

Abstract
Mouse brain slices preincubated with [3H]-noradrenaline or [3H]-serotonin were superfused with medium containing naloxone 10 μM; we studied whether nociceptin (the endogenous ligand at ORL1 receptors) affects monoamine release. Furthermore, the affinities of ORL1 ligands were determined using [3H]-nociceptin binding.
The electrically (0.3 Hz) evoked tritium overflow in mouse cortex slices preincubated with [3H]-noradrenaline was inhibited by nociceptin and [Tyr14]-nociceptin (maximally by 80%; pEC50 7.52 and 8.28) but not affected by [des-Phe1]-nociceptin (pEC50<6). The ORL1 antagonist naloxone benzoylhydrazone antagonized the effect of nociceptin and [Tyr14]-nociceptin.
The effect of nociceptin did not desensitize, was not affected by blockade of NO synthase, cyclo-oxygenase and P1-purinoceptors and was decreased by the α2-adrenoceptor agonist talipexole. Nociceptin also inhibited the evoked overflow in mouse cerebellar, hippocampal and hypothalamic slices in a manner sensitive to naloxone benzoylhydrazone.
The electrically (3 Hz) evoked tritium overflow in mouse cortex slices preincubated with [3H]-serotonin was inhibited by nociceptin; naloxone benzoylhydrazone antagonized this effect.
The affinities (pKi) for [3H]-nociceptin binding to mouse cortex membranes were: nociceptin, 8.71; [Tyr14]-nociceptin, 9.82; [des-Phe1]-nociceptin, <5.5; naloxone benzoylhydrazone, 5.85; naloxone, <4.5.
In conclusion, nociceptin inhibits noradrenaline release in the mouse cortex via ORL1 receptors, which interact with presynaptic α2-autoreceptors on noradrenergic neurones. The effect of nociceptin does not desensitize nor does it involve NO, prostanoids or adenosine. Nociceptin also attenuates noradrenaline release from several subcortical regions and serotonin release from cortical slices by a naloxone benzoylhydrazone-sensitive mechanism.
Keywords: Nociceptin, orphanin FQ, ORL1 receptor, noradrenaline release, serotonin release, mouse brain cortex, presynaptic receptors, [3H]-nociceptin binding
Introduction
A fourth type of opioid receptor, termed opioid receptor-like1 (ORL1) (Mollereau et al., 1994), orphan opioid (Henderson & McKnight, 1997) or, tentatively, OP4 receptor (Hamon, 1998), has been cloned by several groups in 1994 (for review, see Henderson & McKnight, 1997; Meunier, 1997). The heptadecapeptide nociceptin (also termed orphanin FQ) has been identified as endogenous ligand at this receptor (Meunier et al., 1995; Reinscheid et al., 1995). Despite the high structural resemblance of the ORL1 receptor to the classical opioid receptors, most of the ligands at the classical opioid receptors have very low affinity for the ORL1 receptor; another striking difference to the classical opioid receptors is the fact that nociceptin elicits a hyperalgetic rather than an analgetic response in rodents (for review, see Henderson & McKnight, 1997; Meunier, 1997).
On the other hand, ORL1 and classical opioid receptors resemble each other in one important functional aspect; thus, ORL1 receptors, like the classical opioid receptors (for review, see Illes, 1989; Jackisch, 1991; Mulder & Schoffelmeer, 1993; Smith & Leslie, 1993), occur as presynaptic receptors on nerve endings and their activation causes inhibition of transmitter release. Nociceptin inhibits transmitter release (determined directly or via the endorgan response) in a series of peripheral tissues, including the guinea-pig ileum and the rat and mouse vas deferens (for review, see Henderson & McKnight, 1997; Meunier, 1997). With respect to the CNS, Vaughan et al. (1997) found that nociceptin, at a presynaptic site, inhibited the evoked fast GABAergic and glutamatergic postsynaptic currents in rat periaqueductal gray slices. The inhibitory effect of nociceptin on glutamate release was measured directly by Nicol et al. (1996) in rat brain cortex slices. Furthermore, administration of nociceptin into the ventricle or the ventral tegmental area inhibited dopamine release in the nucleus accumbens of the anaesthetized rat (Murphy et al., 1996; 1997). By contrast, nociceptin, administered intrastriatally to conscious rats, increased dopamine release in the striatum (Konya et al., 1998) and, when administered to the ventral tegmental area of the anaesthetized rat, facilitated GABA and glutamate release in this brain region (Murphy et al., 1997). The facilitatory effects of nociceptin in the latter two in vivo studies might be explained by the involvement of presynaptic inhibitory ORL1 receptors located on interneurones.
We have recently shown on superfused cerebrocortical slices from the mouse, rat and guinea-pig that nociceptin inhibits the release also of noradrenaline via presynaptic ORL1 receptors (Schlicker et al., 1998). The aims of the present study were to examine whether this inhibitory effect (1) desensitizes upon prolonged exposure to nociceptin, (2) involves endogenous mediators like adenosine, NO or prostanoids, (3) is influenced by simultaneous activation or blockade of the presynaptic α2-autoreceptor, (4) extends to subcortical brain regions and (5) also extends to the release of serotonin. The affinity of nociceptin and related drugs at ORL1 receptors was determined in binding studies at mouse brain cortex membranes, using the radioligand [3H]-nociceptin.
Methods
Superfusion studies
Slices (0.3 mm thick, diameter 3 mm) were prepared from various brain regions of male NMRI mice. The slices were incubated for 60 min with physiological salt solution (PSS) of 37°C containing [3H]-noradrenaline 25 nM or [3H]-serotonin 100 nM and then superfused with PSS of 37°C for 110 min at a flow rate of 1 ml min−1. The superfusate was collected in 5-min samples. The PSS was composed as follows (mM): NaCl 118, NaHCO3 25, KCl 4.8, CaCl2 1.3, KH2PO4 1.2, MgSO4 1.2, ascorbic acid 0.06, EDTA 0.03, glucose 10; it was aerated with 95% O2 and 5% CO2.
