

Abstract
Nociceptin(NC) is the endogenous ligand for the opioid receptor like-1 receptor (NC-receptor). [Phe1ΨC(CH2-NH)Gly2]Nociceptin(1-13)NH2 ([F/G]NC(1-13)NH2) has been reported to antagonize NC actions in peripheral guinea-pig and mouse tissues. In this study, we investigated the effects of a range of NC C-terminal truncated fragments and [F/G]NC(1-13)NH2 on NC receptor binding, glutamate release from rat cerebrocortical slices (rCX), inhibition of cyclic AMP accumulation in CHO cells expressing the NC receptor (CHONCR) and electrically evoked contractions of the rat vas deferens (rVD).
In radioligand binding assays, a range of ligands inhibited [125I]-Tyr14-NC binding in membranes from rCX and CHONCR cells. As the peptide was truncated there was a general decline in pKi. [F/G]NC(1-13)NH2 was as potent as NC(1-13)NH2.
The order of potency for NC fragments to inhibit cyclic AMP accumulation in whole CHONCR cells was NCNH2⩾NC=NC(1-13)NH2>NC(1-12)NH2>>NC(1-11)NH2. [F/G]NC(1-13)NH2 was a full agonist with a pEC50 value of 8.65.
NCNH2 and [F/G]NC(1-13)NH2 both inhibited K+ evoked glutamate release from rCX with pEC50 and maximum inhibition of 8.16, 48.5±4.9% and 7.39, 58.9±6.8% respectively.
In rVD NC inhibited electrically evoked contractions with a pEC50 of 6.63. Although [F/G]NC(1-13)NH2, displayed a small (instrinsic activity α=0.19) but consistent residual agonist activity, it acted as a competitive antagonist (pA2 6.76) in the rVD.
The differences between [F/G]NC(1-13)NH2 action on central and peripheral NC signalling could be explained if [F/G]NC(1-13)NH2 was a partial agonist with high strength of coupling in the CNS and low in the periphery. An alternative explanation could be the existence of central and peripheral receptor isoforms.
Keywords: Nociceptin/orphanin FQ, nociceptin receptor, binding, cyclic AMP, glutamate release, vas deferens, rat
Introduction
Nociceptin/orphanin FQ (NC) is the endogenous ligand for the guanine nucleotide-binding regulatory protein (G-protein) coupled opioid receptor like-1 (ORL-1) receptor (Meunier et al., 1995; Reinscheid et al., 1995). In the remainder of this manuscript we will use the terminology NC-receptor. Although this receptor has significant (63–65%) sequence homology (see Meunier, 1997 for review) to OP1, OP2 and OP3 opioid receptors it does not bind traditional opioid ligands (see Meunier, 1997 for review). Moreover, NC does not bind to the classical opioid receptors with high affinity (Meunier et al., 1995; Reinscheid et al., 1995), suggesting a distinct divergence between the NC/NC-receptor system and the opioid systems. NC produced hyperalgesia and/or antiopioid actions when injected intracerebroventricularly whilst it exhibits antinociceptive effects when administered intrathecally (see Meunier, 1997 for review). Despite these observations, the cellular actions of NC are similar to those of opioids. NC activates the NC-receptor to either inhibit adenylyl cyclase (Meunier et al., 1995; Reinscheid et al., 1995) and/or voltage-gated calcium channels (Connor et al., 1996b; Abdulla & Smith, 1997) or to stimulate an inwardly rectifying potassium conductance (Matthes et al., 1996; Connor et al., 1996a; Vaughan & Christie, 1996). These actions are expected to reduce neuronal excitability and neurotransmitter release. Indeed, we have recently reported NC induced inhibition of K+ evoked glutamate release from rat cerebrocortical slices (Nicol et al., 1996) and others also reported inhibition of glutamatergic transmission (Faber et al., 1996; Vaughan et al., 1997), GABA-ergic transmission (Vaughan et al., 1997), dopamine (Murphy et al., 1996), noradrenaline (Schlicker et al., 1998), tachykinin (Giuliani & Maggi, 1996; Helyes et al., 1997) and acetylcholine (Patel et al., 1997; Neal et al., 1997) release.
The transcript encoding the NC-receptor is found not only in the central nervous system (Darland & Grandy, 1998) but also in peripheral tissues such as rat intestine, skeletal muscle, vas deferens, spleen and lymphocytes (Wang et al., 1994; Peluso et al., 1998; Wick et al., 1995). NC is reported to inhibit electrically induced contractions in several peripheral isolated preparations (see Meunier, 1997 for review).
In most studies, naloxone did not antagonize the effects of NC excluding an activation of OP1, OP2 and OP3 opioid receptors. However, it had been difficult to demonstrate the direct activation of the NC-receptor by NC due to the absence of a selective antagonist for this receptor. Recently, a selective competitive antagonist for the NC-receptor, [Phe1Ψ(CH2-NH)Gly2]nociceptin(1-13)NH2 ([F/G]NC(1-13)NH2) has been reported by three of us (GC, RG, RB, Guerrini et al., 1998; Calo' et al., 1998a).
The objective of this study was to investigate the pharmacological properties of a range of NC C-terminal truncated fragments that have already been studied in different models (Reinscheid et al., 1996; Dooley & Houghten, 1996; Guerrini et al., 1997; Calo' et al., 1997; Rossi et al., 1997; Butour et al., 1997) and [F/G]NC(1-13)NH2 using preparations expressing NC-receptors from the central nervous system and periphery. We describe binding characteristics of these agents in membranes from rat cerebrocortex and CHO cells expressing the recombinant human NC-receptor (CHONCR). In functional studies, effects on cyclic AMP accumulation and K+ evoked glutamate release (Nicol et al., 1996) were examined in whole CHONCR cells and rat cerebrocortical slices, respectively. The effects of nociceptin and [F/G]NC(1-13)NH2 on electrically stimulated rat vas deferens were also tested to enable a direct comparison to be made.
