

Abstract
The aim of this study was to investigate the effect of N-(3-(aminomethyl)benzyl)acetamidine (1400W), a selective inhibitor of inducible calcium-independent nitric oxide synthase (iNOS), on the functional and histopathological outcomes of experimental transient focal cerebral ischaemia in rats.
Transient ischaemia was produced by the occlusion for 2 h of both the left middle cerebral artery and common carotid artery. Treatments with 1400W (20 mg kg−1) or vehicle were started 18 h after occlusion of the arteries and consisted in seven subcutaneous injections at 8 h interval. Ischaemic outcomes and NOS activities (constitutive and calcium-independent NOS) were evaluated 3 days after ischaemia.
1400W significantly reduced ischaemic lesion volume by 31%, and attenuated weight loss and neurological dysfunction.
1400W attenuated the calcium-independent NOS activity in the infarct by 36% without affecting the constitutive NOS activity.
These findings suggest that iNOS activation contributes to tissue damage and that selective inhibitors of this isoform may be of interest for the treatment of stroke.
Keywords: Focal cerebral ischaemia, 1400W, inducible nitric oxide synthase, neuroprotection, free radicals, stroke, neurodegenerative diseases
Introduction
It is now well established that nitric oxide (NO) plays a complex role in cerebral ischaemia (for review see Dalkara & Moskowitz, 1994). Previous work in our laboratory demonstrated that L-NAME, a non selective inhibitor of NO-synthases, reduced the infarction induced by permanent (Buisson et al., 1992) and transient (Margaill et al., 1997) focal cerebral ischaemia in rats. In the latter study, L-NAME was neuroprotective even when its administration was delayed for 9 h (Margaill et al., 1997). This suggests a delayed production of deleterious NO and raises the question of the isoform involved. Indeed, NO is synthesized by three different types of NO-synthase (NOS) including the constitutive calcium/calmodulin-dependent neuronal (nNOS) and endothelial (eNOS) isoforms and the inducible calcium-independent isoform (iNOS). During ischaemia, eNOS activity is beneficial since knockout mice deficient in eNOS develop larger infarct after middle cerebral artery (MCA) occlusion in comparison to wild-type controls (Huang et al., 1996). In contrast, nNOS appears deleterious in ischaemia since several selective inhibitors of this isoform are neuroprotective (Yoshida et al., 1994; Nagafuji et al., 1995; Zhang et al., 1996b; Escott et al., 1998) and mice bearing a selective nNOS gene alteration show a reduced infarction after permanent (Huang et al., 1994) and transient (Hara et al., 1996) focal ischaemia. The role of iNOS in cerebral ischaemia has been less extensively examined. Expression of iNOS was reported at late stage after ischaemia (Iadecola et al., 1995b; 1996). In our laboratory, we also found substantial appearance of calcium-independent NOS activity associated with an important increase in NO production in a mouse model of ischaemia-reperfusion (Grandati et al., 1997). Evidence for a deleterious role of iNOS in ischaemia was first established with aminoguanidine, a weak and partially selective inhibitor of iNOS. Aminoguanidine was reported to be neuroprotective in models of permanent (Iadecola et al., 1995c; Cockroft et al., 1996) and transient (Iadecola et al., 1996; Zhang et al., 1996a) focal ischaemia in rats, but this compound may have exerted its beneficial effect by mechanisms unrelated to iNOS inhibition, such as inhibition of the polyamine oxydase (Ivanova et al., 1998). The contribution of iNOS to delayed ischaemic injury was however recently confirmed by Iadecola and co-workers with mice lacking the gene of this isoform (Iadecola et al., 1997). Indeed, these iNOS knockout mice develop smaller infarcts compared with wild type control 4 days after permanent focal ischaemia, while lesion size in both groups is similar one day after the onset of ischaemia. These data provide evidence that iNOS plays an important role in the progression of post-ischaemic damage and may explain the beneficial effect we previously observed after a delayed treatment with L-NAME (Margaill et al., 1997). Moreover, they suggest that iNOS may constitute a new attractive target with a large therapeutic window for the treatment of stroke. To further investigate the relevance of this target, we investigated in the present study the effect of the highly selective iNOS inhibitor 1400W (N-(3-(aminomethyl)benzyl)acetamidine) (Garvey et al., 1997) on outcomes associated with transient focal cerebral ischaemia in rats.
Methods
General procedures
Materials
Chemicals were purchased from Sigma Chem ical Corporation. N - ( - 3 - ( aminomethyl ) benzyl ) acetamidine (1400W) was synthesized as a dichlorhydrate salt in the Medicinal Chemistry Department of Rhône-Poulenc Rorer following published methods (Oplinger et al., 1996). 1400W was dissolved in distilled water and the solution was adjusted to pH 7 with NaOH. 1400W was injected subcutaneously (2 ml kg−1) in all the experiments.