Tritium overflow was evoked by two 2-min periods of electrical field stimulation 40 (S1) and 90 min (S2) after onset of superfusion. The stimulation parameters were 0.3 Hz, 50 mA, 2 ms for slices preincubated with [3H]-noradrenaline (‘standard stimulation') and 3 Hz, 100 mA, 2 ms for slices preincubated with [3H]-serotonin. Nociceptin, [Tyr14]-nociceptin, [des-Phe1]-nociceptin or tetrodotoxin was added to (or Ca2+ ions were omitted from) the PSS from 62 min of superfusion onward whereas other drugs were present in the PSS throughout superfusion (Figure 1A).
Figure 1.

Experimental protocol of the superfusion experiments of the present study. (A) The majority of experiments lasted for 110 min and two periods of electrical field stimulation (flash symbols) starting after 40 (S1) and 90 min of superfusion (S2) were administered. Nociceptin, related peptides or tetrodotoxin were added to (and Ca2+ ions were omitted from) the medium from 62 min of superfusion onward (i.e. from 28 min before S2 onward). In few experiments, nociceptin was added from 72 or 82 min of superfusion onward (i.e. from 18 or 8 min before S2 onward, respectively). Other drugs (desipramine, indalpine, metitepine, naloxone, naloxone benzoylhydrazone, L-NAME, naproxen, rauwolscine, 8-(p-sulphophenyl)theophylline and/or talipexole) were present in the medium throughout superfusion. In most of the experimental series, the superfusion medium contained desipramine 1 μM, rauwolscine 1 μM plus naloxone 10 μM (‘standard medium') and the stimulation parameters were 0.3 Hz, 50 mA, 2 ms (duration of 2 min) (‘standard stimulation'). (B) The experiments shown in Figure 3 lasted for 210 min and four 2-min periods of electrical field stimulation (0.3 Hz, 50 mA, 2 ms) starting after 40 (S1), 90 (S2), 140 (S3) and 190 min of superfusion (S4) were administered. Nociceptin was added to the medium either from 62 min to 112 min of superfusion or from 62 min until the end of superfusion. Other drugs (desipramine, rauwolscine plus naloxone) were present in the medium throughout superfusion.
In some of the nine experimental series (see Results), the protocol was modified. In the second series, tritium overflow was evoked at 3 Hz; in order to administer the same number of pulses (i.e. 36) as in the experiments with 0.3 Hz, the duration of stimulation was 12 s. In the third series, nociceptin was added to the PSS from 72 or 82 min of superfusion onward (Figure 1A). In the fourth series of experiments, the superfusion lasted for 210 min and four 2-min periods of electrical field stimulation were administered after 40, 90, 140 and 190 min of superfusion (S1, S2, S3 and S4) (Figure 1B). In the seventh series, the current strength was 12.5 (instead of 50) mA.
At the end of superfusion, the radioactivity of the slices and the superfusate samples was determined by liquid scintillation counting. Tritium efflux was calculated as a fraction of the tritium content of the tissue at the onset of the respective collection period (fractional rate of tritium efflux). For this purpose, tritium in this collection period was divided by the sum of tritium determined in this collection period, all subsequent collection periods and the slice. In order to quantify the effects of drugs on basal tritium efflux, the fractional rates of tritium efflux in the 5-min collection periods from 55–60 (t1), from 85–90 (t2) and, if applicable, from 135–140 (t3) and from 185–190 min of superfusion (t4) were determined. The stimulation-evoked tritium overflow was calculated as the amount of tritium in excess of the basal tritium efflux (the latter was assumed to decline linearly from the 5-min period immediately before Sn to the 5-min sample collected 15–20 min after onset of stimulation) and expressed as per cent of tissue tritium at the onset of the respective stimulation. To quantify drug-induced effects on the stimulated tritium overflow, the ratio of the overflow evoked by S2 and, if applicable, by S3 or S4 over that evoked by S1 was determined or the overflow evoked by S1 obtained in the presence of a given drug was compared to that obtained in its absence.
Binding studies
These were carried out essentially as described by Dooley & Houghten (1996). Cerebral cortex from male NMRI mice or male Wistar rats was homogenized in medium containing Tris-HCl buffer 50 mM (pH 7.4), EDTA 2 mM, sucrose 0.3 M and phenylmethylsulphonyl fluoride (PMSF) 100 μM with a Potter-Elvehjem homogenizer (10 strokes, 1200 r.p.m.) and centrifuged at 1000 g for 10 min (4°C). The supernatant was centrifuged at 35,000×g for 10 min and the final pellet was resuspended in sucrose-free buffer solution and frozen at −80°C.
The binding assay was performed in Tris-HCl buffer (Tris-HCl 50 mM (pH 7.4), EDTA 2 mM, PMSF 100 μM) in a final volume of 0.5 ml containing 70–100 μg protein. Saturation curves were obtained by incubating [3H]-nociceptin at eight concentrations ranging from 0.05–12 nM (25°C). The incubation was terminated after 60 min by rapid vacuum filtration through polyethylenimine (0.3%)-pretreated Whatman GF/C filters followed by rapid washing of the incubation tubes and filters three times with 2 ml Tris buffer. Non-specific binding was determined with nociceptin 5 μM. Competition assays were performed as described for saturation studies; the concentration of [3H]-nociceptin was 0.5 nM (unspecific binding at this concentration was 10%). Ki values were determined using ten concentrations of the drug under study. Protein was assayed according to the method described by Bradford (1976). Saturation and displacement curves were analysed using the programme GraphPadPrism (Prism; GraphPad Software, San Diego, CA, U.S.A.).
Statistical analysis
Results are given as means±s.e.m. of n experiments (superfusion studies) or n experiments in triplicate (binding studies). Student's t-test was used for comparison of mean values; if more than one experimental series was compared to the same control, the Bonferroni correction was applied. In order to evaluate whether the displacement of [3H]-nociceptin binding by the drugs under study is better fitted by a one- or two-site model, the F-test was used.
