

Abstract
The synaptic concentrations of glutamate and γ-aminobutyric acid (GABA) are modulated by their release and re-uptake. The effects of general anaesthetics on these two processes remain unclear. This study evaluates the effects of isoflurane, a clinically important anaesthetic, on glutamate and GABA release and re-uptake in superfused mouse cerebrocortical slices.
Experiments consisted of two 1.5-min exposures to 40 mM KCl in 30 min intervals. During the second exposure, different concentrations of isoflurane with and without 0.3 mM L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC, a competitive inhibitor of glutamate uptake transporter) or 1 mM nipecotic acid (a competitive inhibitor of GABA uptake transporter) were introduced. The ratios of the second to first KCl-evoked increases in glutamate and GABA were used to determine the isoflurane concentration-response curves.
The results can be described as a sum of two independent processes, corresponding to the inhibitions of release and re-uptake, respectively. The EC50 values for the inhibitions of release and re-uptake were 295±16 and 805±43 μM for glutamate, and 229±13 and 520±25 μM for GABA, respectively. Addition of PDC did not significantly affect glutamate release but shifted the re-uptake curve to the left (EC50=315±20 μM). Nipecotic acid completely blocked GABA uptake, rendering isoflurane inhibition of GABA re-uptake undetectable.
Our data suggest that isoflurane inhibits both the release and re-uptake of neurotransmitters and that the inhibitions occur at different EC50's. For GABA, both EC50's are within the clinical concentration range. The net anaesthetic effect on extracellular concentrations of neurotransmitters, particularly GABA, depends on the competition between inhibition of release and that of re-uptake.
Keywords: GABA; glutamate; isoflurane; evoked release; re-uptake; concentration-response curve; L-trans-pyrrolidine-2,4-dicarboxylic acid; nipecotic acid; brain slice; general anaesthetic
Introduction
The effect of general anaesthetics on the release of neurotransmitters from different presynaptic membrane preparations has attracted much attention in recent years (Griffiths & Norman, 1993; Krnjevic, 1992; Langmoen et al., 1995; Richards, 1995). General anaesthetics have been shown to decrease the depolarization- or anoxia-evoked release of glutamate, a major excitatory amino acid in the central nervous system (CNS) (Bickler et al., 1995; Eilers & Bickler, 1996; Liachenko et al., 1998; Miao et al., 1995; Patel et al., 1995; Schlame & Hemmings, 1995). A less predictable behaviour in response to anaesthetics has been found for the inhibitory neurotransmitter, γ-aminobutyric acid (GABA). Whereas some studies show the anaesthetic suppression of the depolarization-evoked GABA release in the CNS, others demonstrate the enhancement of it (for review see Richards, 1995). It remains a subject of debate whether the mechanisms of general anaesthesia are related to the concentration of neurotransmitters in the synaptic clefts. Although new lines of evidence support the notion that the action of general anaesthetics is on the postsynaptic membrane (Adelsberger et al., 1998; Davies et al., 1997; Mihic & Harris, 1996; Weight et al., 1992), the regulation of the extracellular concentration of neurotransmitters is likely to play a significant role in balancing the intricate neuronal events that lead to general anaesthesia (Langmoen et al., 1995; Liachenko et al., 1998; Schlame & Hemmings, 1995).
In addition to the release process, the regulation of concentration of neurotransmitters in the synaptic space involves their re-uptake by specific carriers located in the presynaptic and glial membranes (Borden, 1996; Borowsky & Hoffman, 1995; Gonzalez & Ortega, 1997). This mechanism is essential for the clearance of neurotransmitters from the synapses and possibly for further utilization of neurotransmitters. The influence of the general anaesthetics on this process, however, is not fully understood. Recent studies (Larsen et al., 1997; Miyazaki et al., 1997) suggest that volatile anaesthetics at clinical concentrations can potentiate glutamate uptake into synaptosomes and astrocytes.
In this study, we investigated the concentration-dependent isoflurane effects on the amount of glutamate and GABA outflows from superfused mouse cerebrocortical slices after Ca2+-dependent K+-evoked depolarization (Liachenko et al., 1998). Measurements were made to determine the concentration-response curves in the absence and presence of specific inhibitors for the glutamate and GABA re-uptake transporters, L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC) and nipecotic acid, respectively. It becomes apparent that isoflurane suppresses both the release and re-uptake of neurotransmitters, but does so at different effective concentrations (EC50). The net anaesthetic effects on the synaptic concentrations of neurotransmitters may depend on the balance between the release and re-uptake processes.
Methods
Brain slice preparation
Experimental protocol was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Sixty-eight male adult CD-1 mice, weighing 33.3±0.6 g, had unlimited access to food and water. For each experiment, a mouse brain was rapidly excised and flushed with a cold, oxygenated (95% O2 and 5% CO2), artificial cerebrospinal fluid (oxy-ACSF), which normally contains (in mM): NaCl 123.9, NaHCO3 15.8, glucose 10.0, MgSO4 1.1, KH2PO4 1.2, CaCl2 1.2 and KCl 5.0. For ACSF with higher concentration of KCl, NaCl concentration was adjusted so that the solution remained isotonic. Four, 350 μm-thick, cortical slices, two dorsal and two lateral, were manually cut from the brain, using the method described previously (Espanol et al., 1994; Liachenko et al., 1998). The time required for the procedure was less than 1 min. After cutting, slices were immediately placed into a thermostatic chamber (37±0.5°C) and constantly superfused with non-circulating oxy-ACSF at a rate of 1 ml min−1. The total volume of ACSF in the chamber was maintained at a constant of 0.5 ml.