Animals
All experiments were performed with male Sprague-Dawley rats (300–350 g body weight) in accordance with NIH and French Department of Agriculture guidelines (license no. 01352).
Transient focal cerebral ischaemia
Surgical procedure Arteries occlusion was performed according to Margaill and co-workers (1996). Rats were anaesthetized with chloral hydrate (400 mg kg−1, i.p.) and allowed to breathe spontaneously. The left middle cerebral artery (MCA) was exposed via a temporal craniotomy and occluded with a microclip (15×0.4 mm; Ohwa Tsuho Co., Tokyo, Japan) and the left common carotid artery (CCA) was concomitantly clamped. The microclip was placed on the MCA at a site proximal to the lenticulostriate arteries to induce both cortical and striatal infarction. Two hours after induction of ischaemia, rats were reanaesthetized, the microclip occluding the MCA was removed and circulation within the CCA was restored. Reperfusion in both arteries was checked under microscope. During the surgical procedure and until recovery from anaesthesia, body temperature was maintained at 37–38°C by means of a heating blanket. Sham-operated rats underwent the same surgical procedure but the MCA and the CCA were not occluded. After surgery, rats were returned to their home cage. Animals showing feeding difficulties were fed with mashed lab chow. Functional outcome assessment. Three days after ischaemia, sensorimotor neurological deficits were assessed in a blinded fashion by the neurological scoring system previously described by Wahl et al. (1992). The following parameters were scored for right and left paws: grasping reflex, righting reflex, visual placing and leg hanging reaction. For each side, the sum of scores constitutes the global neurological score which is 5 in normal non ischaemic rats. To assess the body weight loss, ischaemic animals were weighed before ischaemia and 3 days after reperfusion. Measurement of infarct volume. Three days after occlusion of the arteries, rats were killed with an overdose of sodium pentobarbitone. Their brains were removed and 2-mm-thick coronal sections were cut. Sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride, incubated for 20 min at 20°C, and then placed in formalin for one night in the dark. The striatal and cortical areas of infarction (unstained tissue) were measured on each section using an image analyser (Imstar, Paris, France). The distances between respective coronal sections were used to calculate a linear integration for striatal and cortical necrotic volumes. Infarct volume was corrected for brain oedema according to Golanov & Reis (1995).
In some experiments the area of infarction was removed and assayed for NOS activity. A 6-mm thick coronal section was dissected out at the level of the anterior commissure. The infarct tissue, including both cortex and striatum, was isolated from the left hemisphere. A similar segment was isolated from non ischaemic animals. The tissues were frozen (−40°C) and stored at −40°C for later analysis.
NOS activity assay
The catalytic activities of NOS were measured by the conversion of L[14C]-arginine to L-[14C]-citrulline according to the procedure of Bredt & Snyder (1989) modified for detection of constitutive (cNOS) and calcium-independent NOS activity as described previously (Grandati et al., 1997). Calcium-independent NOS activity was used as an index for iNOS activity. Data are expressed as pmol of L-[14C]-citrulline per mg protein, per minute (pmol mg protein−1 min−1). Proteins were assayed by the Bradford method (1976).