Drugs
R-(−)-[Ring-2, 5, 6-3H]-noradrenaline (specific activity (spec. act.) 46.8 Ci mmol−1), [1, 2-3H[N]]-serotonin creatinine sulphate (spec. act. 27.5 Ci mmol−1) (NEN, Zaventem, Belgium); [leucyl-3H]-nociceptin (spec. act. 157–170 Ci mmol−1) (Amersham, Braunschweig, Germany); carbetapentane citrate (Tocris/Biotrend, Köln, Germany); desipramine hydrochloride (Ciba-Geigy, Wehr, Germany); naloxone benzoylhydrazone, rimcazole dihydrochloride, 8-(p-sulphophenyl)theophylline (RBI/Biotrend, Köln, Germany); indalpine (Rhône-Poulenc, Gennevilliers, France); metitepine maleate (Hoffmann-La Roche, Basel, Switzerland); naloxone hydrochloride, naproxen, Nω-nitro-L-arginine methyl ester (L-NAME) (Sigma, München, Germany); nociceptin (Bachem, Heidelberg, Germany or Tocris/Biotrend, Köln, Germany); [des-Phe1]-nociceptin (custom synthesis by Eurogentec, Seraing, Belgium); [Tyr14]-nociceptin (custom synthesis by Eurogentec or Genosys, Cambridge, England); rauwolscine hydrochloride, tetrodotoxin (Roth, Karlsruhe, Germany); talipexole dihydrochloride (former name B-HT 920) (Thomae, Biberach, Germany). Stock solutions of the drugs were prepared with ethanol (naloxone benzoylhydrazone), dimethylsulphoxide (DMSO) (naproxen, 8-(p-sulphophenyl)-theophylline), HCl 0.01 M (indalpine), citrate buffer 0.1 M (pH 4.8) (tetrodotoxin) or water (other drugs) and diluted with PSS (superfusion experiments) or water (binding experiments) to the concentration required. The organic solvents by themselves (ethanol or DMSO up to 0.3%) did not affect tritium efflux or [3H]-nociceptin binding.
Results
Superfusion experiments (general)
Basal tritium efflux was quantified in the 5-min sample from 55–60 min of superfusion (t1) when the drug under study was present throughout superfusion and as the ratio t2/t1 and, if applicable, t3/t1 and t4/t1 when the drug was added to the medium after t1. Basal tritium efflux expressed as the fractional rate of tritium efflux during t1 is given in Table 1 for the various experimental conditions (i.e. slices from various brain regions and preincubated with various [3H]-monoamines). The ratios t2/t1, t3/t1 and t4/t1 in control experiments were uniformly about 0.7–0.8. None of the drugs under study affected basal tritium efflux (results not shown).
Table 1.
Basal and electrically evoked tritium overflow from superfused mouse brain cortex slices preincubated with [3H]-noradrenaline ([3H]-NA) or [3H]-serotonin ([3H]-5-HT), and effect of nociceptin on the evoked tritium overflow
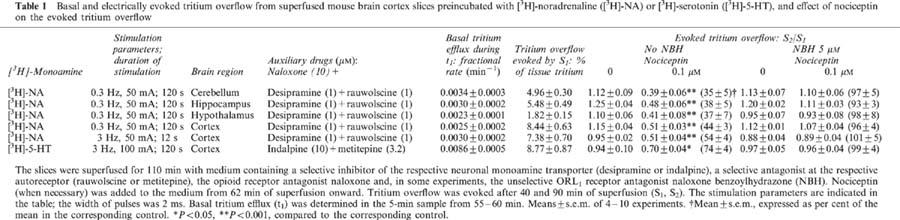
For quantification of the evoked tritium overflow, tritium overflow evoked by S1 (after 40 min of superfusion) was considered when the drug under study was present throughout superfusion and the ratios S2/S1 and, if applicable, S3/S1 and S4/S1 were used when the drug was added to the medium after S1 (Figure 1). S1 values obtained for a variety of experimental conditions are presented in Tables 1 and 5. The Sn/S1 values in control experiments were near unity (see text, Tables 1, 3 and 4 and legends to Figures 3 and 4).
Table 5.
Effect of talipexole and rauwolscine on the electrically evoked tritium overflow from superfused mouse brain cortex slices preincubated with [3H]-noradrenaline

Table 3.
Antagonism of naloxone benzoylhydrazone (NBH) against the effect of nociceptin and [Tyr14]-nociceptin

Table 4.
Influence of Nω;-nitro-L-arginine methyl ester (L-NAME), naproxen and 8-(p-sulphophenyl)-theophylline on the inhibitory effect of nociceptin

Figure 3.
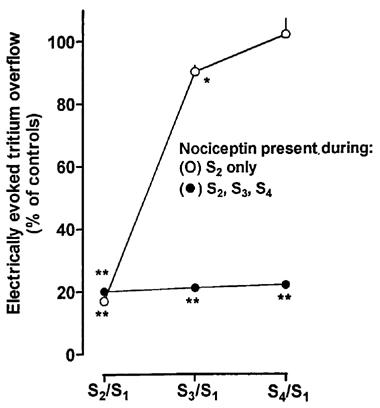
Recovery from, and lack of desensitization to, the nociceptin-induced inhibition of the electrically evoked tritium overflow from superfused mouse brain cortex slices preincubated with [3H]-noradrenaline. Tritium overflow was evoked by four 2-min periods of electrical stimulation (0.3 Hz, 50 mA, 2 ms), after 40, 90, 140 and 190 min of superfusion (S1, S2, S3, S4), and the ratios of the overflow evoked by S2, S3 or S4 over that evoked by S1 were determined. The slices were superfused with medium containing desipramine 1 μM, rauwolscine 1 μM plus naloxone 10 μM throughout superfusion and nociceptin either from 62 to 112 min (i.e. during S2 only) or from 62 to 210 min of superfusion (i.e. during S2, S3 and S4). Results are given as per cent of the S2/S1, S3/S1 and S4/S1 values obtained in the control series (no exposure to nociceptin), which were 1.21±0.03, 1.20±0.03 and 1.16±0.03, respectively (not shown in the graph). Means±s.e.m. of 6–8 experiments; for some data points, s.e.m. is contained within the symbol. *P<0.02, **P<0.001, compared to the corresponding values of the control series.
Figure 4.