Superfusion apparatus
To precisely control the oxygenation of ACSF and the concentration of KCl, isoflurane, PDC, or nipecotic acid in the superfusion chamber, a programmable syringe pump (KD Scientific, Inc., Boston, MA, U.S.A.) with a battery of syringes was used. One of the 60-ml syringes was the primary one and contained fully oxygenated ACSF. Two pairs of secondary syringes were used to switch between: (a) oxy-ACSFs with normal and higher concentrations of KCl (60-ml syringes), and (b) non-oxygenated ACSFs with and without isoflurane in the presence or absence of PDC or nipecotic acid as needed (30-ml syringes). Syringes used in each experiment were all mounted onto the syringe pump and infused simultaneously. The contents of the primary syringe and one of each secondary syringe pair were mixed at a junction immediately before entering the superfusion chamber. Two miniature flow valves were used to switch among the secondary syringes. With this set-up, nearly constant oxygenation in the chamber (∼420 mmHg) was obtained throughout the experiments, without the potential complications caused by the administration of the volatile anaesthetic. The ACSF effluent from the chamber was collected continuously using an automatic sampler (Gilson Microfraction Collector, Model 203, Middleton, WI, U.S.A.) and immediately processed for high performance liquid chromatography (HPLC) measurements (see below).
Anaesthetic solution preparation
An aliquot of 0.5 ml neat isoflurane (Ohmeda, Liberty Corner, NJ, U.S.A.) was added to 35 ml of non-oxygenated ACSF and stirred overnight in a sealed vial, followed by gravity-driven phase separation for at least 1 h at the room temperature. The saturated concentration of isoflurane in the ACSF layer was 12.3±0.4 mM (n=3), as determined by gas chromatography (Perkin-Elmer 8500; Poropak P resin; 150°C) using appropriate gaseous standards. The saturated solution was then diluted with ACSF to the desired isoflurane concentration in a gas-tight syringe and used immediately. The isoflurane concentrations used in all experiments were confirmed by gas chromatography, using the method described previously (Tang et al., 1997; Xu et al., 1996). With our set-up, the maximum isoflurane concentration achievable in the chamber, without affecting the slice oxygenation, was 3 mM.
Neurotransmitter re-uptake inhibitors
Nipecotic acid and PDC were purchased from Sigma (St. Louis, MO, U.S.A.) and used without further purification. Preliminary experiments with varying doses of these agents (data not shown) revealed that the maximum non-toxic PDC concentration without causing excitotoxicity in the slices was 0.3 mM. At a concentration of 0.4 mM, PDC caused irreproducible extracellular glutamate concentration from sample to sample, and the elevation in glutamate outflow lasted much longer than that in the control experiments. When the PDC concentrations of 0.5 mM and higher were used, extracellular glutamate never returned to the basal level after 1.5-min exposure to 40 mM KCl. For nipecotic acid, no toxic effect was found up to 3 mM, and 1 mM was near the saturation concentration for inhibition of GABA re-uptake. Therefore, 0.3 mM of PDC and 1 mM of nipecotic acid were used in this study.
Experimental protocol
The amount of released glutamate and GABA from the slices was measured in the ACSF effluent, using HPLC with fluorescence detection (Palmer et al., 1993). Mice were randomized into the following three experimental groups: (i) no re-uptake inhibitors (n=30), (ii) with 0.3 mM PDC (n=18), and (iii) with 1 mM nipecotic acid (n=20). The experimental protocol consisted of seven time segments in the following sequence: (1) slice stabilization with normal oxy-ACSF (40 min); (2) basal release measurements (3 min); (3) the first K+-evoked depolarization (1.5 min, 40 mM KCl); (4) recovery with normal oxy-ACSF (24 min); (5) exposure to different concentration of isoflurane alone, with 0.3 mM PDC, or with 1 mM nipecotic acid (4.5 min); (6) the second K+-evoked depolarization with the same agents as in period 5 (1.5 min, 40 mM KCl) and (7) final recovery with normal oxy-ACSF (6 min).
During the 40-min stabilization period, slices were superfused with oxy-ACSF but no release measurements were made. To determine the basal release, two ACSF samples were collected over a 3-min period. The beginning of the first sample collection was arbitrarily designated as time 0 (t=0). The first K+-evoked depolarization began by switching the 60-mL secondary syringes to the oxy-ACSF with higher KCl concentration, which was pre-calculated to produce 40 mM KCl in the chamber. Recovery started 1.5 min later with normal oxy-ACSF. At the end of the first recovery period (t=28.5 min) and 1.5 min before the basal measurements for the second K+-evoked depolarisation (t=30 min), isoflurane of different concentrations was introduced with or without PDC or nipecotic acid. This was continued until the end of the second KCl depolarization (t=34.5 min). The continuous outflow of ACSF was collected at a sampling rate of 1.5 min per sample. Samples were collected during the basal, depolarization, and the first 6 min of recovery periods, resulting in 7 samples around each K+-evoked depolarization, or 14 samples for each experiment.