Experimental paradigm
Preliminary studies
Since little published information was available regarding the effects of 1400W at the time the present study was initiated, preliminary experiments were conducted to determine the doses of this substance that inhibit iNOS in vivo. To this end, first we studied the effect of increasing doses of 1400W inhibition on iNOS induced peripherally in a model of lipopolysaccharide (LPS) induced endotoxemia (Tracey et al., 1995). Then, the most efficient dose of 1400W on endotoxemia-induced-iNOS was assessed on ischaemia-induced cerebral iNOS in the infarct tissue. Finally, we verified that this dosage did not alter physiological variables including cerebral blood flow. Effect of 1400W on peripheral calcium-independent NOS Calcium-independent NOS activity was induced in the lung of awake rats by intraperitoneal injection of a solution of bacterial LPS (E. Coli, serotype No 0127:B8) (10 mg kg−1 in saline, 10 ml kg−1) (Tracey et al., 1995). Control rats received saline (n=3). Five hours after LPS injection, rats were treated subcutaneously either with 1400W at 2, 5, 10, 20 or 50 mg kg−1 (n=5–6 per dose) or vehicle (n=5). One hour later (i.e. 6 h after LPS injection), animals were killed with an overdose of sodium pentobarbitone. A segment of 150–200 mg lung tissue was immediately frozen (−40°C) until the measurement of calcium-independent NOS activity. Effect of 1400W on cerebral NOS activity 24 h after ischaemia Twenty-four hours after ischaemia, animals received either 1400W (20 mg kg−1, sc) (n=5) or vehicle (n=5). One hour after injection, rats were killed, and their infarct areas were removed and frozen (−40°C) for assay of NOS activities (calcium-independent and constitutive). NOS activities were also assessed in non-operated rats (control rats, n=3) and in sham-operated rats (n=3)
Effect of 1400W on physiological variables and cerebral blood flow
Animals were anaesthetized with pentobarbitone (50 mg kg−1, 2 ml kg−1, i.p.) in order to achieve a prolonged and stable anaesthesia. Every hour, anaesthesia was maintained by a half dose of pentobarbitone. The left femoral artery was cannulated and rats were placed on a stereotaxic frame (David Kopf®). Body temperature was maintained at 37–38°C by the mean of a heating blanket. The arterial catheter was connected to a pressure transducer (EMKA technologies®) for recording of mean arterial pressure (MAP) and arterial blood samples were collected to measure blood gases and pH (ABL 330, Radiometer®). Cerebral blood flow (CBF) was monitored in the left cerebral cortex with a laser-Doppler Flowmeter (LaserFlo, BPM2, Vasamedics®). The probe was positioned at a site 3–3.5 mm lateral to the middline, 4.5 rostral to the interaural line and 0.5 mm above the dural surface. Once a stable signal (MAP, blood gases, pH and CBF) was obtained (base level), animals were treated subcutaneously either with 1400W (20 mg kg−1) (n=4) or vehicle (n=4) and values were read every 30 min during 3 h.
Effect of 1400W on the outcomes of ischaemia
Effect of 1400W on infarct size, neurological deficits and weight loss Treatments were initiated 18 h after the onset of ischaemia. The rational for beginning the treatment at this time is that in our model, iNOS activity develops more than 18 h after arteries occlusion (unpublished observation). Animals were treated subcutaneously either with 1400W (20 mg kg−1) (n=10) or vehicle (n=10). Then, injections were repeated at 8 h interval until euthanasia, 3 days after arteries occlusion (i.e. a total of seven injections). The 8 h interval between treatments was based upon previous experiments showing that 1400W reduced ischaemia-induced iNOS activity when measurements were taken 6 h after its injection but was inefficient after 9 h (data not shown).
Effect of 1400W on NOS activity 3 days after ischaemia
In these experiments rats were treated either with 1400W (20 mg kg−1) (n=9) or vehicle (n=8) according to the administration schedules described above. NOS activity was also assessed in non-operated rats (control rats, n=5) and in sham-operated rats (n=8). Rats were killed 3 days after ischaemia, and the infarct regions of the cortex and the striatum were removed for NOS activity assay.
Data analysis
Data are expressed as mean±s.e.mean (standard error of mean). Comparisons between two groups were evaluated by a two-tailed unpaired Student's t-test. Comparisons among multiple groups were evaluated by ANOVA with subsequent group comparisons by PLSD Fisher's test. Non parametric Kruskall and Wallis rank analysis was used for the neurological scores with subsequent group comparisons by Mann-Whitney U-test. Differences were considered statistically significant at a value of P<0.05.
Results
Preliminary studies
Effect of 1400W on peripheral calcium-independent NOS
No calcium-independent NOS activity in lung of control rats was detectable. Six hours after LPS injection, there was a massive increase in the calcium-independent NOS activity (Table 1). At 2 mg kg−1, 1400W was inactive, but 5, 10, 20 or 50 mg kg−1 markedly reduced the calcium-independent NOS activity. The maximal reduction of calcium-independent NOS activity in the lung was obtained with 1400W at 20 mg kg−1 (>90%, P<0.001 versus vehicle).
Table 1.
Effect of 1400W on calcium-independent NOS activity in the lung after LPS injection

Effect of 1400W on cerebral NOS activity 24 h after ischaemia
The effect of 1400W at the dose of 20 mg kg−1 was evaluated on NOS activity in the cerebral infarct area 24 h after ischaemia (Figure 1). In sham-operated rats, calcium-independent NOS activity did not differ from control rats (data not shown). Ischaemia induced a substantial increase in calcium-independent NOS activity (P<0.001 versus sham) which was attenuated by 60% with 1400W treatment (P<0.001 versus vehicle) (Figure 1a). In contrast, cNOS activity was substantially reduced after ischaemia (P<0.001 versus sham), and treatment with 1400W did not modify this activity (Figure 1b).