Influence of the α2-adrenoceptor ligands talipexole and rauwolscine on the nociceptin-induced inhibition of the electrically evoked tritium overflow from superfused mouse brain cortex slices preincubated with [3H]-noradrenaline. The slices were superfused with medium containing desipramine 1 μM, naloxone 10 μM and, when necessary, talipexole or rauwolscine throughout superfusion and nociceptin from 62 min of superfusion onward. Tritium overflow was evoked electrically (0.3 Hz, 2 ms, current strength 50 mA (if not stated otherwise) or 12.5 mA (in one series)). Two 2-min periods of stimulation were administered after 40 and 90 min of superfusion (S1, S2), and the ratio of the tritium overflow evoked by S2 over that evoked by S1 was determined. Results are expressed as per cent of the S2/S1 values of the corresponding controls (not shown in the graph), which ranged from 0.82±0.11 to 1.24±0.06. Means±s.e.m. of 4–9 experiments. ≈rcub;P<0.01, compared to the corresponding value without talipexole or rauwolscine.
Addition of tetrodotoxin 1 μM to, or omission of Ca2+ ions from, the medium inhibited (by at least 84%) the electrically evoked tritium overflow (S2/S1) in mouse cerebellar, hippocampal and hypothalamic slices preincubated with [3H]-noradrenaline (and superfused with medium containing desipramine 1 μM and rauwolscine 1 μM) and in mouse cortex slices preincubated with [3H]-serotonin (and superfused with medium containing indalpine 10 μM and metitepine 3.2 μM) (n=4 each, results not shown). Very similar results had been obtained previously for mouse brain cortex slices preincubated with [3H]-noradrenaline (Schlicker et al., 1992).
Superfusion experiments on mouse brain cortex slices preincubated with [3H]-noradrenaline
In most of the experimental series, the slices were superfused with medium containing an inhibitor of the neuronal noradrenaline transporter, desipramine 1 μM, the α2-autoreceptor antagonist rauwolscine 1 μM plus the opioid receptor antagonist naloxone 10 μM (‘standard medium') and the parameters of electrical field stimulation were 0.3 Hz, 50 mA and 2 ms (for 2 min) (‘standard stimulation'). In the first series (performed with standard medium and standard stimulation), the electrically evoked tritium overflow, expressed as S2/S1, was inhibited in a concentration-dependent manner by nociceptin and more potently by [Tyr14]-nociceptin; the maximum inhibitory effect was about 80% for both peptides (Figure 2A). (Identical results were obtained when nociceptin from Tocris/Biotrend (instead of from Bachem) and [Tyr14]-nociceptin from Genosys (instead of from Eurogentec) were used; results not shown). The pEC50 values were graphically determined on the basis of the concentrations causing an inhibition by 40% (Table 2). [des-Phe1]-nociceptin up to 1 μM did not affect the evoked overflow (Figure 2A). The inhibitory effect of nociceptin and [Tyr14]-nociceptin on the evoked overflow was antagonized by naloxone benzoylhydrazone 5 μM (Tables 1 and 3), which, per se, did not affect the evoked overflow (S1) (n=8, not shown).
Figure 2.
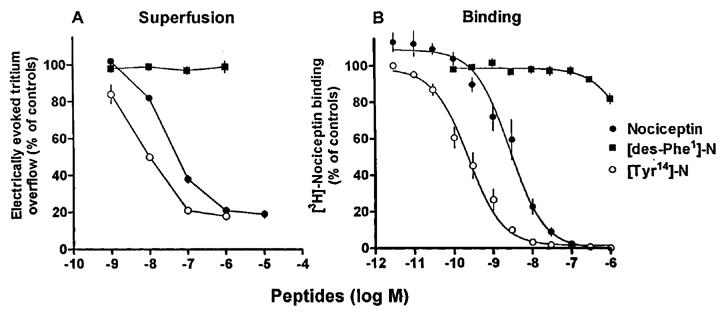
Effects of nociceptin, [Tyr14]-nociceptin ([Tyr14]-N) and [des-Phe1]-nociceptin ([des-Phe1]-N) on (A) the electrically evoked tritium overflow from superfused mouse brain cortex slices preincubated with [3H]-noradrenaline and (B) the specific [3H]-nociceptin binding to mouse brain cortex membranes. (A) The slices were superfused with medium containing desipramine 1 μM, rauwolscine 1 μM plus naloxone 10 μM throughout superfusion and nociceptin or one of the chemically related peptides from 62 min of superfusion onward. Tritium overflow was evoked by two 2-min periods of electrical stimulation (0.3 Hz, 50 mA, 2 ms), after 40 and 90 min of superfusion (S1, S2), and the ratio of the overflow evoked by S2 over that evoked by S1 was determined. (B) Membranes were incubated for 60 min with 0.5 nM [3H]-nociceptin and increasing concentrations of the competitor. The results, which represent means±s.e.m. of four superfusion experiments (A) and 4–6 binding experiments in triplicate (B), are given as per cent of controls (controls not shown); for some data points, s.e.m. is contained within the symbol.
Table 2.
Potencies of nociceptin, related peptides, naloxone and naloxone benzoylhydrazone at the functional ORL1 receptor in mouse brain cortex and their affinities at ORL1 binding sites in the mouse and rat brain

In the second series, slices were superfused with standard medium; however, tritium overflow was evoked at 3 Hz (pulses of 50 mA and 2 ms); the duration of stimulation was 12 s. Nociceptin 0.1 μM inhibited the evoked overflow (S2/S1) also under this condition, in a manner sensitive to naloxone benzoylhydrazone, although the degree of inhibition tended to be lower at 3 than at 0.3 Hz (Table 1).
In the third experimental series, slices were superfused with standard medium; standard stimulation was administered. The electrically evoked tritium overflow (S2/S1) was 1.08±0.03 in eight controls and 0.20±0.01 when nociceptin 1 μM was added to the medium from 62 min of superfusion (usual procedure; n=6, P<0.001). When nociceptin was added from 72 or 82 min of superfusion (Figure 1A), the S2/S1 values were 0.20±0.01 and 0.19±0.01, respectively (n=5–6, P<0.001 for both values, compared to controls).
In the fourth series of experiments, slices were superfused with standard medium. The duration of the experiments was 210 min and four 2 min periods of electrical stimulation (0.3 Hz, 50 mA, 2 ms) were administered after 40, 90, 140 and 190 min (S1, S2, S3 and S4) (Figure 1B). Nociceptin 1 μM, added to the medium at 62 min of superfusion and kept in the medium until the end of the experiments, inhibited the evoked overflow, expressed as Sn/S1, to about the same extent, regardless of whether the time of exposure was 28 min (S2/S1), 78 min (S3/S1) or 128 min (S4/S1) (Figure 3). When nociceptin was present in the medium from 62–112 min of superfusion only (i.e. present during S2 but absent during S3 and S4), its inhibitory effect was readily reversible; the inhibition was only 10±2% 28 min after its withdrawal (S3/S1) and not detectable at all 78 min after its withdrawal (S4/S1) (Figure 3).