Methods
Sources of chemicals, reagents and equipment
The chemicals and their sources were as follows; NC, its amidated fragments and [F/G]NC(1-13)NH2 were prepared at one of our institutes as previously described (Guerrini et al., 1997, Calo' et al., 1998a). Cell culture medium (DMEM, F12), foetal calf serum, glutamine, hygromycin B and G418 were from Gibco (Paisley, U.K.). [125I]-Tyr14-nociceptin (2000 Ci mmol−1) and [2,8-3H]-cAMP (28.4 Ci mmol−1) were from Amersham (Little Chalfont, U.K.) and NEN DuPont (Boston, MA, U.S.A.), respectively. HEPES was from USB (Cleveland, OH, U.S.A.) and MgSO4 was from Fisons Scientific (Loughborough, U.K.). Phosphoramidon was from peptide institute (Osaka, Japan). All other reagents were from Sigma Chemical Co. (Poole, U.K.). CHO cells stably expressing the NCR receptor, CHONCR (transfected using pCIN5) were obtained from Dr F Marshall and Mrs N Bevan of Glaxo-Wellcome, Stevenage, Herts, U.K.
[125I]-Tyr14-NC binding
Female Wistar rats (250–300 g) were decapitated following cervical dislocation. The brain was removed and rapidly transferred to ice-cold buffer (Tris-HCl 50 mM, pH 7.4) and the cerebrocortex dissected. CHONCR cells were maintained in DMEM:F12 (50 : 50) containing 5% foetal calf serum, 2 mM glutamine, 200 μg ml−1 hygromycin B and 200 μg ml−1 G418. Cultures were maintained at 37°C in 5% CO2/humidified air. When confluency was reached (3–4 days), cells were harvested for use by the addition of 0.9% saline containing HEPES (10 mM) and EDTA (0.02%). These tissues were homogenized on ice and the homogenate was centrifuged at 13,500 r.p.m for 10 min at 4°C and the pellet resuspended in Tris-HCl buffer. This procedure was repeated twice more. Membranes were prepared fresh each day. All binding assays were performed in 1 ml of buffer (Tris-HCl 50 mM, MgSO4 5 mM containing 30 μM of peptidase inhibitors; captopril, amastatin, bestatin and phosphoramidon and Bovine serum albumin (BSA) 0.5%, pH 7.4) for 30 min at room temperature with 1–2 pM [125I]-Tyr14-NC and <0 μg of membrane protein from rat cerebrocortex and 3–4 pM [125I]-Tyr14-NC and ∼2 μg membrane protein from CHONCR cells. Non-specific binding (NSB) was defined in the presence of 10−6M nociceptin. These optimal incubation conditions were determined empirically. Following incubation, bound and free radioactivities were separated by vacuum filtration using a Brandel cell harvester. Harvester papers (Whatman GF/B) were soaked in polyethylenimine (0.5%) to reduce NSB, and loaded onto the harvester wet. For determination of the maximal binding capacity (Bmax) and the equilibrium dissociation constant (KD), a pseudo-isotope dilution study was performed (i.e., competition between NC and NC with 125I-Tyr14 substitution as described by Ardati et al., 1997). In both pseudo-isotope dilution and displacement studies, approximately 1–4 pM of [125I]-Tyr14-NC with increasing concentrations of unlabelled displacers were used.
Inhibition of cyclic AMP accumulation
For the measurement of cyclic AMP, whole CHONCR cells were incubated in 0.3 ml Krebs-HEPES buffer containing BSA (0.5%), peptidase inhibitors (30 μM; captopril, amastatin, bestatin, phosphoramidon) 1-isobutyl-4-methylxanthine (IBMX; 1 mM) and forskolin (1 μM). After 15 min at 37°C, the reaction was terminated by the addition of 20 μl HCl (10 M). Following neutralization by the addition of 20 μl NaOH (10 M) and 180 μl of Tris-HCl (1 M, pH 7.4) and centrifugation (13,000×g for 2 min), cyclic AMP was measured in the supernatant by a protein-binding assay using a binding protein from bovine adrenal cortex (Okawa et al., 1998). In some experiments, untransfected CHO cells (CHOwt) and CHONCR cells preincubated with 100 ng ml−1 pertussis toxin (PTX) in culture medium for 24 h were used to examine the involvement of the NC-receptor and pertussis toxin sensitive G proteins, respectively.
Effects on glutamate release from rat cerebrocortical slices
This was performed as described previously (Nicol et al., 1996). Briefly, female Wistar rats (250–300 g) were decapitated following cervical dislocation. The brain was removed and rapidly transferred to ice-cold oxygenated (95% O2 and 5% CO2) Krebs buffer, pH 7.4. Slices were cut and washed three times in fresh Krebs buffer prior to agitation in a shaking water bath at 37°C for 40 min; 1 ml of gravity-packed slices were pipetted into a perfusion chamber. Slices were perfused at 37°C for 60 min at 1 ml min−1 prior to collection of 2 min fractions. A 2 min pulse of 46 mM K+ (Na+ adjusted) was applied (S1) following 6 min of perfusion. Slices were perfused for a further 30 min, prior to the second application of a 2 min pulse of 46 mM K+ (S2). Perfusate glutamate concentrations were measured fluorimetrically, expressed relative to the mean of the first three basal samples and S2/S1 ratios were calculated. To examine the effects of NC-receptor ligands on glutamate release they were applied immediately after S1 until the end of the experiment (also present during S2) in various combinations.
