indicates the zero current level.
Figure 7 summarizes the results of several similar experiments in which various concentrations of EHNA (Figure 7A), cilostamide (Figure 7B) and Ro20-1724 (Figure 7C) were tested for their effect on ICa pre-stimulated by 1 nM isoprenaline. While all these drugs induced an additional increase in ICa, the maximal stimulatory effect was the highest with Ro20-1724 (≈50% at 30–100 nM), intermediate with cilostamide (≈30% at 0.3–1 μM) and the lowest with EHNA (≈20% at 1–10 μM). Under the same conditions (with 1 nM isoprenaline), PDE1 inhibition with 3 μM MIMX produced only a weak but significant (P<0.05) additional stimulation of ICa (8.1±1.2%, n=5).
Figure 7.
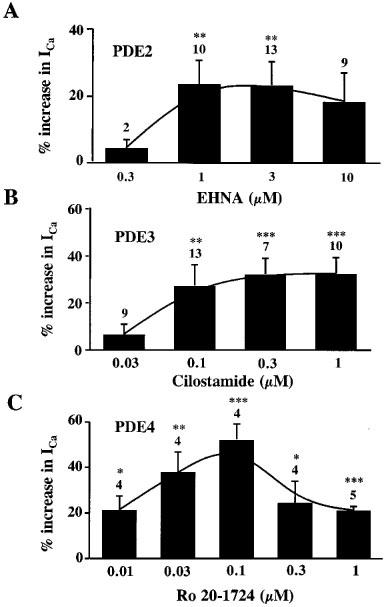
Effects of EHNA, cilostamide and R020-1724 on ICa pre-stimulated by isoprenaline. Summary of the effects of different concentrations of EHNA (A), cilostamide (B) and Ro20-1724 (C) on ICa pre-stimulated by 1 nM isoprenaline in rat ventricular myocytes. The bars indicate the means and the lines the s.e.mean of the numbers of experiments indicated near the bars. The effects are expressed in % variation over the amplitude of ICa in the presence of isoprenaline alone. Statistical significant differences from ICa amplitude in isoprenaline alone are indicated as: *P<0.05; **P<0.01; ***P<0.005.
Effect of PDE inhibitors on intracellular cyclic AMP concentration
To get further insights into the mechanism of action of the different PDE inhibitors tested on ICa, we examined their effects on intracellular cyclic AMP concentration ([cAMP]i) in isolated rat ventricular myocytes. Figure 8 shows the results of cyclic AMP assays performed in ventricular myocytes isolated from three different rat hearts (symbols) together with the mean values (bars). Although one preparation responded by a large increase in [cAMP]i with any of the four PDE inhibitors tested (Figure 8A, filled circles), on average the effects of MIMX (1 μM), EHNA (10 μM), cilostamide (0.1 μM) and Ro20-1724 (0.1 μM) on basal [cAMP]i were small (20–30%) and not statistically significant (Figure 8A). However, when the inhibitors were used in combination to achieve a concomitant inhibition of two PDE subtypes (tested only on PDE2-4), [cAMP]i increased by 40–50% and the effect became statistically significant (P<0.05, Figure 8A). [cAMP]i increased further (to ≈80%) when all three PDE subtypes were inhibited, an effect which was similar to that obtained with a maximal concentration of IBMX (100 μM, Figure 8A). Figure 8B shows the effects of the selective PDE inhibitors on [cAMP]i when applied in the presence of a sub-maximal concentration of isoprenaline (1 nM). Although the preparation that responded to any single PDE inhibitor also responded by a large increase in [cAMP]i with 1 nM isoprenaline (Figure 8B, filled circles), the average stimulatory effect of 1 nM isoprenaline was small (20%) and not statistically significant (Figure 8B). However, in the presence of isoprenaline, any single application of PDE inhibitor (tested only on PDE2-4) induced a large (60–70%) increase in [AMP]i (Figure 8B), which was similar to the effect obtained with 1 μM isoprenaline (Figure 8B) or 100 μM IBMX (Figure 8A).
Figure 8.