Amino acid quantification by HPLC with fluorescence detection
The concentrations of glutamate and GABA in the samples were determined as described previously (Liachenko et al., 1998). Briefly, an aliquot of 250 μl of ACSF was taken from each ACSF sample, vigorously mixed with 500 μl of acidified methanol (8.4 ml of 0.1 M HCl per 100 ml of methanol) and centrifuged at 16,000×g for 20 min. The supernatant layers were then filtered through a 0.2-μm nylon syringe filter and quantified using HPLC with fluorescence detection. A reverse-phase column (150×4.6 mm) and a guard column (15×4.6 mm) packed with octadecylsilane particles (5 μm, Dynamax Microsorb; Rainin Instrument Co., Woburn, MA, U.S.A.) were used to achieve separation. Glutamate and GABA were detected as fluorescent derivatives after precolumn derivatization with o-phthaldialdehyde, using a fluorescence detector (Model 121, Gilson Inc., Middleton, WI, U.S.A.). A curvilinear gradient was delivered at a rate of 1 ml min−1 that was 38% of HPLC-grade methanol in an HPLC-grade phosphate buffer (50 mM H3PO4+50 μM EDTA, pH=5.7) for 9 min, changed in a convex profile from 38 to 51% methanol from 9 to 11 min, remained isocratic (51% methanol) until 18 min, and changed back to 38% methanol at 21 min. Peaks for glutamate and GABA were identified by comparing the retention times with those of the standards. Peak areas were integrated and calibrated against the standard curves for quantification, using Unipoint® software (Gilson Inc., Middleton, WI, U.S.A.).
Calculation of released amounts of amino acids
After subtracting the corresponding baselines, the total K+-evoked increases in amino acids were integrated as a function of time. Introduction of PDC and nipecotic acid to the superfusion media caused considerable increase in the outflow of glutamate and GABA, respectively, even before the ACSF was switched to the higher KCl concentration. Therefore, to measure the K+-evoked amino acid release in the presence of these inhibitors, the baseline was calculated by averaging the two basal samples and the first sample after the switching. This is justified because KCl concentration in the chamber took about 2 min to peak after the switching (Liachenko et al., 1998), and in the absence of the inhibitors, the first sample after switching had similar amino acid amounts to the basal levels (see below). Ratios of the second to the first integrals were used to determine the response curves as a function of isoflurane concentration.
Statistical analysis
Ratios of the second to first integrals of K+-evoked neurotransmitter outflow at different isoflurane concentrations were expressed as a percentage change relative to the control ratio without isoflurane. Nonlinear regression analysis was performed using the SPSS® statistical package (SPSS Inc., Chicago, IL, U.S.A.). Levenberg-Marquardt method was used to determine the best estimates of parameters for the concentration response curves, which are described by the equation:
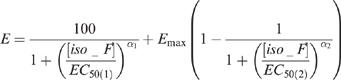 |
where E is the measured ratio (%), Emax is the ratio when the maximum suppression of re-uptake is achieved by isoflurane, [iso_F] is the isoflurane concentration (an independent variable), EC50(1) and EC50(2) are the isoflurane concentrations at which 50% change is observed for the first and the second terms of the equation, respectively, and α1 and α2 are the Hill coefficients. Note that the first and the second terms in the equation describe the isoflurane inhibition of release and re-uptake, respectively. The goodness of fit was tested using the F-statistic and the adjusted coefficient of determination, (Glantz & Slinker, 1990). The EC50s with and without PDC or nipecotic acid were compared using the 95% confidence interval. In the absence of isoflurane, the effects of the re-uptake inhibitors on the K+-evoked release ratios were analysed by one-way ANOVA with the Student-Neuman-Keuls post-hoc multiple comparisons. Calculated parameters are presented as mean±s.e.mean.
Results
In a 30-min interval, two consecutive 1.5-min exposures to 40 mM KCl resulted in nearly the same increase in depolarization-evoked amino acid release. Figures 1 and 2 show respectively the representative plots of glutamate and GABA release in the absence (Figures 1a and 2a) or presence (Figures 1b and 2b) of their respective re-uptake inhibitors. Note the scale difference in the ordinate between Figures 1a and b, and between Figure 2a and b. The shaded areas indicate the K+-evoked increases in the amino acid concentrations in the ACSF effluent. Under the control condition (i.e., without isoflurane or inhibitors during the second KCl exposure), the ratios of the second to first K+-evoked release were 0.976±0.013 (n=6) for glutamate and 1.034±0.008 (n=6) for GABA (Figure 3). Addition of PDC or nipecotic acid before the second KCl administration significantly raised the basal and K+-evoked levels of glutamate or GABA, respectively (see Figures 1b, 2b and 3). In the absence of isoflurane, 0.3 mM PDC increased glutamate ratio to 4.311±0.102 (n=2), but did not change the GABA ratio; whereas 1 mM nipecotic acid increased GABA ratio to 6.550±0.131 (n=3), but did not change the glutamate ratio (Figure 3). After normalizing the ratios at different isoflurane concentrations against that without isoflurane, the concentration-response curves for isoflurane inhibition of 40 mM KCl-evoked glutamate and GABA release are plotted in Figure 4. In the absence of re-uptake inhibitors (open circles), the concentration-response curves exhibit a characteristic ‘bell-shape' biphasic profile. The solid lines in Figure 4a and b are best fit to the data using the equation above, whereas the dashed and dotted lines depict the two contributing components, corresponding to inhibition of release and that of re-uptake, respectively. Addition of 0.3 mM PDC (filled circles in Figure 4a) did not significantly affect the component for the inhibition of release, but shifted the component for the inhibition of re-uptake to the left. Addition of 1 mM nipecotic acid (filled circles in Figure 4b) increased GABA outflow at all isoflurane concentrations and shifted the isoflurane inhibition curve of GABA release to the right. The blockage of GABA uptake by nipecotic acid also renders the component of isoflurane inhibition of the re-uptake completely undetectable (Figure 4b). The EC50 values obtained from the nonlinear regression of the concentration-response curves are listed in Table 1.
Figure 1.