Figure 1.
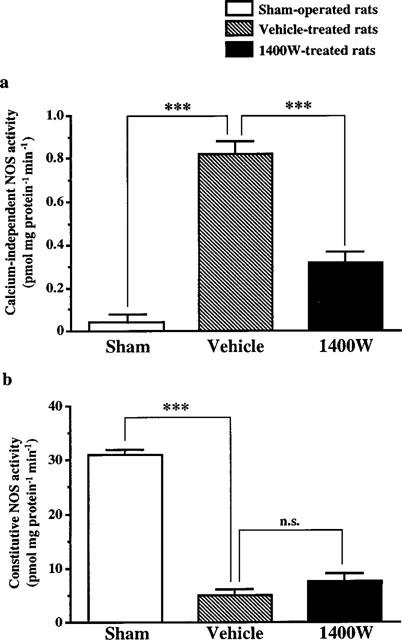
Effect of 1400W on calcium-independent NOS (a) and constitutive NOS (b) enzymatic activities in the infarct area of rats subjected to transient focal cerebral ischaemia. NOS activities were measured in sham-operated rats (n=3) and in ischaemic rats treated either with 1400W (20 mg kg−1, s.c.) (n=5) or vehicle (n=5) 24 h after ischaemia and killed 1 h later. Data are expressed as means± s.e.mean. n.s., non significant; ***P<0.001 versus the vehicle-treated rats.
Effect of 1400W on physiological variables
Over the 3 h investigation periods, administration of 1400W (20 mg kg−1, s.c.) in anaesthetized rats did not modify the values of MAP (87–100 mmHg), CBF (26.5–30.3 ml min−1), PaO2 (92–102 mmHg), PaCO2 (33–36 mmHg) and pH (7.27–7.33) which remained within the physiological range and identical to those of vehicle-treated rats (data not shown).
Effect of 1400W on the outcomes of ischaemia
Effect on infarct size
In vehicle-treated rats, the transient occlusion of the left MCA and CCA caused reproducible infarcts involving the cerebral cortex and the striatum after 3 days of reperfusion (Table 2). Administration of 1400W reduced infarct size in cerebral cortex by 34% (P<0.05 versus vehicle) but not in the striatum (Table 2). The tissue rescued from infarction was located at the level of sections lying between 9.7 and 5.7 mm anterior to the interaural line which corresponded to the central region of infarction (Figure 2).
Table 2.
Effect of 1400W on infarct size 3 days after transient focal cerebral ischaemia

Figure 2.
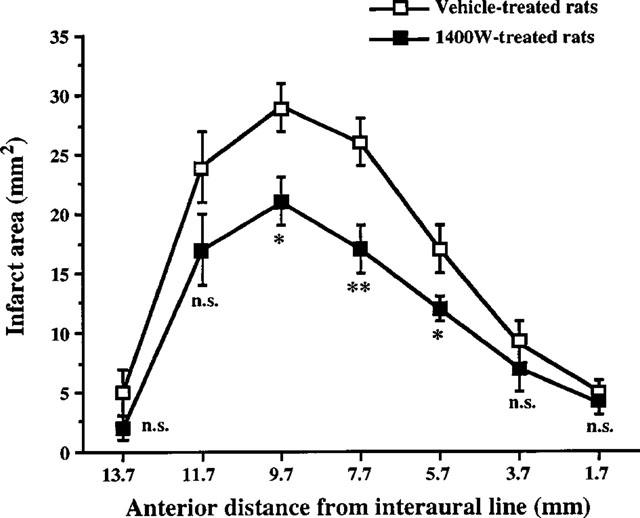
Effect of 1400W on the infarct area measured 3 days after transient focal cerebral ischaemia. Treatments were started 18 h after arteries occlusion and consisted in seven subcutaneous injections at 8 h interval. Ischaemic rats were treated either with 1400W (20 mg kg−1) (n=10) or vehicle (n=10). The figure represents the rostrocaudal distribution of infarct areas plotted as a function of the anterior distance from interaural line. Data are expressed as means±s.e.mean. n.s., non significant; *P<0.05; **P<0.01 versus vehicle-treated rats.
Effect on motor deficits and weight loss
In vehicle-treated rats, ischaemia caused at 3 days a significant neurological deficit on the right side (P<0.001) without affecting the left side (Figure 3a). This neurological deficit was associated with a marked loss of weight (Figure 3b). A correlation analysis revealed that both the neurological deficits and the loss of weight were significantly correlated with the total infarct volumes (neurological deficit: r=0.941, n=10, P<0.001; loss of weight: r=0.837, n=10, P<0.01). Treatment with 1400W reduced the neurological deficit (Figure 3a) and the loss of weight (Figure 3b) in comparison with vehicle (P<0.05).