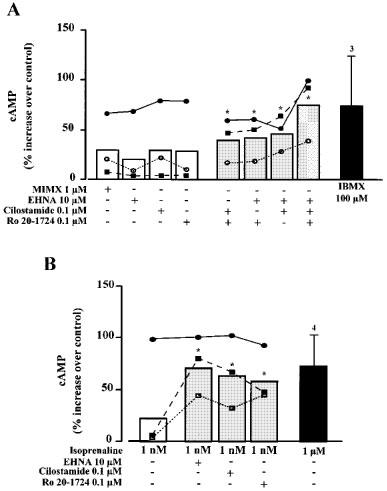
Effects of PDE inhibitors and isoprenaline on [cAMP]i in isolated rat ventricular myocytes. The selective PDE inhibitors MIMX (1 μM), EHNA (10 μM), cilostamide (0.1 μM) and Ro20-1724 (0.1 μM) were tested as indicated in three different preparations of rat ventricular myocytes (corresponding to the different symbols used in A and B) in the absence (A) or presence (B) of 1 nM isoprenaline. The effects are expressed in % increase over basal cyclic AMP concentration which was, respectively, 59 (○), 52 (▪) and 22 (•) pmoles per mg protein, assuming that each myocyte contained 15 ng protein. Each individual measurement was done in triplicate. The bars indicate the average response in each condition. Separate experiments were performed with IBMX (100 μM, A) and isoprenaline (1 μM, B), and the corresponding bars indicate the mean and the s.e.mean of the number of preparations indicated near the bars. Statistical significant differences from basal cyclic AMP concentration are indicated as: *P<0.05.
Discussion
In the recent years, it has become well established that cardiac ICa as well as myocardial contractility is modulated by cyclic AMP-dependent phosphorylation. An increase in cyclic AMP concentration activates cyclic AMP-dependent protein kinase (PKA), which phosphorylates various proteins, including the L-type Ca2+ channels, leading to an increase in the mean open probability of individual channels (McDonald et al., 1994; Hartzell et al., 1991; Hove-Madsen et al., 1996). The cyclic AMP signal, which is generated at the level of the sarcolemmal membrane by adenylyl cyclase, is terminated by cyclic nucleotide phosphodiesterases, a class of enzymes that hydrolyse cyclic AMP into 5′-AMP (Beavo, 1995).
At present, four different subtypes of PDE (PDE1-4) have been characterized in mammalian hearts (Taira et al., 1993; Engels et al., 1994; Kostic et al., 1997). Their intracellular localization and/or respective activities may vary depending on the animal species and/or the cardiac tissue (for reviews, see Beavo, 1995; Stoclet et al., 1995). In our study, we have confirmed by RT–PCR the presence of mRNA transcripts encoding for all four PDE subtypes in isolated rat ventricular myocytes.
Investigation of their functional role has been made possible by the development of pharmacological agents that inhibit selectively their activity. A number of compounds, such as the bypiridine derivatives amrinone and milrinone, have long been used as selective PDE3 inhibitors with therapeutic applications. Among these, cilostamide is by far the most potent inhibitor of PDE3 (Reeves & England, 1990). The antidepressants Ro-20-1724 or rolipram potentiate the positive inotropic effect of forskolin on the heart by acting as selective inhibitors of PDE4 (Muller et al., 1990). The adenosine deaminase inhibitor EHNA was recently characterised as a potent PDE2 inhibitor, which is also selective of this PDE subtype as far as other PDEs are concerned (Méry et al., 1995; Podzuveit et al., 1995). Potent and selective inhibitors of PDE1 are still to come, although the 8-methoxymethyl-derivative of IBMX, MIMX, has been shown to possess some selectivity at the PDE1 subtype (Wells & Miller, 1988).