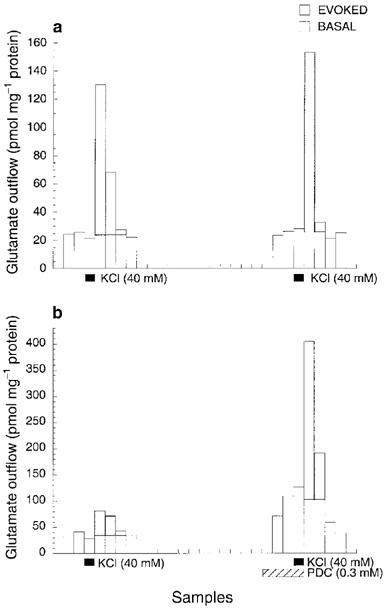
Representative profiles of glutamate outflow from mouse cerebrocortical slices after two consecutive 1.5-min exposures to 40 mM KCl in the absence (a) and presence (b) of 0.3 mM L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC), a selective glutamate re-uptake inhibitor. All samples were collected in 1.5 min. The time interval between the two KCl exposures was 30 min. The shaded areas represent K+-evoked increases in glutamate outflow above the mean basal level. The integration of the shaded areas is used to calculate the ratio of the second to first K+-evoked glutamate outflow.
Figure 2.
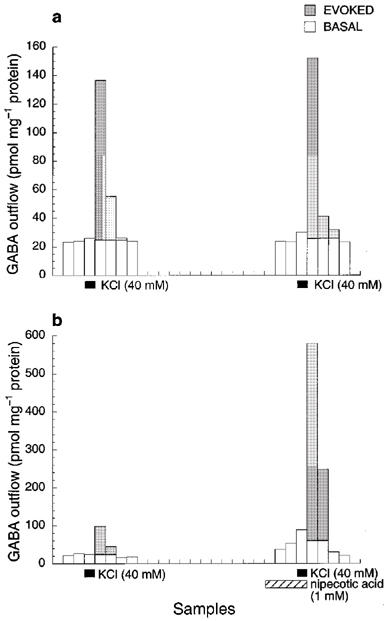
Representative profiles of GABA outflow from mouse cerebrocortical slices after two consecutive 1.5-min exposures to 40 mM KCl in the absence (a) and presence (b) of 1 mM nipecotic acid, a GABA re-uptake inhibitor. All samples were collected in 1.5 min. The time interval between the two KCl exposures was 30 min. The shaded areas represent K+-evoked increases in GABA outflow above the mean basal level. The integration of the shaded areas is used to calculate the ratio of the second to first K+-evoked GABA outflow.
Figure 3.

Effects of the re-uptake inhibitors on the ratios of the second to first K+-evoked glutamate and GABA outflow in superfused mouse cerebrocortical slices. L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC, 0.3 mM) or nipecotic acid (1 mM) was introduced into the superfusion chamber 4.5 min before the second 1.5-min exposure to 40 mM KCl. *Significantly different from other groups (P<0.01, one-way ANOVA with Student-Neuman-Keuls multiple comparisons).
Figure 4.
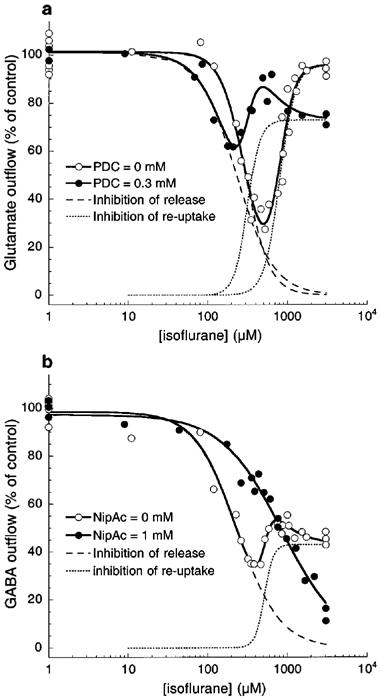
Effects of isoflurane on the K+-evoked outflow of glutamate (a) and GABA (b) in the absence (open circles) and presence (filled circles) of their respective re-uptake inhibitors: 0.3 mM of L-trans-pyrrolidine-2,4-dicarboxylic acid (PDC) for glutamate, and 1 mM of nipecotic acid for GABA. Solid lines are best fit to the data using the equation, which consists of two independent components corresponding to the inhibition of release (dashed lines) and inhibition of re-uptake (dotted lines). The EC50 values are listed in Table 1. Note that PDC reduced the isoflurane EC50 for the inhibition of glutamate re-uptake, whereas nipecotic acid eliminated the component for the isoflurane inhibition of GABA re-uptake.
Table 1.
Nonlinear regression parameters for isoflurane inhibition of glutamate and GABA release and re-uptake

Discussion
Using the same brain slice preparation, we showed previously (Liachenko et al., 1998) that 1.5 min depolarization by 40 mM KCl caused a Ca2+-dependent increase in glutamate and GABA release, suggesting that the process was of neuronal origin. We also showed that at a physiologically relevant concentration, a volatile anaesthetic, 1-chloro-1,2,2-trifluorocyclobutane (F3), and a structurally similar nonimmobilizer, 1,2-dichlorohexafluorocyclobutane (F6), could suppress the glutamate release. In contrast, only F3, but not F6, could reduce the net amount of GABA in the ACSF effluent after K+-evoked depolarization.