Effects on electrically evoked contractions of the rat vas deferens (rVD)
Male Sprague Dawley rats (350–400 g) were decapitated, the vas deferens isolated and suspended in 10 ml organ baths containing Krebs solution ((in mM): NaCl 118.5, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, CaCl2 1.8, glucose 10) oxygenated with 95% O2 and 5% CO2 at 37°C. In some experiments the peptidase inhibitor cocktail (30 μM; captopril, amastatin, bestatin, phosphoramidon) was added to the incubation buffer 30 min before peptide application. A resting tension of 1 g was applied to the tissue. The vas deferens were continuously stimulated through two platinum ring electrodes with supramaximal voltage rectangular pulses of 1 msec duration and 0.1 Hz frequency. The electrically evoked contractions were measured isotonically with a strain gauge transducer (Basile 7006) and recorded on a Linseis multichannel chart recorder (model 2005). After an equilibration period of about 60 min the contractions induced by electrical field stimulation (EFS) were stable; at this time, cumulative concentration-response curves (crc) to NC were performed (0.5 log unit steps). When required, the NC receptor antagonist, [F/G]NC(1-13)NH2, or the non selective opioid receptor antagonist, naloxone, were added to the medium 15 min before performing crc to NC.
Data analysis
Data are presented as the mean±s.e.mean (n, independent experiments). In binding studies, Bmax and pKD were obtained from pseudo-isotope dilution studies. Concentration of displacers producing 50% displacement of specific binding (pIC50) was corrected for the competing mass of [125I]-Tyr14-NC to yield pKi. All curve fitting was performed using PRISM-V2.0 (GraphPAD software, San Diego, CA, U.S.A.). Statistical comparisons of paired samples were made using Freidmans analysis of variance and Wilcoxon Rank sum test as appropriate and considered significant when P<0.05.
Results
[125I]-Tyr14-NC binding
The binding of NC assessed using [125I]-Tyr14-NC (pseudo-isotope dilution) was concentration-dependent and saturable. Bmax (fmol mg protein−1) and pKD values in rat cerebrocortex (from Okawa et al., 1998) and CHONCR cells were 179.7±15.3, 10.26±0.09 (n=5) and 1716±363, 9.70±0.23 respectively (n=3). Binding slope factors in both membrane preparations were close to unity (1.04±0.05 and 0.93±0.07 in rat cerebrocortical and CHONCR membranes, respectively). NC and a range of NC fragments/analogues examined all produced concentration-dependent inhibition of [125I]-Tyr14-NC binding in both membrane preparations, with pKi values shown in Table 1. There were no differences between NC and NCNH2 in either preparation. In both preparations as the peptide was truncated from 13 to 9 amino acids there was a general decline in pKi. There were no significant differences between [F/G]NC(1-13)NH2 and NC(1-13)NH2 in either preparation (P<0.05) (Figure 1). In both preparations naloxone <10 μM did not inhibit [125I]-Tyr14-NC binding (data not shown).
Table 1.
Inhibition of [125I]-Tyr14-nociceptin (for graphical representation see Figure 1)

Figure 1.
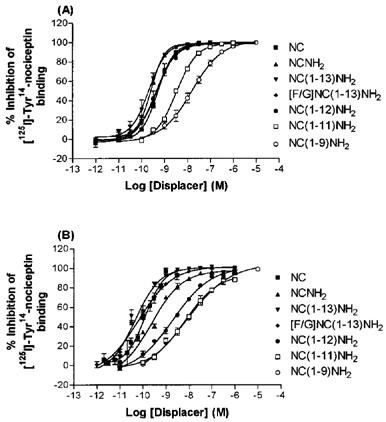
Inhibition of [125I]-Tyr14-nociceptin binding by nociceptin fragments and [F/G]NC(1-13)NH2 in membranes from rat cerebrocortex (A) and from CHO cells expressing the human nociceptin receptor (B). All agents produced dose-dependent inhibition of [125I]-Tyr14-nociceptin binding. For pKi values, see Table 1, (n=4–5).
Inhibition of cyclic AMP accumulation
All agents examined inhibited forskolin-stimulated cyclic AMP accumulation in CHONCR cells in a concentration-dependent manner (Figure 2) with pEC50 and maximal inhibition values shown in Table 2. In experiments using CHOwt cells and CHONCR cells preincubated with PTX, neither NC nor [F/G]NC(1-13)NH2 inhibited forskolin stimulated cyclic AMP accumulation (Figure 3). These results demonstrate the involvement of the NC-receptor and a pertussis toxin sensitive G protein(s) in the inhibition of cyclic AMP accumulation. To examine further a possible antagonism of NC induced inhibition of cyclic AMP formation by [F/G]NC(1-13)NH2, cells were incubated in the presence or absence of 3×10−9 M [F/G]NC(1-13)NH2. This treatment produced no appreciable shift in the concentration response curve compared to control (pEC50: control=9.82±0.04; +[F/G]NC(1-13)NH2=9.67± 0.06). In two additional experiments using 10−7 and 10−6 M [F/G]NC(1-13)NH2 (and despite marked agonist activity) we were also unable to demonstrate any antagonist activity (data not shown).
Figure 2.

Effects of nociceptin fragments and [F/G]NC(1-13)NH2 on forskolin stimulated cyclic AMP accumulation in whole CHO cells expressing the human nociceptin receptor. All agents produced dose-dependent inhibition of cyclic AMP acculumation. For pIC50 and Imax values, see Table 2, (n=5–7).
Table 2.
Inhibition of cyclic AMP formation in CHONCR cells (for graphical representation see Figure 2)

Figure 3.
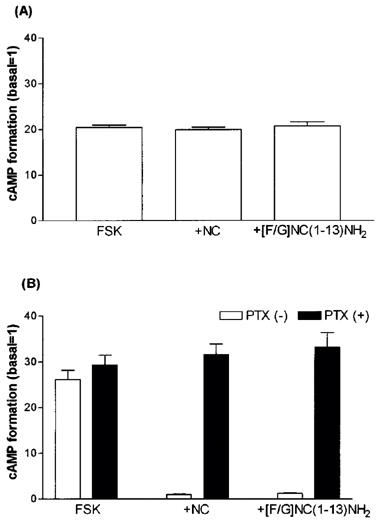
cyclic AMP assay carried out in untransfected CHO cells (A) and in CHO cells expressing the human nociceptin receptor preincubated with 100 ng ml−1 of pertussis toxin (B). Neither NC nor [F/G]NC(1-13)NH2 inhibited forskolin stimulated cyclic AMP accumulation (n=5).