In the present study, we used the broad-spectrum PDE inhibitor IBMX as well as the selective inhibitors MIMX, EHNA, cilostamide and Ro20-1724 to assess the respective roles of PDE1-4 in the regulation of basal and stimulated ICa and [cAMP]i in rat ventricular myocytes. Some of these PDE inhibitors have been shown earlier to increase ICa by elevating [cAMP]i. For instance, application of IBMX leads to a large stimulation of basal ICa in guinea-pig (Mubagwa et al., 1997), rabbit (Akita et al., 1994; Kajimoto et al., 1997) and human cardiomyocytes (Kajimoto et al., 1997). Similarly, we found that IBMX also increased basal ICa in rat ventricular myocytes. This indicates that, in all these preparations: (i) there is a substantial basal activity of adenylyl cyclase which determines a basal cyclic AMP synthesis, and (ii) there is a significant basal PDE activity which limits the degree of cyclic AMP-dependent phosphorylation of L-type Ca2+-channels. The situation in mammals differs from what was found in frog ventricular myocytes, where IBMX had no effect on ICa unless the current had been pre-stimulated by isoprenaline, forskolin or cyclic AMP (Fischmeister & Hartzell, 1990; 1991). Thus, in frog cardiomyocytes, unlike in most mammalian species, basal adenylyl cyclase activity is insufficient to generate enough cyclic AMP to lead to a stimulation of ICa even when all PDE activity is blocked.
For this reason, selective PDE inhibitors, such as EHNA or milrinone, have no effect on basal ICa in frog myocytes (Fischmeister & Hartzell, 1990; Méry et al., 1995) while they do stimulate basal ICa in human atrial myocytes (Kirstein et al., 1995; Kajimoto et al., 1997; Rivet-Bastide et al., 1997). Similarly, PDE3 inhibitors stimulate basal ICa in rabbit cardiomyocytes (Kajimoto et al., 1997) and exert a positive inotropic effect in guinea-pig left atria (Muller et al., 1990). However, in this study, we found that none of the selective PDE inhibitors used alone had any significant effect on basal ICa or [cAMP]i in rat ventricular myocytes. Thus, inhibition of a single PDE subtype is insufficient to raise cyclic AMP concentration near Ca2+ channels to the threshold concentration necessary for ICa stimulation. The likely explanation for this is that other PDE subtypes accounted for cyclic AMP hydrolysis under such condition. To test this hypothesis, we tried to block two PDE subtypes at the same time using combinations of PDE inhibitors. The only ‘duo' of inhibitors that was found to enhance basal ICa was cilostamide and Ro20-1724, indicating that PDE3 and PDE4 were the dominant PDE subtypes regulating basal cyclic AMP concentration and ICa in rat ventricular myocytes. These results are in agreement with a number of biochemical studies showing a much higher specific activity of PDE3 and PDE4 compared with PDE1 and PDE2 in rat heart (Shahid & Nicholson, 1990; Bode et al., 1991; Dubois et al., 1993; Picq et al., 1996). Also, functional experiments have shown that inhibition of PDE3 or PDE4 alone had no effect on the contractile activity of rat heart while a dual PDE3 and PDE4 inhibition produced a strong positive inotropic effect (Shahid & Nicholson, 1990).
Surprisingly, however, we found that not only cilostamide and Ro20-1724 but also any other ‘duo' combination of PDE2-4 inhibitors induced a substantial elevation in [cAMP]i in rat ventricular myocytes. This difference between ICa and [cAMP]i measurements may indicate the presence of different pools of cyclic AMP inside cardiac myocytes, which are not all associated with L-type Ca2+ channels. Such a hypothesis is supported by recent findings of a strong cyclic AMP compartmentation near L-type Ca2+ channels in frog ventricular myocytes exposed to isoprenaline (Jurevicius & Fischmeister, 1996). It is also supported by a large body of evidence for distinct intracellular localization of the different PDE subtypes. For instance, a substantial amount of PDE3 and PDE4 activity was found in a membrane-bound fraction of rat cardiomyocytes (Weishaar et al., 1987; Shahid et al., 1990; Kaasic & Ohisalo, 1996; Okruhlicova et al., 1996) which may accentuate the role of these PDE subtypes in the control of sarcolemmal processes, such as cyclic AMP-dependent phosphorylation of L-type Ca2+ channels. PDE3 and PDE4 have also a 5–30 fold lower Km for cyclic AMP than PDE2 (Bode et al., 1991; Beavo, 1995), which should attenuate the role of PDE2 in ICa regulation when [cAMP]i near the membrane is low. This may explain why EHNA did not affect ICa unless PDE3 and PDE4 were blocked or when their activity was saturated by an enhanced cyclic AMP synthesis in the presence of isoprenaline.