Figure 3.
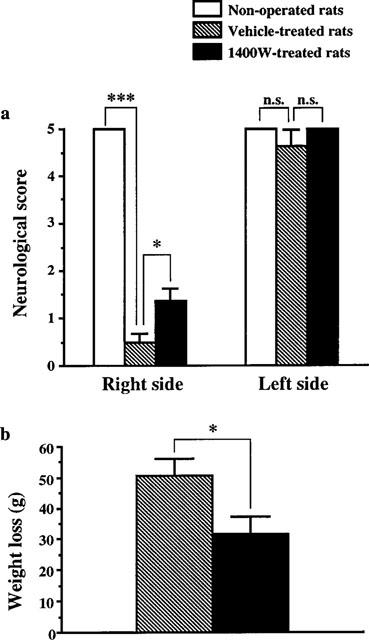
Effect of 1400W on neurological deficits (a) and weight loss (b) measured 3 days after transient focal cerebral ischaemia. Treatments were started 18 h after arteries occlusion and consisted in seven subcutaneous injections at 8 h interval. Ischaemic rats were treated either with 1400W (20 mg kg−1) (n=10) or vehicle (n=10). Neurological deficits (a) for the left and right paws were assessed by four neurological tests as described in Methods. Non-operated rats were used as control (n=5). The weight loss (b) was assessed as the difference before ischaemia and 3 days after reperfusion. Data are expressed as means±s.e.mean. n.s., non significant; *P<0.05; ***P<0.001 versus vehicle-treated rats.
Effect of 1400W on cerebral NOS activities 3 days after ischaemia
In sham-operated rats as in control rats, calcium-independent NOS activity was very weak (less than 0.03 pmol mg protein−1 min−1). Three days after ischaemia, there was a massive increase in calcium-independent NOS activity (P<0.001 versus control) which was much higher than that observed at 24 h. This increase after 3 days of reperfusion was attenuated by 36% in animals treated with 1400W, according to the protocol used for the study on infarct size (P<0.01 versus vehicle) (Figure 4a). Simultaneously, ischaemia led to a dramatic reduction in cNOS activity which was not influenced by 1400W treatment (Figure 4b).
Figure 4.
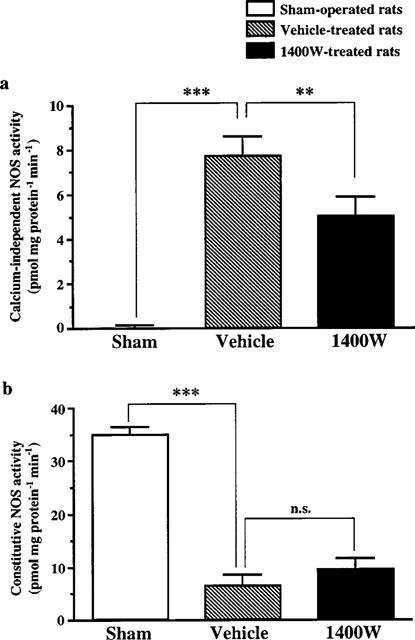
Effect of 1400W on calcium-independent NOS (a) and constitutive NOS (b) enzymatic activities in the infarct area 3 days after transient focal cerebral ischaemia. Treatments were started 18 h after arteries occlusion and consisted in seven subcutaneous injections at 8 h interval. NOS activities were measured in sham-operated rats (n=8) and in ischaemic rats treated either with 1400W (20 mg kg−1) (n=9) or vehicle (n=8). Data are expressed as means±s.e.mean. n.s., non significant; **P<0.01; ***P<0.001 versus vehicle-treated rats.
Discussion
In the present study, we demonstrate that the highly selective iNOS inhibitor 1400W reduces the infarct size, ameliorates the neurological score and attenuates weight loss of rats submitted to transient focal cerebral ischaemia. These effects correlate with a decrease in inducible calcium independent NOS activity (iNOS) induced by ischaemia. These data suggest that iNOS induction contributes to postischaemic brain injury.