Effects on glutamate release from rat cerebrocortical slices
NCNH2 produced a concentration-dependent inhibition of K+ evoked glutamate release (Figure 4) from rat cerebrocortical slices with maximal inhibition of 48.5±4.9% and a pEC50 of 8.16. These values were not markedly different from our previous report using NC (Nicol et al., 1996). [F/G]NC(1-13)NH2 produced a concentration-dependent inhibition of K+ evoked glutamate release with maximal inhibition of 58.9±6.8 and a pEC50 of 7.39. The inhibition produced by [F/G]NC(1-13)NH2 was naloxone insensitive (Control S2/S1 ratio (n=5) 0.82±0.17, +100 nM [F/G]NC(1-13)NH2 0.48±0.07, +[F/G]NC(1-13)NH2 and 10 μM Naloxone 0.36±0.07). In addition NC(1-13)NH2 (the template for [F/G]NC(1-13)NH2) at 300 nM inhibited release by 43.1±9.9% which was similar to that of NCNH2 and NC (Nicol et al., 1996).
Figure 4.
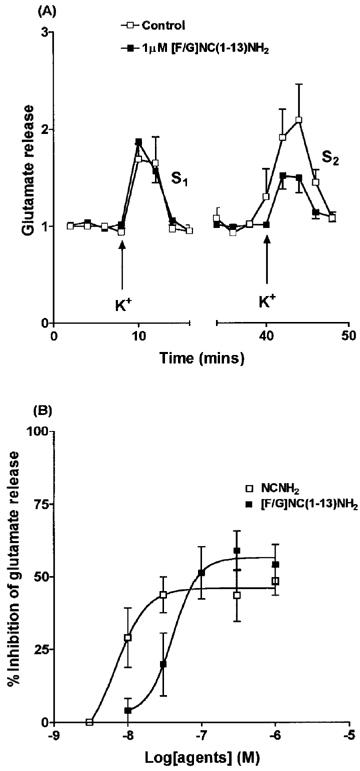
Effects of NCNH2 and [F/G]NC(1-13)NH2 on K+ evoked glutamate release from rat cerebrocortical slices. Time course for effects of [F/G]NC(1-13)NH2 is shown in (A). In (B) both agents produced dose-dependent inhibition of K+ evoked glutamate release. (n=5 except 3 nM NCNH2 where no inhibition was observed in two experiments). In (A) data are expressed relative to the mean of the first three fractions collected during S1 or S2.
Electrically stimulated rat vas deferens
NC inhibits the electrically induced contractions of the rVD with a pEC50 value of 6.63±0.2 and a maximal effect of about −80% of control values. Application of 1 μM naloxone did not modify the concentration response curve (crc) to the peptide (data not shown). Application of 1 μM [F/G]NC(1-13)NH2 was followed by an inhibition of the response of the rVD which reached about −15% of control values. In the presence of the pseudopeptide the crc to NC was shifted to the right by approximately one log unit, without any significant modification of the maximal effect induced by NC. A pA2 value of 6.85 was estimated for [F/G]NC(1-13)NH2 by means of the Gaddum Schild equation from this series of data. These experiments were repeated in the presence of the cocktail of peptidase inhibitors (30 μM amastatin, captopril, phosphoramidon and bestatin). The inhibitors caused per se a slight (about −10% of control values) but consistent inhibition of the electrically induced twitches; in addition in their presence the potency of NC was significantly enhanced (pEC50 7.37) while that of [F/G]NC(1-13)NH2 was unchanged (pA2 6.90).
We have also performed cumulative concentration response curves to [F/G]NC(1-13)NH2 (1 nM–10 μM). [F/G]NC(1-13)NH2 produced a concentration dependent inhibition of electrically induced twitches with a pEC50 of 6.84 and a maximal effect of −13±5% (corresponding to an intrinsic activity α of 0.19). The antagonistic properties of [F/G]NC(1-13)NH2 have also been evaluated over an extended range of concentrations (0.3–10 μM) to obtain data for a Schild analysis. As shown in Figure 5A, [F/G]NC(1-13)NH2 displaced to the right the crc to NC in a concentration dependent manner, the curves being parallel to the control, and reaching the same maximal effects. Figure 5B shows the corresponding Schild plot which was linear (r=0.99) with a slope of 0.91 which was not significantly different from unity. The extrapolated pA2 value was 6.76.
Figure 5.
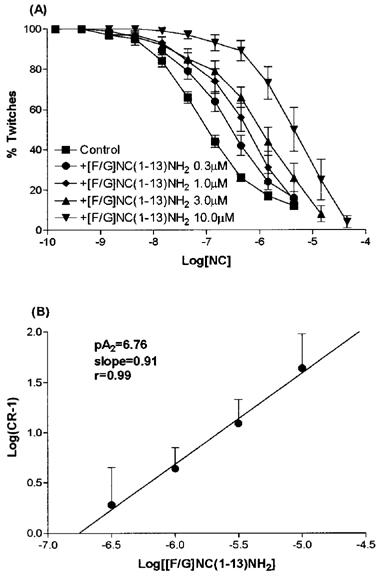
Effects of [F/G]NC(1-13)NH2 on NC induced inhibition of contractile response in rVD. [F/G]NC(1-13)NH2 produced a parallel rightward shift in the concentration response curve (A) which is depicted as a Schild plot (B). Data are mean±s.e.mean (n=5). In (B) the estimated pA2 for [F/G]NC(1-13)NH2 was 6.76.