Regulation of ICa by PDE1 in heart remains unclear. In this study, we found that the PDE1 inhibitor MIMX (Wells & Miller, 1988) had negligible effects on basal ICa and increased only by 8% isoprenaline stimulated ICa. However, a major drawback of our experiments is that the solution used in the patch-pipette was adjusted to a free Ca2+ concentration of pCa 8.5 using 5 mM EGTA. Although the Ca2+ buffering capacity of the pipette solution should only weakly affect the Ca2+ concentration near the membrane, as evidenced by the presence of a large Ca2+-mediated inactivation of ICa under these conditions, it might abolish a Ca2+-calmodulin dependent activation of PDE1 in a remote part of the cell. Nystatin-perforated patch-clamp experiments would be necessary to elucidate the role of PDE1 in the control of ICa. However, it should be mentioned that biochemical studies have shown that rat heart possesses a much lower PDE1 activity as compared to other PDE subtypes (Dubois et al., 1993, Shahid & Nicholson, 1990, Bode et al., 1991).
To the exception of MIMX, all other PDE inhibitors tested exerted a clear stimulation of ICa after application of 1 nM isoprenaline. This low concentration of the β-adrenoceptor agonist moderately increased the activity of adenylyl cyclase localized in the membrane, bringing the cyclic AMP concentration above threshold for cyclic AMP-dependent phosphorylation of L-type Ca2+ channels. In these conditions, EHNA, cilostamide or Ro20-1724 were found to further enhance ICa as well as [cAMP]i. Thus, unlike what was observed under basal conditions, inhibition of any single PDE2-4 subtype stimulates Ca2+ channel activity when the intracellular cyclic AMP level is above threshold. At their maximal effects, the rank order of potency of the different PDE inhibitors on isoprenaline stimulated ICa was Ro20-1724>cilostamide>EHNA>MIMX. However, in the case of Ro20-1724, increasing the concentration above the maximal effective concentration (100 nM) produced lower stimulatory effects on ICa (Figure 7C). While, we have no clear explanation for this effect, a similar phenomenon was observed in contractile experiments on guinea-pig heart by Muller et al. (1990) with another PDE4 inhibitor, rolipram. An interesting possibility is that the increase in cyclic AMP and PKA activity that results from PDE4 inhibition leads to phosphorylation and activation of PDE4 as has been shown recently in thyroid cells (Sette & Conti, 1996).
In summary, our results demonstrate that PDE2, PDE3 and PDE4 regulate ICa in rat ventricular myocytes. PDE3 and PDE4 are the dominant PDE subtypes involved in the regulation of basal ICa. Each of these two PDEs possesses a sufficient enzymatic activity to fully hydrolyse alone the cyclic AMP produced by the basal activity of adenylyl cyclase. This double hydrolytic system may prevent intracellular cyclic AMP from rising immoderately in the absence of stimulatory hormones or neuromediators. When the cell is challenged by a stimulus activating cyclic AMP production, such as a β-adrenoceptor agonist, all PDE subtypes become determinant in regulating the intracellular cyclic AMP concentration. Hence, the response of ICa to a sympathetic stimulation will be determined by the activity of each PDE subtype, with the rank order of potency PDE4>PDE3>PDE2>PDE1.
Acknowledgments
We thank Patrick Lechêne for excellent technical help, and Florence Lefebvre and Isabelle Paic for preparation of the myocytes. Ignacio Verde was supported by a fellowship from the Fundacion Caixa-Galicia.