Involvement of iNOS in ischaemic damage was initially studied by Iadecola and coworkers. These authors observed an increase in the expression of iNOS mRNA and enzymatic activity in models of permanent and transient focal ischaemia in rats (Iadecola et al., 1995b; 1996). Furthermore, aminoguanidine, a weak and partially selective iNOS inhibitor, was neuroprotective in these models (Iadecola et al., 1995c; 1996; Zhang et al., 1996a). Finally, knockout mice that do not express iNOS were protected from focal ischaemia (Iadecola et al., 1997). Taken together these data support the hypothesis that iNOS is deleterious during cerebral ischaemia. Of particular interest is the fact that appearance of iNOS is delayed by 24 h after ischaemia (Iadecola et al., 1995a; 1995b; 1996) suggesting that iNOS inhibitors may have a large therapeutic window. Accordingly it was found that aminoguanidine remains neuroprotective when given 24 h after ischaemia (Iadecola et al., 1995c; Zhang et al., 1996a).
Hitherto, pharmacological evidence for the deleterious role of iNOS in cerebral ischaemia was based only on studies with aminoguanidine. It has been reported that aminoguanidine may however protect ischaemic brain by mechanisms unrelated to the inhibition of iNOS because this substance was also protective when given 15 min after the onset of ischaemia (Cockroft et al., 1996), that is a time point where iNOS is hardly induced. Among these mechanisms, a recent study indicated that reduction in ischaemic brain infarction with aminoguanidine may be related to the inhibition of polyamine oxydase activity (Ianova et al., 1998). Thus, it appeared necessary to examine the effects of more potent and selective iNOS inhibitor to further ascertain the deleterious pathophysiological role of NO produced by iNOS in the consequences of ischaemia.
Among the available iNOS inhibitors, the newly synthetized N-(3-(aminomethyl)benzyl)acetamidine (1400W) has been described as the most potent and selective (Babu & Griffith, 1998). 1400W is an irreversible or extremely slowly reversible inhibitor of iNOS. In contrast, inhibition of the neuronal and endothelial constitutive NOS is weaker and rapidly reversible (Garvey et al., 1997). This led us to examine the effect of 1400W in our model of transient focal cerebral ischaemia. Preliminary studies were carried out to determine the efficient dose of 1400W that inhibits iNOS activity in vivo. Induction of peripheral iNOS by LPS was first used as a rapid procedure to evaluate 1400W inhibiting activity (Tracey et al., 1995). We found that 20 mg kg−1 of 1400W is able to markedly inhibit LPS-induced iNOS activity in the lung. In a second experiment, this dose was shown to also produce a significant although less marked inhibition of the brain iNOS activity induced by ischaemia. This last result suggests that 1400W at the dose of 20 mg kg−1 is able to access and inhibit brain iNOS activity. The lesser extent to which 1400W inhibited iNOS activity in ischaemic brain with respect to lung tissue may be likely related to some limitation in brain penetration. A higher dose might have been more efficacious to reduce iNOS activity. However, this would have occurred at the expenses of haemodynamic side effects (Garvey et al., 1997). On the contrary, 20 mg kg−1 of 1400W did not affect basal MAP and cerebral blood flow. This led us to investigate the neuroprotective potential of this dose on ischaemic brain injury.
We report here for the first time that 1400W is able to reduce cortical infarction without affecting striatal lesion after ischaemia in rats. Accordingly, it has been recently shown that brain slices exposed to oxygen-glucose deprivation were rescued by 1400W (Cardenas et al., 1998). The reduction of cortical lesion is associated with an amelioration of neurological deficit and a reduction of weight loss, indicating that the neuroprotection obtained has positive functional consequences. It must also be put forward that the protective effect was obtained with a treatment initiated 18 h after ischaemia. This delay was chosen according to data from our laboratory indicating that in our model, iNOS appears between 18 and 24 h after the onset of ischaemia. According to this kinetic, it is likely that the therapeutic window of iNOS inhibitors may even be a little wider. Such a reduction in brain infarction and wide therapeutic window with 1400W are similar to those obtained with aminoguanidine by Iadecola and coworkers (see above). However, to our knowledge, the functional protection observed with 1400W has not been reported with aminoguanidine.
Inhibition of the iNOS activity by 1400W is timely correlated with the neuroprotective effect. However, it must be noticed that iNOS inhibition is determined on sample taken from both the ischaemic core in the striatum and the penumbra in the cortex, whereas the neuroprotection is limited to the cortex. The repeated treatments with 1400W for 48 h reduces iNOS activity by 36% after 3 days of reperfusion, at the time of the measurement of ischaemic outcomes. This apparent reduction in inhibition of iNOS activity as compared with the one observed at 24 h do not reflect a loss of efficacy of 1400W, as the 3 day measurements were taken 6 h after the last injection, whilst the 24 h observations were made after only 1 h. Actually, the inhibition is still significant, although at 3 days after ischaemia iNOS activity levels were greater than those at 24 h. These observations suggest that 1400W was able to inhibit, at least partially, iNOS activity during the 48 h period of treatment. In contrast, 1400W does not modify the postischaemic cNOS activity. Thus, the beneficial effect of 1400W is not the consequence of the inhibition of the deleterious neuronal NOS. Taken together, these data support the hypothesis that iNOS inhibition is the mechanism by which 1400W reduces ischaemic damage. However, it can not be excluded that 1400W may act via other protective mechanism(s) unknown to date.