Discussion
Binding experiments were performed in membranes from rat cerebrocortex with the native rat brain NC-receptor and compared with those from CHO cells expressing the recombinant human NC-receptor. Bmax and KD values in rat cerebrocortical membranes (from our previous report, Okawa et al., 1998) were consistent with others using the same radioligand (Ardati et al., 1997). Slope factors were close to unity in both reports and suggest that [125I]-Tyr14-NC is binding to a single class of receptors although Mathis et al. (1997) reported both high and low affinity binding sites in mouse brain membranes. Bmax in CHONCR cell membranes was consistent with others (Fawzi et al., 1997) while it was slightly higher than those of Reinscheid et al. (1996) and Ardati et al. (1997) using the same radioligand. These differences are merely due to transfection but it is important to compare our data with those using similar expression levels. Butour et al. (1997) and Adapa & Toll (1997) reported values similar to ours using [3H]-NC and [3H]-Tyr14-NC, respectively. In this study we observed binding slope factors close to unity in CHONCR cell membranes, again suggesting a single binding site in accordance with others (Reinscheid et al., 1996; Ardati et al.,1997, Adapa & Toll, 1997; Mathis et al., 1997). Adapa & Toll (1997) conducted binding experiments using intact CHONCR cells in physiological buffer similar to that used for our cyclic AMP assay. They observed high and low affinity binding sites. The latter sites were approximately two orders of magnitude weaker than that of CHONCR cell membranes in Tris buffer, making up 95% of the binding. This sensitivity of NC binding to Na+, in the same manner as opioid binding to the μ-opioid receptor (Yabaluri & Medzihradsky, 1997), must be borne in mind when comparing binding characteristics in Na+ free buffer with functional activities in physiological media. However, comparative studies using one system may yield potency information for a range of compounds. The ability to inhibit [125I]-Tyr14-NC binding was similar for NC, NCNH2, [F/G]NC(1-13)NH2, NC(1-13)NH2 and NC(1-12)NH2 in membranes from both rat cerebrocortex and CHONCR cells. Among NC fragments tested, NC(1-11)NH2 and NC(1-9)NH2 had lower affinity for the NC-receptor. Dooley & Houghten (1996) reported that NC(1-13)NH2 displaced [3H]-NC as potently as NC or NCNH2 in rat brain membranes while NC(1-12)NH2 and NC(1-9)NH2 were one and three orders of magnitude weaker than the natural peptide, respectively. We recently confirmed this order of affinity for NC fragments using mouse brain membranes (Varani et al., 1998). Butour et al. (1997) observed significant differences in Ki values between NC, NC(1-13), NC(1-12) and NC(1-9) (0.13, 4.0, 145 and 4300 nM, respectively) in CHONCR cell membranes. However in this study [3H]-NC and unamidated fragments were used. In our present studies with rat cerebrocortex and CHONCR we did not observe this large difference in potency between NC(1-12)NH2 and NC(1-13)NH2 in binding assays (but see below for cyclic AMP studies). These differences are difficult to explain but could potentially result from a number of factors including, the presence of peptidase inhibitors in our studies, differences in radioligand used, different buffer systems or a combination of these.
In functional studies, the ability to inhibit forskolin stimulated cyclic AMP accumulation was used as an indicator of NC-receptor activation in CHONCR cells. The use of cells transfected with a target receptor that couples with a particular cellular process enables investigation of drug interaction with the expressed receptor. NCNH2, NC(1-13)NH2 were as potent as NC, NC(1-12)NH2 and [F/G]NC(1-13)NH2 were 17 and 13 fold less potent respectively. NC(1-11)NH2, NC(1-9)NH2 were 200 fold and 1096 fold less potent than NC, respectively. IC50 of NC was similar to that previously reported (Fawzi et al., 1997; Adapa & Toll, 1997; Butour et al., 1997; Reinscheid et al., 1996). Butour et al. (1997) reported decreasing potency of NC by C-terminal truncation for inhibition of adenylyl cyclase (EC50 value of 0.8, 7.8, 67, >10,000 nM for NC, NC(1-13), NC(1-12) and NC(1-9) respectively) again using unamidated fragments. Reinscheid et al. (1996) reported the inability of NC(1-11) to inhibit adenylyl cyclase, claiming that the entire structure is necessary for biological activity. In the mouse vas deferens and the guinea-pig ileum, NC(1-13)NH2 was the shortest fragment to retain activity similar to NC (Guerrini et al., 1997; Calo' et al., 1997) and is consistent with our data. Similar results were also obtained in electrically stimulated rat vas deferens (Calo' et al., 1998c) where NC(1-12)NH2 was less potent (about 10 fold) than NC(1-13)NH2. In addition, in the rat in vivo NC(1-13)NH2 was as potent as NC in stimulating food intake while NC(1-12)NH2 was inactive (Polidori et al., 1998).
In our hands [F/G]NC(1-13)NH2 behaved as a full agonist in glutamate release and cyclic AMP studies. In a recent short report Butour et al. (1998) have also reported agonist action of [F/G]NC(1-13)NH2 at the level of cyclic AMP accumulation in transfected CHO cells. This is in contrast to the original (Guerrini et al., 1998) inhibitory effects of this compound reported in mouse and guinea-pig peripheral tissues and with the data we obtained in the rat vas deferens (see below). Reduced cellular cyclic AMP levels produced by opioids inhibit the Ih current and is speculated to decrease action potential frequency (Ingram & Williams, 1994). Although inhibition of cyclic AMP is only one of the assumed mechanisms of action of NC following its binding to the NC-receptor, it provides valuable information on specific activation of the this receptor. The observed inhibition is PTX sensitive indicating the coupling to the Gi/Go class of G-proteins. Consistent with these data we have shown that both NC and [F/G]NC(1-13)NH2 actions on cyclic AMP formation are completely reversed by PTX pretreatment.