Abbreviations
- EHNA
Erythro-9-[2-Hydroxy-3-nonyl]adenine
- IBMX
3-isobutyl-1-metylxanthine
- ICa
L-type calcium current
- MIMX
8-methoxymethyl-3-isobutyl-1-methylxanthine
- PDE
phosphodiesterase
- PDE1
Ca2+/calmodulin-activated PDE
- PDE2
cyclic GMP-stimulated PDE
- PDE3
cyclic GMP-inhibited PDE
- PDE4
low Km cyclic GMP-independent PDE
- PKA
cyclic AMP-dependent protein kinase
References
- AKITA T., JOYNER R.W., LU C.B., KUMAR R., HARTZELL H.C. Developmental changes in modulation of calcium currents of rabbit ventricular cells by phosphodiesterase inhibitors. Circulation. 1994;90:469–478. doi: 10.1161/01.cir.90.1.469. [DOI] [PubMed] [Google Scholar]
- BEAVO J.A. Cyclic nucleotide phosphodiesterases : functional implications of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- BODE D.C., KANTER J.R., BRUNTON L.L. Cellular distribution of phosphodiesterase isoforms in rat cardiac tissue. Circ. Res. 1991;68:1070–1079. doi: 10.1161/01.res.68.4.1070. [DOI] [PubMed] [Google Scholar]
- DUBOIS M., PICQ M., NÉMOZ G., LAGARDE M., PRIGENT A.F. Inhibition of different phosphodiesterase isoforms of rat heart cytosol by free fatty acids. J. Cardiovasc. Pharmacol. 1993;21:522–529. doi: 10.1097/00005344-199304000-00003. [DOI] [PubMed] [Google Scholar]
- ENGELS P., FICHTEL K., LUBBERT H. Expression and regulation of human and rat phosphodiesterase type IV isogenes. FEBS Lett. 1994;350:291–295. doi: 10.1016/0014-5793(94)00788-8. [DOI] [PubMed] [Google Scholar]
- FISCHMEISTER R., HARTZELL C. Regulation of calcium current by low-Km cyclic AMP phosphodiesterases in cardiac cells. Mol. Pharmacol. 1990;38:426–433. [PubMed] [Google Scholar]
- FISCHMEISTER R., HARTZELL H.C. Cyclic AMP phosphodiesterases and Ca2+ current regulation in cardiac cells. Life Sci. 1991;48:2365–2376. doi: 10.1016/0024-3205(91)90369-m. [DOI] [PubMed] [Google Scholar]
- GAO T.Y., YATANI A., DELLACQUA M.L., SAKO H., GREEN S.A., DASCAL N., SCOTT J.D., HOSEY M.M. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- GRIMALDI B., BONNIN A., FILLION M.P., RUAT M., TRAIFFORT E., FILLION G. Characterization of 5-HT6 receptor and expression of 5-HT6 mRNA in the rat brain during ontogenetic development. Naunyn-Schmied. Arch. Pharmacol. 1998;357:393–400. doi: 10.1007/pl00005184. [DOI] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HARTZELL H.C., MÉRY P.F., FISCHMEISTER R., SZABO G. Sympathetic regulation of cardiac calcium current is due exclusively to cAMP-dependent phosphorylation. Nature. 1991;351:573–576. doi: 10.1038/351573a0. [DOI] [PubMed] [Google Scholar]
- HOVE-MADSEN L., MÉRY P.-F., JUREVICIUS J., SKEBERDIS A.V., FISCHMEISTER R. Regulation of myocardial calcium channels by cyclic AMP metabolism. Basic Res. Cardiol. 1996;91 Suppl. 2:101–108. doi: 10.1007/BF00795355. [DOI] [PubMed] [Google Scholar]
- JUREVICIUS J., FISCHMEISTER R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl Acad. Sci. USA. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAASIC A., OHISALO J.J. Membrane-bound phosphodiesterases in rat. J. Pharm. Pharmacol. 1996;48:962–964. doi: 10.1111/j.2042-7158.1996.tb06012.x. [DOI] [PubMed] [Google Scholar]