In conclusion, we have demonstrated that administration of the iNOS inhibitor 1400W, inhibits iNOS activity in the postischaemic brain, reduces ischaemia-induced cerebral infarction and functional deficits. These beneficial effects are observed 3 days after transient focal ischaemia when repeated treatments with 1400W for 48 h are begun 18 h after the induction of ischaemia. This wide window of therapeutic opportunity underscores the complementary of iNOS inhibition based strategies with respect to other anti-ischaemic strategies such as antagonism at glutamate receptors (Margaill et al., 1997). Thus, selective iNOS inhibitors may in the future provide an effective therapeutic option for the treatment of stroke.
Abbreviations
- CBF
cerebral flood flow
- CCA
common carotid artery
- cNOS
constitutive NO-synthase
- eNOS
endothelial NO-synthase
- iNOS
inducible NO-synthase
- L-NAME
NW-nitro-L-arginine methyl ester
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- MCA
middle cerebral artery
- nNOS
neuronal NO-synthase
- NO
nitric oxide
- PaCO2
arterial pressure in CO2
- PaO2
arterial pressure in O2
- s.c.
subcutaneously
References
- BABU R.B., GRIFFITH O.W. Design of isoform-selective inhibitors of nitric oxide synthase. Curr. Opin. Chem. Biol. 1998;2:491–500. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- BRADFORD M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- BREDT D.S., SNYDER S.H. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in cerebellum. Proc. Natl. Acad. Sci. U.S.A. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUISSON A., PLOTKINE M., BOULU R.G. The neuroprotective effect of a nitric oxide synthase inhibitor in a rat model of focal cerebral ischaemia. Br. J. Pharmacol. 1992;106:766–767. doi: 10.1111/j.1476-5381.1992.tb14410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDENAS A., DE ALBA J., MORO M.A., LORENZO P., LIZASOAIN I. Protective effect of N-(3-aminomethylbenzyl)acetamidine, an inducible nitric oxide synthase inhibitor, in brain slices exposed to oxygen-glucose deprivation. Eur. J. Pharmacol. 1998;354:161–165. doi: 10.1016/s0014-2999(98)00458-0. [DOI] [PubMed] [Google Scholar]
- COCKROFT K.M., MEISTRELL M., ZIMMERMAN G.A., RISUCCI D., BLOOM O., CERAMI A., TRACEY K.J. Cerebroprotective effects of aminoguanidine in a rodent model of stroke. Stroke. 1996;27:1393–1398. doi: 10.1161/01.str.27.8.1393. [DOI] [PubMed] [Google Scholar]
- DALKARA T., MOSKOWITZ M.A. The complex role of nitric oxide in the pathophysiology of focal cerebral ischaemia. Brain Pathology. 1994;4:49–57. doi: 10.1111/j.1750-3639.1994.tb00810.x. [DOI] [PubMed] [Google Scholar]
- ESCOTT K.J., BEECH J.S., HAGA K.K., WILLIAM S.C.R., MELDRUM B.S., BATH P.M.W. Cerebroprotective effect of the nitric oxide synthase inhibitors, 1-(2-trifluoromethylphenyl) imidazole and 7-nitro indazole, after transient focal cerebral ischaemia. J. Cereb. Blood Flow Metab. 1998;18:281–287. doi: 10.1097/00004647-199803000-00006. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J.R., KNOWLES R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GOLANOV E.V., REIS D.J. Contribution of cerebral edema to the neuronal salvage elicited by stimulation of cerebral fastiglial nucleus after occlusion of the middle cerebral artery in rat. J. Cereb. Blood Flow Metab. 1995;15:172–173. doi: 10.1038/jcbfm.1995.19. [DOI] [PubMed] [Google Scholar]
- GRANDATI M., VERRECCHIA C., REVAUD M.L., ALLIX M., BOULU R.G., PLOTKINE M. Calcium-independent NO-synthase activity and nitrites/nitrates production in transient focal cerebral ischaemia in mice. Br. J. Pharmacol. 1997;122:625–630. doi: 10.1038/sj.bjp.0701427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARA H., HUANG P.L., PANAHIAN N., FISHMAN M.C., MOSKOWITZ M.A. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J. Cereb. Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- HUANG Z., HUANG P.L., MA J., MENG W., AYATA C., FISHMAN M.C., MOSKOWITZ M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-arginine. J. Cereb. Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- HUANG Z., HUANG P.L., PANAHIAN N., DALKARA T., FISHMAN M.C., MOSKOWITZ M.A. Effects of cerebral ischaemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- IADECOLA C., ZU X.H., ZHANG F.Y., EL-FAKAHANY E., ROSS M.E. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischaemia. J. Cereb. Blood Flow Metab. 1995a;15:52–59. doi: 10.1038/jcbfm.1995.6. [DOI] [PubMed] [Google Scholar]