In the fifth experimental series, slices were superfused with standard medium and standard stimulation was administered. Table 4 shows that the inhibitory effect of nociceptin 0.1 μM on the evoked tritium overflow (S2/S1) was not affected by L-NAME 100 μM (an inhibitor of NO synthase), naproxen 10 μM (an inhibitor of cyclooxygenase) and 8-(p-sulphophenyl)theophylline 100 μM (a P1 purinoceptor antagonist). L-NAME, naproxen and 8-(p-sulphophenyl)theophylline per se did not affect the evoked overflow (S1) (n=5–6, not shown).
In the sixth series of experiments, slices were superfused with medium containing desipramine 1 μM plus naloxone 10 μM; the standard stimulation protocol was used. Figure 4 shows that nociceptin inhibited the evoked overflow (S2/S1) and that its concentration-response curve was shifted to the left by the α2-adrenoceptor antagonist rauwolscine 1 μM and to the right by the α2-adrenoceptor agonist talipexole 0.2 μM. Given alone, rauwolscine increased and talipexole decreased the evoked overflow (S1) (Table 5).
As shown in Table 5, rauwolscine 1 μM by itself increased the evoked overflow (S1). In order to adjust the level of tritium overflow to that obtained in the absence of rauwolscine, the current strength was reduced from 50 to 12.5 mA in the seventh series. Under this experimental condition, nociceptin 0.1 μM reduced the evoked overflow (S2/S1) at least as strong as at the usual current strength of 50 mA (Figure 4).
Superfusion experiments on slices from various brain regions of the mouse preincubated with [3H]-noradrenaline or [3H]-serotonin
In the eighth series of experiments, cortical, cerebellar, hippocampal and hypothalamic slices were preincubated with [3H]-noradrenaline and superfused with standard medium; standard stimulation was administered. The evoked overflow (S2/S1) was inhibited by nociceptin 0.1 μM not only in cortical slices but also in cerebellar, hippocampal and hypothalamic slices (Table 1). The effect in the latter three brain regions even tended to be more marked than, but did not significantly differ from, that in the cortex (Table 1). This inhibitory effect was abolished by naloxone benzoylhydrazone 5 μM, which, by itself, did not affect the evoked overflow (S1) (n=4 each, not shown; for S1 in controls see Table 1).
In the ninth experimental series, cortex slices were preincubated with [3H]-serotonin and superfused with medium containing an inhibitor of the neuronal serotonin transporter, indalpine 10 μM, the serotonin autoreceptor antagonist metitepine 3.2 μM plus naloxone 10 μM; tritium overflow was evoked electrically (3 Hz, 100 mA, 2 ms; 2 min). The evoked overflow (S2/S1) was inhibited by nociceptin 0.1 μM by 26±4%; the inhibitory effect of nociceptin was abolished by naloxone benzoylhydrazone 5 μM (Table 1), which, given alone, did not affect the evoked overflow (S1) (n=5, not shown; for S1 in controls see Table 1).
Binding studies
In saturation binding studies on mouse brain cortex membranes, using [3H]-nociceptin at various concentrations from 0.05–12 nM, a dissociation constant (KD) of 1.35±0.20 nM with a maximum number of binding sites of 0.52±0.04 pmol mg−1 protein was determined (n=3). Scatchard analysis (not shown) revealed a straight line with a Hill coefficient (nH) of unity. Nociceptin and even more potently [Tyr14]-nociceptin displaced binding of [3H]-nociceptin 0.5 nM to mouse brain cortex membranes (monophasic displacement, nH values near unity), whereas [des-Phe1]-nociceptin hardly affected binding (Figure 2B, Table 2). (Identical results were obtained when nociceptin from Tocris/Biotrend (instead of from Bachem) and [Tyr14]-nociceptin from Genosys (instead of from Eurogentec) were used; not shown). Naloxone benzoylhydrazone also displaced [3H]-nociceptin binding (monophasic displacement, nH values near unity; for pKi values see Table 2) whereas two putative ORL1 receptor antagonists, carbetapentane and rimcazole (Kobayashi et al., 1997), failed to do so (pKi<4.5; n=4 each, results not shown). [3H]-Nociceptin binding was also not affected by naloxone (Table 2) and the following compounds, used as auxiliary drugs in the present study (pKi<4.5): desipramine, indalpine, metitepine, L-NAME, naproxen, rauwolscine, 8-(p-sulphophenyl)theophylline, talipexole and tetrodotoxin (n=4 each, results not shown).
Saturation binding studies on cerebral cortex membranes from the rat revealed a KD of 0.73±0.20 nM and a Bmax of 0.42±0.01 pmol mg−1 protein; Scatchard analysis yielded a straight line with a nH value of unity (n=3, results not shown). Binding of [3H]-nociceptin 0.5 nM to rat brain cortex membranes was monophasically displaced by nociceptin and, again more potently, by [Tyr14]-nociceptin (nH near unity; for pKi values see Table 2).
Discussion
The electrically evoked tritium overflow in our superfusion experiments represents quasi-physiological noradrenaline and serotonin release (Schlicker et al., 1992 and present results). The neuronal transporter of the respective monoamine was blocked routinely and the respective autoreceptor was blocked in most experiments to avoid an interaction of the test drugs with the respective neuronal transporter and autoreceptor (another reason for addition of an autoreceptor antagonist to the medium will be discussed below). Naloxone was present in the medium for blockade of the classical opioid receptors. Since the degradation of nociceptin in the mouse brain cortex is slow (Montiel et al., 1997) and the inhibitory effect of nociceptin on noradrenaline release in mouse brain cortex slices was not increased by the peptidase inhibitor PMSF (unpublished), peptidase inhibitors were not used for superfusion experiments. In our binding studies, however, PMSF was used, as in the study by Dooley & Houghten (1996). Although we determined [3H]-nociceptin binding to rat cortex membranes according to almost the same protocol as in the latter study, our KD value was lower, possibly due to the fact that we used membranes from the cortex rather than from the whole brain minus cerebellum. [3H]-nociceptin binding in mouse cortex membranes resembled that in rat cortex membranes with respect to the monophasic saturation binding curve and the KD and Bmax values.