- KAJIMOTO K., HAGIWARA N., KASANUKI H., HOSODA S. Contribution of phosphodiesterase isoenzymes to the regulation of the L-type calcium current in human cardiac myocytes. Br. J. Pharmacol. 1997;121:1549–1556. doi: 10.1038/sj.bjp.0701297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KATANO Y., ENDOH M. Cyclic AMP metabolism in intact rat ventricular cardiac myocytes: interaction of carbachol with isoproterenol and 3-isobutyl-1-methylxanthine. Mol. Cell. Biochem. 1993;119:195–201. doi: 10.1007/BF00926871. [DOI] [PubMed] [Google Scholar]
- KIRSTEIN M., RIVET-BASTIDE M., HATEM S., BENARDEAU A., MERCADIER J.J., FISCHMEISTER R. Nitric oxide regulates the calcium current in isolated human atrial myocytes. J. Clin. Invest. 1995;95:794–802. doi: 10.1172/JCI117729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSTIC M.M., ERDOGAN S., RENA G., BORCHERT G., HOCH B., BARTEL S., SCOTLAND G., HUSTON E., HOUSLAY M.D., KRAUSE E.G. Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart. J. Mol. Cell. Cardiol. 1997;29:3135–3146. doi: 10.1006/jmcc.1997.0544. [DOI] [PubMed] [Google Scholar]
- LEVI R.C., ALLOATI G., FISCHMEISTER R. Cyclic GMP regulates the Ca-channel current in guinea pig ventricular myocytes. Pflügers Arch. 1989;413:685–687. doi: 10.1007/BF00581823. [DOI] [PubMed] [Google Scholar]
- LOUGHNEY K., FERGUSON K.Identification and quantification of PDE isoenzymes and subtypes by molecular biological methods Phosphodiesterase inhibitors 1996San Diego: Academic Press Inc; 1–19.En, S., Schudt, C., Dent, G. & Rabe, K.F., ed. pp [Google Scholar]
- MCDONALD T.F., PELZER S., TRAUTWEIN W., PELZER D.J. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MÉRY P.F., LOHMANN S.M., WALTER U., FISCHMEISTER R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc. Natl. Acad. Sci. USA. 1991;88:1197–1201. doi: 10.1073/pnas.88.4.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MÉRY P.F., PAVOINE C., PECKER F., FISCHMEISTER R. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP-stimulated phosphodiesterases in isolated cardiac myocytes. Mol. Pharmacol. 1995;48:121–130. [PubMed] [Google Scholar]
- MUBAGWA K., SHIRAYAMA T., MOREAU M., PAPPANO A.J. Effects of PDE inhibitors and carbachol on the L-type Ca current in guinea pig ventricular myocytes. Am. J. Physiol. 1997;264:H1323–H1363. doi: 10.1152/ajpheart.1993.265.4.H1353. [DOI] [PubMed] [Google Scholar]
- MULLER B., LUGNIER C., STOCLET J.C. Involvement of rolipram-sensitive cyclic AMP phosphodiesterase in the regulation of cardiac contraction. J. Cardiovasc. Pharmacol. 1990;16:796–803. doi: 10.1097/00005344-199011000-00016. [DOI] [PubMed] [Google Scholar]
- OKRUHLICOVA L., TRIVULOBA N., ECKLY A., LUGNIER C., SLEZAK J. Cytochemical distribution of cyclic AMP-dependent 3′,5′-nucleotide phosphodiesterase in the rat myocardium. Histochem. J. 1996;28:165–172. doi: 10.1007/BF02331440. [DOI] [PubMed] [Google Scholar]
- ONO K., TRAUTWEIN W. Potentiation by cyclic GMP of β-adrenergic effect on Ca2+ current in guinea pig ventricular cells. J. Physiol. 1991;443:387–404. doi: 10.1113/jphysiol.1991.sp018839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PICQ M., DUBOIS M., GRYNBERG A., LAGARDE M., PRIGENT A.F. Specific effects of n-3 fatty acids and 8-bromo-cGMP on the cyclic nucleotide phosphodiesterase activity in neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 1996;28:2151–2161. doi: 10.1006/jmcc.1996.0207. [DOI] [PubMed] [Google Scholar]