- IADECOLA C., ZHANG F.Y., CASEY R., CLARK H.B., ROSS M.E. Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischaemia. Stroke. 1996;27:1373–1380. doi: 10.1161/01.str.27.8.1373. [DOI] [PubMed] [Google Scholar]
- IADECOLA C., ZHANG F.Y., CASEY R., NAGAYAMA M., ROSS M.E. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IADECOLA C., ZHANG F.Y., XU X.H. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am. J. Physiol. 1995c;268:R286–R292. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- IADECOLA C., ZHANG F.Y., XU S., CASEY R., ROSS M.E. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J. Cereb. Blood Flow Metab. 1995b;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- IVANOVA S., BOTCHKINA G.I., AL-ABED Y., MEISTRELL M., BATLIWALLA F., DUBINSKY J.M., IADECOLA C., WANG H., GREGERSEN P.K., EATON J.W., TRACEY K.J. Cerebral ischaemia enhances polyamine oxidation: identification of enzymatically formed 3-aminopropanal as an endogenous mediator of neuronal and glial cell death. J. Exp. Med. 1998;188:327–340. doi: 10.1084/jem.188.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARGAILL I., ALLIX M., BOULU R.G., PLOTKINE M. Dose- and time-dependence of L-NAME neuroprotection in transient focal cerebral ischaemia in rats. Br. J. Pharmacol. 1997;120:160–163. doi: 10.1038/sj.bjp.0700889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARGAILL I., PARMENTIER S., CALLEBERT J., ALLIX M., BOULU R.G., PLOTKINE M. Short therapeutic window for MK801 in transient focal cerebral ischemia in normotensive rats. J. Cereb. Blood Flow Metab. 1996;16:107–113. doi: 10.1097/00004647-199601000-00013. [DOI] [PubMed] [Google Scholar]
- NAGAFUJI T., SUGIYAMA M., MUTO A., MAKINO T., MIYAUCHI T., NABATA H. The neuroprotective effect of a potent and selective inhibitor of type I NOS (L-MIN) in a rat model of focal cerebral ischaemia. Neuroreport. 1995;6:1541–1545. doi: 10.1097/00001756-199507310-00019. [DOI] [PubMed] [Google Scholar]
- OPLINGER J.A., GARVEY E.P., FURFINE E.S., SHEARER B.G., COLLINS J.L.Acetamidine derivatives and their use as inhibitors for the nitric oxide synthase 1996. International Patent Application Number WO 96/19440 (published June 27, 1996)
- TRACEY W.R., TSE J., CARTERE G. Lipopolysaccharide-induced changes in plasma nitrite and nitrate concentrations in rats and mice: pharmacological evaluation of nitric oxide synthase inhibitors. J. Pharmacol. Exp. Ther. 1995;272:1011–1015. [PubMed] [Google Scholar]
- WAHL F., ALLIX M., PLOTKINE M., BOULU R.G. Neurological and behavioral outcomes of focal cerebral ischemia in the rat. Stroke. 1992;23:267–272. doi: 10.1161/01.str.23.2.267. [DOI] [PubMed] [Google Scholar]
- YOSHIDA T., LIMMROTH V., IRIKURA K., MOSKOWITZ M.A. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J. Cereb. Blood Flow Metab. 1994;14:924–929. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- ZHANG F.Y., CASEY R.M., ROSS M.E., IADECOLA C. Aminoguanidine ameliorates and L-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke. 1996a;27:317–323. doi: 10.1161/01.str.27.2.317. [DOI] [PubMed] [Google Scholar]
- ZHANG Z.G., REIF D., MACDONALD J., TANG W.X., GENTILE R.J., SHAKESPEAR W.C., MURRAY R.J., CHOPP M. ARL 17477, a potent and selective neuronal NOS inhibitor decreases infarct volume after transient middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 1996b;16:599–604. doi: 10.1097/00004647-199607000-00009. [DOI] [PubMed] [Google Scholar]