Our previous superfusion study on mouse cortex slices (Schlicker et al., 1998) had shown that the inhibitory effect of nociceptin on noradrenaline release was antagonized by the unselective ORL1 receptor antagonist naloxone benzoylhydrazone (described by Dunnill et al., 1996; Nicholson et al., 1998) and the selective partial ORL1 agonist [Phe1ψ(CH2-NH)Gly2]-nociceptin(1–13)NH2 (described by Guerrini et al., 1998), suggesting that nociceptin acts via ORL1 receptors. This view is further supported by our present finding that [Tyr14]-nociceptin (identified as a potent ORL1 receptor ligand by Reinscheid et al., 1995; 1996; Berzetei-Gurske et al., 1996; Shimohigashi et al., 1996; Adapa & Toll, 1997; Mathis et al., 1997) mimicked the effect of nociceptin, in a manner sensitive to naloxone benzoylhydrazone. As expected, [des-Phe1]-nociceptin, a degradation product of nociceptin (Montiel et al., 1997), failed to mimic the effect of nociceptin.
The 10 fold higher potency of [Tyr14]-nociceptin, relative to nociceptin, both in our superfusion and binding studies (also found when both peptides were purchased from other manufacturers) contrasts with results from the literature. Thus, the affinity/potency of [Tyr14]-nociceptin did not exceed that of nociceptin both at the ORL1 receptor of the mouse vas deferens (Berzetei-Gurske et al., 1996) and in a binding study on mouse brain membranes, using [125I]-[Tyr14]-nociceptin (Mathis et al., 1997). The reason for the discrepancies is unclear but one has to consider that the functional ORL1 receptors in the cerebral cortex and in the vas deferens of the mouse differ with respect to the liability to desensitization (see later) and were identified under very different experimental conditions (i.e. direct (present study) vs indirect determination of noradrenaline release (Berzetei-Gurske et al., 1996; Dooley et al., 1997)). Our binding study and that by Mathis et al. (1997) markedly differ with respect to the tissue (cerebral cortex vs whole brain), the radioligand (see above) and the shape of the saturation and displacement curves (mono- vs biphasic curves).
Nociceptin inhibited noradrenaline release in the mouse brain cortex also when tritium overflow was evoked at 3 Hz, i.e. at a stimulation frequency closer to that occurring in noradrenergic neurones in freely moving animals (Jacobs, 1986) than 0.3 Hz used in most of our experiments. The inhibitory effect of nociceptin tended to be lower at 3 than at 0.3 Hz. This is not unexpected since there is an inverse relationship between the extent of effect mediated via presynaptic receptors and the stimulation frequency (for review, see Starke et al., 1989).
ORL1 receptors were shown to desensitize upon prolonged exposure to nociceptin in neuroblastoma×glioma NG108-15 hybrid cells (Ma et al., 1997; Morikawa et al., 1998) and in the isolated mouse vas deferens (Dooley et al., 1997). In our model, the effect of nociceptin on noradrenaline release did not change when the time of exposure, usually 28 min, was reduced to 18 or 8 min or extended to 78 or 128 min. The effect of nociceptin was rapidly reversible after its withdrawal from the medium, excluding that the events interposed between activation of the ORL1 receptor and the inhibition of noradrenaline release exhibit a long time course, as e.g. alteration of protein synthesis.
In other experiments, we studied whether mediators like adenosine, nitric oxide or prostanoids might be involved in the effect of nociceptin. This possibility, however, can be discarded since 8-(p-sulphophenyl)theophylline, L-NAME and naproxen (drugs blocking the formation or the effect of the three mediators at the concentrations used in the present study; von Kügelgen et al., 1997; Chu & Etgen, 1996; Molderings et al., 1994) failed to influence the effect of nociceptin.
Next, the question was examined whether the extent of inhibition mediated via the ORL1 receptor is affected when the presynaptic α2-adrenoceptor on the noradrenergic neurones (α2-autoreceptor) is simultaneously activated (by talipexole) or blocked (by rauwolscine). Interactions with the α2-autoreceptor have been described for several types of non-adrenergic receptors (‘heteroreceptors') causing inhibition of noradrenaline release (see Schlicker & Göthert (1998) for review). Usually, activation of the autoreceptor blunts the inhibitory effect mediated via the heteroreceptor and this was found for the ORL1 receptor examined in the present study as well. Conversely, blockade of the autoreceptor increased the ORL1 receptor-mediated effect. The most likely explanation for the latter phenomenon is that the α2-autoreceptor is tonically activated by endogenously released noradrenaline; thus, rauwolscine prevents endogenous noradrenaline from blunting the ORL1 receptor-mediated effect and thereby increases this effect. This property of rauwolscine was an additional reason to include the drug in the medium in most superfusion experiments.
The possibility that the influence of talipexole and rauwolscine on the effect of nociceptin is influenced by an (accidental) affinity of both drugs for ORL1 receptors could be excluded by our binding experiments (pKi<4.5 for either drug). The possibility that the increase of the ORL1 receptor-mediated effect by rauwolscine is merely due to the simultaneous increase in noradrenaline release from 3.5 to 8.5% (and not the consequence of the blockade of the α2-autoreceptor) had to be considered as well. In experiments in which the rauwolscine-related increase in noradrenaline release was compensated for (i.e. adjusted to a level of 3%) by reducing the current strength from 50 to 12.5 mA, rauwolscine still increased the ORL1 receptor-mediated effect, suggesting that α2-autoreceptor blockade (and not facilitation of noradrenaline release) is the decisive event. The interaction between the α2 and ORL1 receptor described here might occur at the level of the receptors themselves or at a step behind the receptors (e.g. at the level of G proteins or ion channels, which may be shared by both receptors).