We have previously demonstrated that NC inhibits K+ evoked glutamate release from rat cerebrocortical slices in a concentration-dependent and naloxone insensitive manner (Nicol et al., 1996). This assay system was used to further investigate the central effects of [F/G]NC(1-13)NH2. [F/G]NC(1-13)NH2 was as efficacious as NCNH2 or NC for inhibition of K+ evoked glutamate release. In addition, there was no difference between the inhibition produced by 300 nM [F/G]NC(1-13)NH2, 300 nM NCNH2 and 300 nM NC(1-13)NH2. Agonist activity of [F/G]NC(1-13)NH2 at central NC functional sites has also been described in other experimental models. In particular, [F/G]NC(1-13)NH2 mimicked the action of NC (i) when injected intrathecally in the rat inducing analgesia (Xu et al., 1998; Candeletti et al., 1998), (ii) when injected intracerebroventricularly also in the rat inducing stimulation of food intake (Polidori et al., 1998); and finally (iii) when injected intracerebroventricularly in the mouse (Calo' et al., 1998b) inducing hyperalgesia and antimorphine actions. On the other hand, in the periphery [F/G]NC(1-13)NH2 exhibits antagonist activity (Guerrini et al., 1998). Specifically, in the rat vas deferens, [F/G]NC(1-13)NH2 antagonized NC effects with a pA2 value of 6.76. Worthy of mention is the fact that the peptidase inhibitor cocktail increases the apparent affinity of NC (Nicholson et al; 1998b) but not that of the pseudopeptide, indicating (in line with the findings by Xu et al., 1998) that [F/G]NC(1-13)NH2 is more metabolically stable compared with the natural peptide.
The early indications of [F/G]NC(1-13)NH2 agonist activity at central NC functional sites and antagonist activity at peripheral NC functional sites is now not so well defined. Nicholson et al. (1998a) reported in rat cortex [35S]GTPγS assay that [F/G]NC(1-13)NH2 possessed some efficacy but also inhibited the response to NC with a pA2 of 8.6. Similar results (intrinsic activity α=0.45, pA2 7.2) were obtained by Schlicker et al. (1998) studying the inhibitory effect of NC on [3H]-noradrenaline release from mouse cerebral cortex slices. Of particular interest to the present study is the report of Toll et al. (1998) in which [F/G]NC(1-13)NH2 inhibited cyclic AMP formation and enhanced [35S]-GTPγS binding in CHO and SH-SY5Y cells transfected with the human NC receptor. However, the authors reported that [F/G]NC(1-13)NH2 was an antagonist at low levels of receptor expression and a partial agonist at high levels of expression.
What is the nature of the reported differences between central and peripheral NC functional sites? The most likely explanation is that [F/G]NC(1-13)NH2 is a partial agonist at the NC receptor and that the strength of NC signaling is very high in the CNS and in high expressing transfected systems, and low in the periphery. If these suppositions are true, the estimated intrinsic activity of [F/G]NC(1-13)NH2 will be near 0 in the periphery and near 1 in the CNS. An alternative but much more speculative explanation is that the differences may result from different NC receptor subtypes. However, the evidence in support of this hypothesis is scant and to date only a single gene encoding the NC receptor has been identified. Certainly, the currently identified splice variants show distribution patterns that encompass both central and peripheral tissues (Wang et al., 1994; Wick et al., 1995; Peluso et al., 1998). Addressing these questions will represent an important area for future studies.
Acknowledgments
The authors would like to thank Mrs N Bevan and Dr F Marshall of Glaxo-Wellcome, Stevenage, Herts for providing CHO cells expressing the NC receptor and Professor Regoli for helpful discussions. Supported by a Project grant (DGL) from The Wellcome Trust.
Abbreviations
- CHO
Chinese hamster ovary
- NC
Nociceptin/orphanin FQ
- PTX
pertussis toxin
References
- ABDULLA F.A., SMITH P.A. Nociceptin inhibits T-type Ca2+ channel current in rat sensory neurons by a G-protein-independent mechanism. J. Neurosci. 1997;17:8721–8728. doi: 10.1523/JNEUROSCI.17-22-08721.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADAPA I.D., TOLL L. Relationship between binding affinity and functional activity of nociceptin/orphanin FQ. Neuropeptides. 1997;31:403–408. doi: 10.1016/s0143-4179(97)90032-9. [DOI] [PubMed] [Google Scholar]
- ARDATI A., HENNINGSEN R.A., HIGELIN J., REINSCHEID R.K., CIVELLI O., MONSMA F.J., JR Interaction of [3H] orphanin FQ and 125I-Tyr14-orphanin FQ with the orphanin FQ receptor: kinetics and modulation by cations and guanine nucleotides. Mol. Pharmacol. 1997;51:816–824. doi: 10.1124/mol.51.5.816. [DOI] [PubMed] [Google Scholar]
- BUTOUR J.L., MOISAND C., MAZARGUIL H., MOLLEREAU C., MEUNIER J.C. Recognition and activation of the opioid receptor-like ORL1 receptor by nociceptin, nociceptin analogs and opioids. Eur. J. Pharmacol. 1997;321:97–103. doi: 10.1016/s0014-2999(96)00919-3. [DOI] [PubMed] [Google Scholar]
- BUTOUR J.L., MOISAND C., MOLLEREAU C., MEUNIER J.C. [Phe1ψ(CH2-NH)Gly2]nociceptin-(1-13)-NH2 is an agonist of the nociceptin (ORL1) receptor. Eur. J. Pharmacol. 1998;349:R5–R6. doi: 10.1016/s0014-2999(98)00273-8. [DOI] [PubMed] [Google Scholar]