- PODZUWEIT T., NENNSTIEL P., MULLER A. Isozyme selective inhibition of cGMP-stimulated cyclic nucleotide phosphodiesterases by erythro-9-(2-hydroxy-3-nonyl) adenine. Cell. Signal. 1995;7:733–738. doi: 10.1016/0898-6568(95)00042-n. [DOI] [PubMed] [Google Scholar]
- REEVES M.L., ENGLAND P.J. Cardiac Phosphodiesterases and the Functional Effects of Selective Inhibition. Cyclic nucleotide phosphodiesterases: structure, regulation and drug action. 1990;2:299–316. [Google Scholar]
- RIVET-BASTIDE M., VANDECASTEELE G., VERDE I., HATEM S., BENARDEAU A., MERCADIER J.J., FISCHMEISTER R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J. Clin. Invest. 1997;99:2710–2718. doi: 10.1172/JCI119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETTE C., CONTI M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase – Involvement of serine 54 in the enzyme activation. J. Biol. Chem. 1996;271:16526–16534. doi: 10.1074/jbc.271.28.16526. [DOI] [PubMed] [Google Scholar]
- SHAHID M., NICHOLSON C.D. Comparison of cyclic nucleotide phosphodiesterase isoenzymes in rat and rabbit ventricular myocardium, positive inotropic and phosphodiesterase inhibitory effects of Org 30029, milrinone and rolipram. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990;342:698–705. doi: 10.1007/BF00175715. [DOI] [PubMed] [Google Scholar]
- SHAHID M., NICHOLSON C.D., MARSHALL R.J. Species-dependent differences in the properties of particulate cyclic nucleotide phosphodiesterase from rat and rabbit ventricular myocardium. J. Pharm. Pharmacol. 1990;42:283–284. doi: 10.1111/j.2042-7158.1990.tb05409.x. [DOI] [PubMed] [Google Scholar]
- SHIRIYAMA T., PAPPANO A.J. Bifasic effects of intrapipette cyclic guanosine monophosphate on L-type calcium current and contraction of guinea pig ventricular myocytes. J. Pharmacol. Exp. Ther. 1996;279:1274–1281. [PubMed] [Google Scholar]
- STOCLET J.C., KERAVIS T., KOMAS N., LUGNIER C. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiovasculasr diseases. Exp. Opin. Invest. Drugs. 1995;4:1081–1100. [Google Scholar]
- TAIRA M., HOCKMAN S.C., CALVO J.C., TAIRA M., BELFRAGE P., MANGANIELLO V.C. Molecular Cloning of the Rat Adipocyte Hormone-Sensitive Cyclic GMP-Inhibited Cyclic Nucleotide Phosphodiesterase. J. Biol. Chem. 1993;268:18573–18579. [PubMed] [Google Scholar]
- VERDE I., VANDECASTEELE G., FISCHMEISTER R. PDE4 regulates the L-type calcium current in human atrial and rat ventricular myocytes. J. Mol. Cell. Cardiol. 1997;29:A36. [Google Scholar]
- WEISHAAR R.E., KOBYLARZ-SINGER D.C., KAPLAN H.R. Subclasses of cyclic AMP phosphodiesterase in cardiac muscle. J. Mol. Cell. Cardiol. 1987;19:1025–1036. doi: 10.1016/s0022-2828(87)80574-6. [DOI] [PubMed] [Google Scholar]
- WELLS J.N., MILLER J.R. Methylxanthine inhibitors of phosphodiesterases. Meth. Enzymol. 1988;159:489–496. doi: 10.1016/0076-6879(88)59048-1. [DOI] [PubMed] [Google Scholar]
- WIECHEN K., YUE D.T., HERZIG S. Two distinct functional effects of protein phosphatase inhibitors on guinea-pig cardiac L-type Ca2+ channels. J. Physiol. (Lond.) 1995;484:583–592. doi: 10.1113/jphysiol.1995.sp020688. [DOI] [PMC free article] [PubMed] [Google Scholar]