Finally, nociceptin also inhibited noradrenaline release in mouse cerebellar, hippocampal and hypothalamic slices and serotonin release in mouse cortex slices; the effect of nociceptin was counteracted by naloxone benzoylhydrazone, suggesting that ORL1 receptors are involved. The extent of inhibition of noradrenaline release elicited by nociceptin in the cerebral cortex and the three subcortical brain regions was similar. The effect of nociceptin on serotonin release was relatively small and this may be, at least partially, due to the high stimulation frequency of 3 Hz (which, however, had to be used since the amount of serotonin release is not sufficiently high at lower frequencies). Our binding experiments exclude the possibility that the effect of nociceptin on serotonin and noradrenaline release is influenced by an accidental affinity of the auxiliary drugs (desipramine, rauwolscine, indalpine, metitepine) for ORL1 receptors.
In conclusion, inhibition of transmitter release in the CNS appears to be a common property associated with the ORL1 receptor in terms of the chemical identity of the transmitter (see also references in the Introduction) and the brain region. It is tempting to assume that inhibition of neurotransmitter release can explain at least some of the behavioural effects elicited by nociceptin.
Acknowledgments
This study was supported by grants from the Deutsche Forschungsgemeinschaft and the State of Nordrhein-Westfalen. We wish to thank Mrs D. Petri for her skilled technical assistance and the companies Ciba-Geigy, Rhône-Poulenc, Hoffmann-La Roche and Thomae for gifts of drugs.
Abbreviations
- [des-Phe1]-N
[des-Phe1]-nociceptin
- DMSO
dimethylsulphoxide
- 5-HT
5-hydroxytryptamine (serotonin)
- L-NAME
Nω-nitro-L-arginine methyl ester
- NA
noradrenaline
- NBH
naloxone benzoylhydrazone
- ORL1
opioid receptor-like1
- PMSF
phenylmethylsulphonyl fluoride
- PSS
physiological salt solution
- S
electrical stimulation
- spec. act.
specific activity
- t
collection period in which basal tritium efflux was determined
- [Tyr14]-N
[Tyr14]-nociceptin
References
- ADAPA I.D., TOLL L. Relationship between binding affinity and functional activity of nociceptin/orphanin FQ. Neuropeptides. 1997;31:403–408. doi: 10.1016/s0143-4179(97)90032-9. [DOI] [PubMed] [Google Scholar]
- BERZETEI-GURSKE I.P., SCHWARTZ R.W., TOLL L. Determination of activity for nociceptin in the mouse vas deferens. Eur. J. Pharmacol. 1996;302:R1–R2. doi: 10.1016/0014-2999(96)00238-5. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- CHU H.P., ETGEN A.M. Effects of nitric oxide on stimulated release of norepinephrine from female rat hypothalamic slices. Brain Res. 1996;741:60–67. doi: 10.1016/s0006-8993(96)00896-7. [DOI] [PubMed] [Google Scholar]
- DOOLEY C.T., HOUGHTEN R.A. Orphanin FQ: receptor binding and analog structure relationships in rat brain. Life Sci. 1996;59:PL23–PL29. doi: 10.1016/0024-3205(96)00261-5. [DOI] [PubMed] [Google Scholar]
- DOOLEY C.T., SPAETH C.G., BERZETEI-GURSKE I.P., CRAYMER K., ADAPA I.D., BRANDT S.R., HOUGHTEN R.A., TOLL L. Binding and in vitro activities of peptides with high affinity for the nociceptin/orphanin FQ receptor, ORL1. J. Pharmacol. Exp. Ther. 1997;283:735–741. [PubMed] [Google Scholar]
- DUNNILL R.J., KAKIZAWA K., MCKNIGHT A.T., HENDERSON G. Characterization of the actions of naloxone benzoylhydrazone at μ-opioid, κ-opioid and ORL1 receptors in isolated tissues from rat and guinea-pig. Br. J. Pharmacol. 1996;119:275P. [Google Scholar]
- GUERRINI R., CALO G., RIZZI A., BIGONI R., BIANCHI C., SALVADORI S., REGOLI D. A new selective antagonist of the nociceptin receptor. Br. J. Pharmacol. 1998;123:163–165. doi: 10.1038/sj.bjp.0701640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMON M. The new approach to opioid receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358 suppl. 2:R581. [Google Scholar]
- HENDERSON G., MCKNIGHT A.T. The orphan opioid receptor and its endogenous ligand–nociceptin/orphanin FQ. Trends Pharmacol. Sci. 1997;18:293–300. [PubMed] [Google Scholar]
- ILLES P. Modulation of transmitter and hormone release by multiple neuronal opioid receptors. Rev. Physiol. Biochem. Pharmacol. 1989;112:139–233. doi: 10.1007/BFb0027497. [DOI] [PubMed] [Google Scholar]
- JACKISCH R.Regulation of neurotransmitter release by opiates and opioid peptides in the central nervous system Presynaptic regulation of neurotransmitter release: a handbook 1991Tel Aviv: Freund Publishing House; 551–592.eds. Feigenbaum, J. & Hanani, M. pp [Google Scholar]
- JACOBS B.L. Single unit activity of brain monoamine-containing neurons in freely moving animals. Ann. N.Y. Acad. Sci. 1986;473:70–77. doi: 10.1111/j.1749-6632.1986.tb23604.x. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI T., IKEDA K., TOGASHI S., ITOH N., KUMANISHI T. Effects of σ ligands on the nociceptin/orphanin FQ receptor coexpressed with the G-protein-activated K+ channel in xenopus oocytes. Br. J. Pharmacol. 1997;120:986–987. doi: 10.1038/sj.bjp.0701068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONYA H., MASUDA H., ITOH K., NAGAI K., KAKISHITA E., MATSUOKA A. Modification of dopamine release by nociceptin in conscious rat striatum. Brain Res. 1998;788:341–344. doi: 10.1016/s0006-8993(98)00075-4. [DOI] [PubMed] [Google Scholar]
- MA L., CHENG Z.J., FAN G.H., CAI Y.C., JIANG L.Z., PEI G. Functional expression, activation and desensitization of opioid receptor-like receptor ORL1 in neuroblastoma×glioma NG 108-15 hybrid cells. FEBS Lett. 1997;403:91–94. doi: 10.1016/s0014-5793(97)00031-8. [DOI] [PubMed] [Google Scholar]