- CALO' G., GUERRINI R., BIGONI R., RIZZI A., BIANCHI C., REGOLI D., SALVADORI S. Structure-Activity Study of the Nociceptin(1-13)-NH2 N-Terminal Tetrapeptide and Discovery of a Nociceptin Receptor Antagonist. J. Med. Chem. 1998a;41:3360–3366. doi: 10.1021/jm970805q. [DOI] [PubMed] [Google Scholar]
- CALO' G., RIZZI A., BODIN M., NEUGEBAUER W., SALVADORI S., GEURRINI R., BIANCHI C., REGOLI D. Pharmacological characterization of nociceptin receptor. Can. J. Physiol. Pharmacol. 1997;75:713–718. [PubMed] [Google Scholar]
- CALO' G., RIZZI A., MARZOLA G., GUERRINI R., SALVADORI S., BEANI L., REGOLI D., BIANCHI C. Pharmacological characterization of the nociceptin receptor mediating hyperalgesia in the mouse tail withdrawal assay. Br. J. Pharmacol. 1998b;125:373–378. doi: 10.1038/sj.bjp.0702087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALO' G., RIZZI A., REGOLI D.Pharmacology of nociceptin receptors Peptidergic G-protein coupled receptors: from basic research to clinical applications 1998cAmsterdam, Netherlands: IOS press; eds Geppetti, P., Swartz, T.W., Regoli, D. & Muller-Esterl, W(in press) [Google Scholar]
- CANDELETTI S., GUERRINI R., CALO' G., FERRI S.Effects of the nociceptin receptor antagonist Phe1ψ(CH2NH)Gly2NC(1-13)NH2 on nociception in rats 29th International Narcotic Research Conference 1998A170Garmisch-Partenkirchen: Germany; 20–25 July, 1998 [Google Scholar]
- CONNOR M., VAUGHAN C.W., CHIENG B., CHRISTIE M.J. Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus in vitro. Br. J. Pharmacol. 1996a;119:1614–1618. doi: 10.1111/j.1476-5381.1996.tb16080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONNOR M., YEO A, HENDERSON G. The effect of nociceptin on Ca2+ channel current and intracellular Ca2+ in the SH-SY5Y human neuroblastoma cell line. Br. J. Pharmacol. 1996b;118:205–207. doi: 10.1111/j.1476-5381.1996.tb15387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DARLAND T., GRANDY D.K. The orphanin FQ system: an emerging target for the management of pain. Br. J. Anaesthesia. 1998;81:29–37. doi: 10.1093/bja/81.1.29. [DOI] [PubMed] [Google Scholar]
- DOOLEY C., HOUGHTEN R.A. Orphanin FQ: receptor binding and analog structure activity relationships in rat brain. Life. Sci. 1996;59:23–29. doi: 10.1016/0024-3205(96)00261-5. [DOI] [PubMed] [Google Scholar]
- FABER E.S.L., CHAMBERS J.P., EVANS R.H., HENDERSON G. Depression of glutamatergic transmission by nociceptin in the neonatal rat hemisected spinal cord preparation in vitro. Br. J. Pharmacol. 1996;119:189–190. doi: 10.1111/j.1476-5381.1996.tb15969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAWZI A.B., ZHANG H., WEIG B., HAWES B., GRAZIANO M.P. Nociceptin activation of the human ORL1 receptor expressed in Chinese hamster ovary cells: functional homology with opioid receptors. Eur. J. Pharmacol. 1997;336:233–242. doi: 10.1016/s0014-2999(97)01227-2. [DOI] [PubMed] [Google Scholar]
- GIULIANI S., MAGGI C.A. Inhibition of tachykinin release from peripheral endings of sensory nerves by nociceptin, a novel opioid peptide. Br. J. Pharmacol. 1996;118:1567–1569. doi: 10.1111/j.1476-5381.1996.tb15576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUERRINI R., CALO' G., RIZZI A., BIANCHI C., LAZARUS L.H., SALVADORI S., TEMUSSI P.A., REGOLI D. Address and message sequences for the nociceptin receptor: A structure-activity study of nociceptin-(1-13)-peptide amide. J. Med. Chem. 1997;40:1789–1793. doi: 10.1021/jm970011b. [DOI] [PubMed] [Google Scholar]
- GUERRINI R., CALO' G., RIZZI A., BIGONI R., BIANCHI C., SALVADORI S., REGOLI D. A new selective antagonist of the nociceptin receptor. Br. J. Pharmacol. 1998;123:163–165. doi: 10.1038/sj.bjp.0701640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELYES Z., NEMETH J., PINTER E., SZOLCSANYI J. Inhibition by nociceptin of neurogenic inflammation and the release of SP and CGRP from sensory nerve terminals. Br. J. Pharmacol. 1997;121:613–615. doi: 10.1038/sj.bjp.0701209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INGRAM S.L., WILLIAMS J.T. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–186. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- MATHIS J.P., RYAN-MORO J., CHANG A., HOM J.S.H., SCHEINBERG D.A., PASTERNAK G.W. Biochemical evidence for orphanin FQ/nociceptin receptor heterogeneity in mouse brain. Biochem. Biophys. Res. Commun. 1997;230:462–465. doi: 10.1006/bbrc.1996.5867. [DOI] [PubMed] [Google Scholar]
- MATTHES H., SEWARD E.P., KIEFFER B., NORTH R.A. Functional selectivity of orphanin FQ for its receptor coexpressed with potassium channel subunit in Xenopus laevis oocytes. Mol. Pharmacol. 1996;50:447–450. [PubMed] [Google Scholar]
- MEUNIER J.C. Nociceptin/orphanin FQ and the opioid receptor-like ORL1 receptor. Eur. J. Pharmacol. 1997;340:1–15. doi: 10.1016/s0014-2999(97)01411-8. [DOI] [PubMed] [Google Scholar]
- MEUNIER J.C., MOLLEREAU C., TOLL L., SUAUDEAU C., MOISAND C., ALVINERIE P., BUTOUR J.L., GUILLEMOT J.C., FERRARA P., MONSARRAT B., MAZARGUIL H., VASSART G., PARMENTIER M., COSTENTIN J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- MURPHY N.P., LY H.T., MAIDMENT N.T. Intracerebroventricular orphanin FQ nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetised rats. Neuroscience. 1996;75:1–4. doi: 10.1016/0306-4522(96)00322-3. [DOI] [PubMed] [Google Scholar]