In the present study with varying concentrations of isoflurane, it becomes clear that the anaesthetic effects on the K+-evoked outflow of glutamate and GABA are more complicated than previously appreciated. Our new experiments revealed that in the concentration range of 0–3 mM, which covers the clinical concentrations, isoflurane exerts two distinct actions on the regulation of extracellular glutamate and GABA. The net amount of amino acid neurotransmitters present in the synapses is regulated by at least three mechanisms: release, re-uptake, and diffusion (Watkins & Evans, 1981). In our set-up, it was difficult to evaluate the isoflurane influence on diffusion. It was also difficult to assess the synaptic concentrations of glutamate and GABA solely based on the amount in the outflow. Nevertheless, the obtained results in the effluent reflected the isoflurane effects on the cellular events that control the extracellular concentrations of neurotransmitters. The fact that the concentration-response curves can be well described by the balance of the release and re-uptake, as suggested by the excellent fitting of the data with equation (Table 1), indicates that these two processes are dominant. To confirm that the second component in the equation is indeed the inhibition of re-uptake rather than the potentiation of release by isoflurane, specific re-uptake inhibitors were used. As expected, the re-uptake inhibitors interfere mostly with the second component in the equation (Figure 4, dotted lines). Thus, PDC shifted the isoflurane inhibition of glutamate re-uptake to the left without significantly altering the inhibition of release, and nipecotic acid eliminated the second component in GABA outflow. It is concluded that isoflurane can inhibit both the release and re-uptake of glutamate and GABA, and that the inhibition of release occurs at lower isoflurane concentration than does the inhibition of re-uptake.
The importance of anaesthetic effects on the neurotransmitter regulation by the re-uptake mechanism has not been fully recognized. A number of studies, focusing on the anaesthetic influence on the depolarization- or anoxia-evoked release of neurotransmitters, failed to separate the re-uptake processes from the release itself (Bickler et al., 1995; Eilers & Bickler, 1996; Larsen et al., 1994). The data shown in Figures 123 suggest that the re-uptake contribution to reducing the extracellular concentration of neurotransmitters appears to be nontrivial. In our experiments, 1 mM of nipecotic acid, which has been shown to inhibit GABA re-uptake in rat striatal synaptosomes by more than 95% (Mantz et al., 1995), increased the amount of GABA outflow by 6.55 times. Assuming that nipecotic acid did not significantly augment the GABA release, it can be estimated that when nipecotic acid is absent, the amount of GABA diffused from the slices into the ACSF was less than 15% of the actual amount released from the presynaptic membrane. The same is probably also true for glutamate, as indicated by the elevation in glutamate outflow when isoflurane concentration is increased to ∼1 mM (Figure 4). Around this concentration, glutamate re-uptake is steeply inhibited by isoflurane while there remains residue glutamate release. The increase in net glutamate outflow at reduced release and suppressed re-uptake suggests that under the control condition when re-uptake mechanism works normally, the majority of released glutamate is taken up before diffusing into the ACSF. Thus, although some direct measurements of neurotransmitter re-uptake have concluded that uptake of glutamate or GABA may not play a significant role in the action of thiopentone, pentobarbitone, methohexitone, altesin, ketamine, halothane, or urethane (Griffiths & Norman, 1993; Kendall & Minchin, 1982; Minchin, 1981), it seems too early at present time to rule out the possible involvement of neurotransmitter re-uptake in the intricate balance of neuronal events that lead to general anaesthesia (Miyazaki et al., 1997).
As discussed above, the purpose of using glutamate and GABA re-uptake inhibitors in the present study was to confirm that the increase in glutamate and GABA outflow at high isoflurane concentrations was indeed due to isoflurane inhibition of re-uptake, rather than isoflurane potentiation of release. Therefore, a single non-toxic concentration of PDC or nipecotic acid was used. In our preliminary studies (data not shown), we found that 1 mM nipecotic acid was near the saturation concentration to block the GABA re-uptake in our slice preparation. In contrast, excitotoxicity effects of PDC (Amin & Pearce, 1997; Lievens et al., 1997) precludes its use at concentrations higher than 0.4 mM, above which irreversible damages was found in the slices. One interesting finding in Figure 4 was that 0.3 mM PDC clearly shifted the curve of isoflurane inhibition of glutamate re-uptake to the left, without greatly changing the inhibition amplitude and Hill coefficient.
Depolarization-evoked release of neurotransmitters involves the participation of the voltage-gated calcium channels, which are transmembrane proteins that regulate the calcium influx into the nerve terminals to trig the release process. Although these channels seems to be largely insensitive to general anaesthetics at clinical concentrations (Franks & Lieb, 1993), the sharp dependence of synaptic transmission on calcium entry leaves open the possibility that anaesthetic action is at these channels. At the submolecular level, anaesthetics at clinical concentrations preferentially distribute to the membrane interface (Tang et al., 1997; Xu & Tang, 1997), where they may also interact with some unknown critical domains on the Ca2+ channels to influence the neurotransmitter release. The re-uptake processes, in contrast, are mediated by a diverse number of protein carriers, which act as shuttles and traverse through the membrane core (Arriza et al., 1994; Borden, 1996; Rauen et al., 1998; Wadiche et al., 1995). Therefore, it is conceivable that as the anaesthetic concentration increases, the release process is affected first, followed by the re-uptake process at higher anaesthetic concentrations when the membrane core is inundated with anaesthetic molecules.
It should be pointed out that sodium and potassium channels involved in action potential generation may also affect the release of neurotransmitters. In hippocampal slices, 0.1 μM tetrodotoxin produced a 30–40% decrease in the potassium-evoked release of glutamate, aspartate and GABA (Martin et al., 1991; 1993). Although the sensitivity of Na+ and K+ channels to general anaesthetics lies beyond the region of clinical relevance, and the suppression of axonal conduction is unlikely related to the production of general anaesthesia (Griffiths & Norman, 1993; Franks & Lieb, 1994), it remains possible that isoflurane produces some of its effects through inhibition of action potential propagation in our set-up. This might also explain why the release is more sensitive to isoflurane than is the re-uptake.