- MATHIS J.P., RYAN-MORO J., CHANG A., HOM J.S.H., SCHEINBERG D.A., PASTERNAK G.W. Biochemical evidence for orphanin FQ/nociceptin receptor heterogeneity in mouse brain. Biochem. Biophys. Res. Commun. 1997;230:462–465. doi: 10.1006/bbrc.1996.5867. [DOI] [PubMed] [Google Scholar]
- MEUNIER J.C. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. Eur. J. Pharmacol. 1997;340:1–15. doi: 10.1016/s0014-2999(97)01411-8. [DOI] [PubMed] [Google Scholar]
- MEUNIER J.C., MOLLEREAU C., TOLL L., SUAUDEAU C., MOISAND C., ALVINERIE P., BUTOUR J.L., GUILLEMOT J.C., FERRARA P., MONSARRAT B., MAZARGUIL H., VASSART G., PARMENTIER M., COSTENTIN J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- MOLDERINGS G.J., COLLING E., LIKUNGU J., JAKSCHIK J., GÖTHERT M. Modulation of noradrenaline release from the sympathetic nerves of the human saphenous vein and pulmonary artery by presynaptic EP3- and DP-receptors. Brit. J. Pharmacol. 1994;111:733–738. doi: 10.1111/j.1476-5381.1994.tb14799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOLLEREAU C., PARMENTIER M., MAILLEUX P., BUTOUR J.L., MOISAND C., CHALON P., CAPUT D., VASSART G., MEUNIER J.C. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341:33–38. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- MONTIEL J.L., CORNILLE F., ROQUES B.P., NOBLE F. Nociceptin/orphanin FQ metabolism: role of aminopeptidase and endopeptidase 24.15. J. Neurochem. 1997;68:354–361. doi: 10.1046/j.1471-4159.1997.68010354.x. [DOI] [PubMed] [Google Scholar]
- MORIKAWA H., FUKUDA K., MIMA H., SHODA T., KATO S., MORI K. Nociceptin receptor-mediated Ca2+ channel inhibition and its desensitization in NG108-15 cells. Eur. J. Pharmacol. 1998;351:247–252. doi: 10.1016/s0014-2999(98)00306-9. [DOI] [PubMed] [Google Scholar]
- MULDER A.H., SCHOFFELMEER A.N.M.Multiple opioid receptors and presynaptic modulation of neurotransmitter release in the brain Opioids I. Handbook of experimental pharmacology 1993Berlin: Springer; 125–144.Vol. 104/I. eds. Herz, A., Akil, H. & Simon, E.J. pp [Google Scholar]
- MURPHY N.P., ACKERSON L., MAIDMENT N.T. Orphanin FQ modulation of mesolimbic dopamine release. Soc. Neurosci. Abstr. 1997;23:1208. [Google Scholar]
- MURPHY N.P., LY H.T., MAIDMENT N.T. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience. 1996;75:1–4. doi: 10.1016/0306-4522(96)00322-3. [DOI] [PubMed] [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MENZIES J.R.W., CORBETT A.D., MCKNIGHT A.T. Pharmacological studies on the ‘orphan' opioid receptor in central and peripheral sites. Can. J. Physiol. Pharmacol. 1998;76:304–313. [PubMed] [Google Scholar]
- NICOL B., LAMBERT D.G., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T. Nociceptin induced inhibition of K+ evoked glutamate release from rat cerebrocortical slices. Br. J. Pharmacol. 1996;119:1081–1083. doi: 10.1111/j.1476-5381.1996.tb16007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REINSCHEID R.K., ARDATI A., MONSMA F.J., CIVELLI O. Structure-activity relationship studies on the novel neuropeptide orphanin FQ. J. Biol. Chem. 1996;271:14163–14168. doi: 10.1074/jbc.271.24.14163. [DOI] [PubMed] [Google Scholar]
- REINSCHEID R.K., NOTHACKER H.P., BOURSON A., ARDATI A., HENNINGSEN R.A., BUNZOW J.R., GRANDY D.K., LANGEN H., MONSMA F.J., CIVELLI O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., BEHLING A., LÜMMEN G., MALINOWSKA B., GÖTHERT M. Mutual interaction of histamine H3-receptors and α2-adrenoceptors on noradrenergic terminals in mouse and rat brain cortex. Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;345:639–646. doi: 10.1007/BF00164577. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., GÖTHERT M. Interactions between the presynaptic α2-autoreceptor and presynaptic inhibitory heteroreceptors on noradrenergic neurones. Brain Res. Bull. 1998;47:129–132. doi: 10.1016/s0361-9230(98)00068-9. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., WERTHWEIN S., KATHMANN M., BAUER U. Nociceptin inhibits noradrenaline release in the mouse brain cortex via presynaptic ORL1 receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:418–422. doi: 10.1007/pl00005273. [DOI] [PubMed] [Google Scholar]
- SHIMOHIGASHI Y., HATANO R., FUJITA T., NAKASHIMA R., NOSE T., SUJAKU T., SAIGO A., SHINJO K., NAGAHISA A. Sensitivity of opioid receptor-like receptor ORL1 for chemical modification on nociceptin, a naturally occurring nociceptive peptide. J. Biol. Chem. 1996;271:23642–23645. doi: 10.1074/jbc.271.39.23642. [DOI] [PubMed] [Google Scholar]
- SMITH J.A.M., LESLIE F.M.Use of organ systems for opioid bioassay Opioids I. Handbook of experimental pharmacology 1993Berlin: Springer; 53–78.Vol. 104/I. eds. Herz, A., Akil, H. & Simon, E.J. pp [Google Scholar]
- STARKE K., GÖTHERT M., KILBINGER H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 1989;69:864–989. doi: 10.1152/physrev.1989.69.3.864. [DOI] [PubMed] [Google Scholar]
- VAUGHAN C.W., INGRAM S.L., CHRISTIE M.J. Actions of the ORL1 receptor ligand nociceptin on membrane properties of rat periaqueductal gray neurons in vitro. J. Neurosci. 1997;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VON KÜGELGEN I., KOCH H., STARKE K. P2-receptor-mediated inhibition of serotonin release in the rat brain cortex. Neuropharmacology. 1997;36:1221–1227. doi: 10.1016/s0028-3908(97)00101-9. [DOI] [PubMed] [Google Scholar]