- NEAL M.J., CUNNINGHAM J.R., PATERSON S.J., MCKNIGHT A.T. Inhibition by nociceptin of the light-evoked release of Ach from retinal cholinergic neurones. Br. J. Pharmacol. 1997;120:1399–1400. doi: 10.1038/sj.bjp.0701135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLSON J.R., MASON S.L., LEE K., MCKNIGHT A.T.The effect of the agonist AC-RYYRWKNH2 and the antagonist Phe1ψ(CH2NH)Gly2NC(1-13)NH2 at the ORL1 receptor of central and peripheral sites 29th International Narcotic Research Conference 1998aA129Garmisch-Partenkirchen: Germany; 20-25 July, 1998 [Google Scholar]
- NICHOLSON J.R., PATERSON S.J., MENZIES J.R.W., CORBETT A.D., MCKNIGHT A.T. Pharmacological studies on the ‘orphan' opioid receptor in central and peripheral sites. Can. J. Physiol. Pharmacol. 1998b;76:304–313. [PubMed] [Google Scholar]
- NICOL B, , LAMBERT D.G., ROWBOTHAM D.J., SMART D., MCKNIGHT A.T. Nociceptin induced inhibition of K+ evoked glutamate release from rat cerebrocortical slices. Br. J. Pharmacol. 1996;119:1081–1083. doi: 10.1111/j.1476-5381.1996.tb16007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKAWA H., HIRST R.A., SMART D., MCKNIGHT A.T., LAMBERT D.G. Rat central ORL-1 receptor uncouples from adenylyl cyclase during membrane preparation. Neurosci. Lett. 1998;246:49–52. doi: 10.1016/s0304-3940(98)00228-6. [DOI] [PubMed] [Google Scholar]
- PATEL H.J., GIEMBYCZ M.A., SPICUZZA L., BARNES P.J., BELVISI M.G. Naloxone-insensitive inhibition of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by the novel opioid, nociceptin. Br. J. Pharmacol. 1997;120:735–736. doi: 10.1038/sj.bjp.0701013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PELUSO J., LAFORGE K.S., MATTHES H.W., KREEK M.J., KIEFFER B.L., GAV'ERIAUX-RUFF C. Distribution of nociceptin/orphanin FQ receptor transcript in human central nervous system and immune cells. J. Neuroimmunol. 1998;81:184–192. doi: 10.1016/s0165-5728(97)00178-1. [DOI] [PubMed] [Google Scholar]
- POLIDORI C., CALO' G., GUERRINI R., REGOLI D., MASSI M.Pharmacological characterization of the nociceptin receptor mediating hyperphagia. 12th International Conference on the Physiology of Food and Fluid Intake (ICPFFI XII) Joint meeting with the Society for the Study of Ingestive Behaviour (SSIB) 1998Pecs: Hungary; 5–8 July, 1998pg. 31 [Google Scholar]
- REINSCHEID R.K., ARDATI A., MONSMA F.J. , JR, CIVELLI O. Structure-activity relationship studies on the novel neuropeptide orphanin FQ. J. Biol. Chem. 1996;271:14163–14168. doi: 10.1074/jbc.271.24.14163. [DOI] [PubMed] [Google Scholar]
- REINSCHEID R.K., NOTHACKER H.P., BOURSON A., ARDATI A., HENNINGSEN R.A., BUNZOW J.R., GRANDY D.K., LANGEN H., MONSMA F.J. , JR, CIVELLI O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- ROSSI G.C., LEVENTHAL L., BOLAN E., PASTERNAK G.W. Pharmacological characterization of orphanin FQ/nociceptin and its fragments. J. Pharmacol. Exp. Ther. 1997;282:858–865. [PubMed] [Google Scholar]
- SCHLICKER E., WERTHWEIN S., KATHMANN M., BAUER U. Nociceptin inhibits noradrenaline release in the mouse brain cortex via presynaptic ORL1 receptors. N.S. Arch. Pharmacol. 1998;358:418–422. doi: 10.1007/pl00005273. [DOI] [PubMed] [Google Scholar]
- TOLL L., BURNSIDE J., BERZETEI-GURSKE I.Agonist activity of ORL1 antagonists is dependent upon receptor number 29th International Narcotic Research Conference 1998A89Garmisch-Partenkirchen: Germany; 20–25 July, 1998 [Google Scholar]
- VARANI K., CALO G., RIZZI A., MERIGHI S., TOTH G., GUERRINI R., SALVADORI S., BOREA P.A., REGOLI D. Nociceptin receptor binding in mouse forebrain membranes: thermodynamic characteristics and structure activity relationships. Br. J. Pharmacol. 1998;125:1485–1490. doi: 10.1038/sj.bjp.0702226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAUGHAN C.W., CHRISTIE M.J. Increase by the ORL1 receptor (opioid receptor-like1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br. J. Pharmacol. 1996;117:1609–1611. doi: 10.1111/j.1476-5381.1996.tb15329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAUGHAN C.W., INGRAM S.L., CHRISTIE M.J. Actions of the ORL1 receptor ligand nociceptin on membrane properties of rat periaqueductal gray neurons in vitro. J. Neuroscience. 1997;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J.B., JOHNSON P.S., IMAI Y., PERSICO A.M., OZENBERGER B.A., EPPLER C.M., UHL G.R. cDNA cloning of an orphan opiate receptor gene family member and its splice variant. FEBS. Lett. 1994;348:75–79. doi: 10.1016/0014-5793(94)00557-5. [DOI] [PubMed] [Google Scholar]
- WICK M.J., MINNERATH S.R., ROY S., RAMAKRISHNAN S., LOH H.H. Expression of alternate forms of brain orphan receptor mRNA in activated human peripheral blood lymphocytes and lymphocytic cell lines. Brain. Res. Mol. Brain. Res. 1995;32:342–347. doi: 10.1016/0169-328x(95)00096-b. [DOI] [PubMed] [Google Scholar]
- XU I.S., WIESANFELD-HALLIN Z., XU X-J. [Phe1ψ(CH2-NH)Gly2]-nociceptin-(1-13)NH2, a proposed antagonist of the nociceptin receptor, is a potent and stable agonist in the rat spinal cord. Neurosci. Lett. 1998;249:127–130. doi: 10.1016/s0304-3940(98)00411-x. [DOI] [PubMed] [Google Scholar]
- YABALURI N., MEDZIHRADSKY F. Regulation of μ-opioid receptor in neural cells by extracellular sodium. J. Neurochem. 1997;68:1053–1061. doi: 10.1046/j.1471-4159.1997.68031053.x. [DOI] [PubMed] [Google Scholar]