The increases in glutamate and GABA outflow in the presence of re-uptake inhibitors support the notion that the depolarization of neuronal membrane does not block the activity of re-uptake (Balcar & Johnston, 1972). The mechanisms of re-uptake involve amino acid transporters, the function of which depends on ion gradients (Borden, 1996; Danbolt, 1994). GABA re-uptake involves a family of Na+- and Cl−-coupled transporters, whereas glutamate uptake depends on Na+ co-transport and K+ counter-transport (Danbolt, 1994). Thus, the increase in extracellular K+ concentration and the decrease in extracellular Na+ during the 1.5-min exposure to 40 mM KCl may lead to certain degree of inhibition of glutamate and GABA re-uptake. This effect, however, is the same for all experiments and hence does not affect the interpretation of the results with re-uptake inhibitors.
The EC50 values for the inhibition of glutamate and GABA release (295±16 μM and 229±13 μM, respectively) do not differ significantly from each other. They are also in a good agreement with the EC50 values for isoflurane anaesthesia, which occurs at 290 μM for tadpoles (Firestone et al., 1986). Comparable aqueous concentrations can also be calculated based on the minimum alveolar concentration (MAC) for mice (320 μM), rats (350 μM), and humans (310 μM) (Franks & Lieb, 1993). In contrast, the isoflurane EC50 for the inhibition of glutamate and GABA re-uptake (805±43 and 520±25 μM, respectively) differ significantly. Only the inhibition of GABA re-uptake, not that of glutamate re-uptake, has an EC50 that is within the clinical concentration range for isoflurane. This may suggest that the regulation of synaptic GABA is more critical to general anaesthesia. In the presence of 1 mM of nipecotic acid, the EC50 for isoflurane inhibition of GABA release increased rather dramatically (Table 1). The mechanism of this increase is unclear, but is probably related to the GABA autoreceptors. Under the normal conditions, the autoreceptors mediate the GABA inhibition of evoked neurotransmitter release (Bonanno et al., 1997; Bonanno & Raiteri, 1993; Ennis & Minchin, 1993). However, at high synaptic concentrations of GABA when nipecotic acid is present, these autoreceptors may become desensitized, leading to the enhancement of GABA release and, thus to a large inhibition EC50 for isoflurane. Other mechanisms, including direct enhancement of the evoked release of GABA by nipecotic acid, cannot be ruled out. Generalization of our finding, that general anaesthetics can suppress both the release and re-uptake of neurotransmitters but do so at different EC50 and Emax, has significant implications. Although the cellular mechanisms of general anaesthesia remain unknown, some suggest that the regulation of neurotransmitter concentrations at synaptic clefts may be involved. If this is true, then our finding suggests that the same anaesthetic molecules can have multiple mechanisms of action at any given site. This adds a new dimension (and possibly new complication) to the ‘pluralistic' theory of general anaesthesia: not only may there be different sites of action for different anaesthetics, there may also be the same or different sites for the same anaesthetic with different actions.
In conclusion, the regulation of extracellular neurotransmitter concentration during the 40 mM K+-evoke depolarization is a dynamic process, involving both release and re-uptake mechanisms. Isoflurane exerts distinct action on both mechanisms. Although the release appears to be more sensitive to isoflurane than does the re-uptake, the net anaesthetic effect on the extracellular concentration of neurotransmitters, particularly GABA, depends on the competition between the inhibition of release and that of re-uptake.
Acknowledgments
The authors thank Dr Leonard Firestone for continuing encouragement and support. This work was supported by grants from the National Institute of General Medical Sciences, GM49202 (YXu) and GM56257 (PTang), and from the University of Anesthesiology and Critical Care Medicine Foundation, University of Pittsburgh.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CNS
central nervous system
- EC50
50% effective concentration
- GABA
γ-aminobutyric acid
- HPLC
high performance liquid chromatography
- PDC
L-trans-pyrrolidine-2,4-dicarboxylic acid
References
- ADELSBERGER H., WILDE J., FRANKE C., DUDEL J. Multiple mechanisms of block by the anesthetic isoflurane of a gamma-aminobutyric acid activated chloride channel in crayfish. J. Comp. Physiol. [A]-Sensory Neural & Behavioral Physiology. 1998;182:51–58. doi: 10.1007/s003590050157. [DOI] [PubMed] [Google Scholar]
- AMIN N., PEARCE B. Glutamate toxicity in neuron-enriched and neuron-astrocyte co-cultures: effect of the glutamate uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylate. Neurochem. Int. 1997;30:271–276. doi: 10.1016/s0197-0186(96)00092-7. [DOI] [PubMed] [Google Scholar]
- ARRIZA J.L., FAIRMAN W.A., WADICHE J.I., MURDOCH G.H., KAVANAUGH M.P., AMARA S.G. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALCAR V.J., JOHNSTON G.A. Glutamate uptake by brain slices and its relation to the depolarization of neurones by acidic amino acids. J. Neurobiol. 1972;3:295–301. doi: 10.1002/neu.480030403. [DOI] [PubMed] [Google Scholar]
- BICKLER P.E., BUCK L.T., FEINER J.R. Volatile and intravenous anesthetics decrease glutamate release from cortical brain slices during anoxia. Anesthesiology. 1995;83:1233–1240. doi: 10.1097/00000542-199512000-00014. [DOI] [PubMed] [Google Scholar]
- BONANNO G., FASSIO A., SCHMID G., SEVERI P., SALA R., RAITERI M. Pharmacologically distinct GABAB receptors that mediate inhibition of GABA and glutamate release in human neocortex. Br. J. Pharmacol. 1997;120:60–64. doi: 10.1038/sj.bjp.0700852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BONANNO G., RAITERI M. γ-Aminobutyric acid (GABA) autoreceptors in rat cerebral cortex and spinal cord represent pharmacologically distinct subtypes of the GABAB receptor. J Pharmacol. Exp. Ther. 1993;265:765–770. [PubMed] [Google Scholar]
- BORDEN L.A. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem. Int. 1996;29:335–356. doi: 10.1016/0197-0186(95)00158-1. [DOI] [PubMed] [Google Scholar]
- BOROWSKY B., HOFFMAN B.J. Neurotransmitter transporters: molecular biology, function, and regulation. Int. Rev. Neurobiol. 1995;38:139–199. doi: 10.1016/s0074-7742(08)60526-7. [DOI] [PubMed] [Google Scholar]
- DANBOLT N.C. The high affinity uptake system for excitatory amino acids in the brain. Prog. Neurobiol. 1994;44:377–396. doi: 10.1016/0301-0082(94)90033-7. [DOI] [PubMed] [Google Scholar]
- DAVIES P.A., KIRKNESS E.F., HALES T.G. Modulation by general anaesthetics of rat GABAA receptors comprised of alpha 1 beta 3 and beta 3 subunits expressed in human embryonic kidney 293 cells. Br. J. Pharmacol. 1997;120:899–909. doi: 10.1038/sj.bjp.0700987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EILERS H., BICKLER P.E. Hypothermia and isoflurane similarly inhibit glutamate release evoked by chemical anoxia in rat cortical brain slices. Anesthesiology. 1996;85:600–607. doi: 10.1097/00000542-199609000-00020. [DOI] [PubMed] [Google Scholar]
- ENNIS C., MINCHIN M.C. Modulation of the GABAA-like autoreceptor by barbiturates but not by steroids. Neuropharmacol. 1993;32:355–357. doi: 10.1016/0028-3908(93)90156-w. [DOI] [PubMed] [Google Scholar]
- ESPANOL M.T., XU Y., LITT L., YANG G.Y., CHANG L.H., JAMES T.L., WEINSTEIN P., CHAN P.H. Modulation of glutamate-induced intracellular energy failure in neonatal cerebral cortical slices by kynurenic acid, dizocilpine, and NBQX. J. Cereb. Blood Flow Metab. 1994;14:269–278. doi: 10.1038/jcbfm.1994.34. [DOI] [PubMed] [Google Scholar]
- FIRESTONE L.L., SAUTER J.F., BRASWELL L.M., MILLER K.W. Actions of general anesthetics on acetylcholine receptor-rich membranes from Torpedo californica. Anesthesiology. 1986;64:694–702. doi: 10.1097/00000542-198606000-00004. [DOI] [PubMed] [Google Scholar]
- FRANKS N.P., LIEB W.R. Selective actions of volatile general anaesthetics at molecular and cellular levels. Br. J. Anaesthes. 1993;71:65–76. doi: 10.1093/bja/71.1.65. [DOI] [PubMed] [Google Scholar]
- FRANKS N.P., LIEB W.R. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- GLANTZ S.A., SLINKER B.K. Primer of Applied Regression and Analysis of Variance. New York: McGraw-Hill, Inc; 1990. [Google Scholar]
- GONZALEZ M.I., ORTEGA A. Regulation of the Na+-dependent high affinity glutamate/aspartate transporter in cultured Bergmann glia by phorbol esters. J. Neurosci. Res. 1997;50:585–590. doi: 10.1002/(SICI)1097-4547(19971115)50:4<585::AID-JNR9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- GRIFFITHS R., NORMAN R.I. Effects of anaesthetics on uptake, synthesis and release of transmitters. Br. J. Anaesthes. 1993;71:96–107. doi: 10.1093/bja/71.1.96. [DOI] [PubMed] [Google Scholar]
- KENDALL T.J., MINCHIN M.C. The effects of anaesthetics on the uptake and release of amino acid neurotransmitters in thalamic slices. Br. J. Pharmacol. 1982;75:219–227. doi: 10.1111/j.1476-5381.1982.tb08776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRNJEVIC K. Cellular and synaptic actions of general anaesthetics. Gen. Pharmacol. 1992;23:965–975. doi: 10.1016/0306-3623(92)90274-n. [DOI] [PubMed] [Google Scholar]
- LANGMOEN I.A., LARSEN M., BERG-JOHNSEN J. Volatile anaesthetics: cellular mechanisms of action. Eur. J. Anaesthesiol. 1995;12:51–58. [PubMed] [Google Scholar]
- LARSEN M., GRONDAHL T.O., HAUGSTAD T.S., LANGMOEN I.A. The effect of the volatile anesthetic isoflurane on Ca(2+)-dependent glutamate release from rat cerebral cortex. Brain Res. 1994;663:335–337. doi: 10.1016/0006-8993(94)91282-3. [DOI] [PubMed] [Google Scholar]
- LARSEN M., HEGSTAD E., BERG-JOHNSEN J., LANGMOEN I.A. Isoflurane increases the uptake of glutamate in synaptosomes from rat cerebral cortex. Br. J. Anaesthes. 1997;78:55–59. doi: 10.1093/bja/78.1.55. [DOI] [PubMed] [Google Scholar]
- LIACHENKO S., TANG P., SOMOGYI G.T., XU Y. Comparison Of Anaesthetic and Non-Anaesthetic Effects On Depolarization-Evoked Glutamate and GABA Release From Mouse Cerebrocortical Slices. Br. J. Pharmacol. 1998;123:1274–1280. doi: 10.1038/sj.bjp.0701728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEVENS J.C., DUTERTRE M., FORNI C., SALIN P., KERKERIAN-LE G.L. Continuous administration of the glutamate uptake inhibitor L-trans-pyrrolidine-2,4-dicarboxylate produces striatal lesion. Brain Res. Mol. Brain Res. 1997;50:181–189. doi: 10.1016/s0169-328x(97)00182-4. [DOI] [PubMed] [Google Scholar]
- MANTZ J., LECHARNY J.B., LAUDENBACH V., HENZEL D., PEYTAVIN G., DESMONTS J.M. Anesthetics affect the uptake but not the depolarization-evoked release of GABA in rat striatal synaptosomes. Anesthesiol. 1995;82:502–511. doi: 10.1097/00000542-199502000-00020. [DOI] [PubMed] [Google Scholar]
- MARTIN D., BUSTOS G.A., BOWE M.A., SHERRILYNN D.B., NADLER J.V. Autoreceptor regulation of glutamate and aspartate release from slices of the hippocampal CA1 area. J. Neurochem. 1991;56:1647–1655. doi: 10.1111/j.1471-4159.1991.tb02063.x. [DOI] [PubMed] [Google Scholar]
- MARTIN D., THOMPSON M.A., NADLER J.V. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur. J. Pharmacol. 1993;250:473–476. doi: 10.1016/0014-2999(93)90037-i. [DOI] [PubMed] [Google Scholar]
- MIAO N., FRAZER M.J., LYNCH C.R. Volatile anesthetics depress Ca2+ transients and glutamate release in isolated cerebral synaptosomes. Anesthesiol. 1995;83:593–603. doi: 10.1097/00000542-199509000-00019. [DOI] [PubMed] [Google Scholar]
- MIHIC S.J., HARRIS R.A. Inhibition of rho1 receptor GABAergic currents by alcohols and volatile anesthetics. J. Pharmacol. Exp. Ther. 1996;277:411–416. [PubMed] [Google Scholar]
- MINCHIN M.C. The effect of anaesthetics on the uptake and release of γ-aminobutyrate and D-aspartate in rat brain slices. Br. J. Pharmacol. 1981;73:681–689. doi: 10.1111/j.1476-5381.1981.tb16803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAZAKI H., NAKAMURA Y., ARAI T., KATAOKA K. Increase of glutamate uptake in astrocytes: a possible mechanism of action of volatile anesthetics. Anesthesiol. 1997;86:1359–1366. doi: 10.1097/00000542-199706000-00018. [DOI] [PubMed] [Google Scholar]
- PALMER A.M., MARION D.W., BOTSCHELLER M.L., SWEDLOW P.E., STYREN S.D., DEKOSKY S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- PATEL P.M., DRUMMOND J.C., COLE D.J., GOSKOWICZ R.L. Isoflurane reduces ischemia-induced glutamate release in rats subjected to forebrain ischemia. Anesthesiol. 1995;82:996–1003. doi: 10.1097/00000542-199504000-00024. [DOI] [PubMed] [Google Scholar]
- RAUEN T., TAYLOR W.R., KUHLBRODT K., WIESSNER M. High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998;291:19–31. doi: 10.1007/s004410050976. [DOI] [PubMed] [Google Scholar]
- RICHARDS C.D. The synaptic basis of general anaesthesia. Eur. J. Anaesthesiol. 1995;12:5–19. [PubMed] [Google Scholar]
- SCHLAME M., HEMMINGS H.C., JR Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiol. 1995;82:1406–1416. doi: 10.1097/00000542-199506000-00012. [DOI] [PubMed] [Google Scholar]
- TANG P., YAN B., XU Y. Different distribution of fluorinated anesthetics and nonanesthetics in model membrane: a 19F NMR study. Biophys. J. 1997;72:1676–1682. doi: 10.1016/S0006-3495(97)78813-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WADICHE J.I., ARRIZA J.L., AMARA S.G., KAVANAUGH M.P. Kinetics of a human glutamate transporter. Neuron. 1995;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- WATKINS J.C., EVANS R.H. Excitatory amino acid transmitters. Annu. Rev. Pharmacol. Toxicol. 1981;21:165–204. doi: 10.1146/annurev.pa.21.040181.001121. [DOI] [PubMed] [Google Scholar]
- WEIGHT F.F., AGUAYO L.G., WHITE G., LOVINGER D.M., PEOPLES R.W. GABA- and glutamate-gated ion channels as molecular sites of alcohol and anesthetic action. Adv. Biochem. Psychopharmacol. 1992;47:335–347. [PubMed] [Google Scholar]
- XU Y., TANG P. Amphiphilic sites for general anesthetic action? Evidence from 129Xe-[1H] intermolecular nuclear Overhauser effects. Biochim. Biophys. Acta. 1997;1323:154–162. doi: 10.1016/s0005-2736(96)00184-8. [DOI] [PubMed] [Google Scholar]
- XU Y., TANG P., FIRESTONE L., ZHANG T.T. 19F nuclear magnetic resonance investigation of stereoselective binding of isoflurane to bovine serum albumin. Biophys. J. 1996;70:532–538. doi: 10.1016/S0006-3495(96)79599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]