

Abstract
We have studied the effects of μ and δ opioids on intestinal function (permeability, PER; gastrointestinal transit, GIT), and their antagonism after the intracerebroventricular (i.c.v.) administration of specific antibodies (ABs) or antisense oligodeoxynucleotides (ODN) to μ-receptors (OR). Central versus peripheral site/s of action of subcutaneous (s.c.) μ-opioids, were also assessed.
Male Swiss CD-1 mice were used. GIT was measured with charcoal and PER by the passage of 51Cr-EDTA from blood to lumen.
Morphine and fentanyl (i.c.v. and s.c.) inhibited GIT and PER in a dose-related manner; they were more potent by i.c.v. route, both on GIT and PER (70 and 17 times for morphine and fentanyl). They also had a greater effect on GIT than PER (4.3 and 1.6 times). DPDPE had a lower potency than μ-agonists in all experiments, and no dose-response could be obtained after s.c. administration on GIT.
Pretreatment with i.c.v. ABs (24 h) or antisense ODN (5 days), decreased the effects (GIT and PER) of i.c.v. morphine and fentanyl, while those of DPDPE remained unchanged. The ABs did not alter the peripheral effects of μ-opioids.
The results show that (i.c.v. or s.c.) μ opioids produce dose-related inhibitions of PER and GIT, being more potent by the i.c.v. route. Delta-opioids had a greater effect on PER than GIT, while the opposite occurred for μ-agonists. Pretreatment with ABs or ODN to μ-OR, blocked the central effects of μ (but not δ) agonists on GIT and PER.
Keywords: Gastrointestinal transit, intestinal permeability, μ-receptor antibodies, μ-antisense oligodeoxynucleotide, opioids, peripheral effects
Introduction
Cloning of the δ (Evans et al., 1992) μ (Thompson et al., 1993) and κ (Chen et al., 1993) OR has provided new tools to study the pharmacological effects of opioids at the molecular level (Meng et al., 1993). Although the structure of the three OR is highly homologous, they have discrete regions which contain sequences unique to each receptor (Mansour et al., 1995), that have been particularly useful for generating ABs (Garzón et al.,1994; 1995) and target the specific receptors using antisense ODNs (Lai et al., 1994; Bilsky et al., 1996). Both probes have been used to study receptor mediated functions in vitro and in vivo (Wahlested et al., 1993). Thus, the i.c.v. administration of antisera generated against the amino-terminal portion or the peptide sequence of the second extracellular loop of the cloned μ-OR, decreases the antinociceptive effects of μ-specific opioid agonists in mice (Garzón & Sánchez-Blázquez, 1995). Similarly, the i.c.v. administration of ODNs targeting specific regions of mRNA for the μ-OR, also block the supraspinal antinociceptive effects of morphine (Rossi et al., 1994; Sánchez-Blázquez et al., 1997), while random sequences ODN are inactive. In addition, ODNs have been reported to block opioid induced inhibition of GIT in mice (Rossi et al., 1995).
The inhibitory effects of systemic opioids on gastrointestinal function, are mediated by OR located at central (CNS) and peripheral sites (gut) (Shook et al., 1987). In mice, inhibition of GIT by opioids is produced by binding predominately to μ and δ-OR (Porreca et al., 1984; Pol et al., 1994) located at both sites; however, the precise implication of each anatomical site is not well established. Other intestinal effects of opioids such as the inhibition of water and electrolyte permeability have not been completely characterized.
The primary aim of our investigation was to characterize the effects of μ and δ opioid agonists on intestinal PER and their predominant site of action (central vs peripheral). In addition, we wanted to determine if the i.c.v. administration of specific μ-OR ABs or antisense ODN to μ-OR, could reverse the effects of μ agonists on GIT and intestinal PER.
Methods
Animals
Experiments were performed in male Swiss CD-1 mice, weighting 20–25 g. Animals were housed under 12 h light and 12 h dark conditions in a room with controlled temperature (22°C) and humidity (66%). Animals had free access to food and water and were allowed to become acclimated to their housing conditions for a least 1 week before the study. All experiments were conducted between 09.00 h and 14.00 h. The study protocol was approved by the local Committee of Animal Use and Care of our Institution and in accordance with the guidelines of European Community on Care and Use of Laboratory Animals.
Gastrointestinal transit (GIT)
Gastrointestinal transit was measured according to the procedures used in our laboratory (Pol et al., 1996b; Puig et al., 1996). Briefly, food was removed 18 h before the experiment but animals had free access to water. At this time, a charcoal meal (0.25 ml of 10% charcoal in 5% gum acacia) was administered intragastrically and GIT was evaluated 20 min later. Animals were then sacrificed and the small intestine separated from omentum avoiding stretching. The length of intestine from the pyloric sphincter to the ileocecal junction, and the distance travelled by the charcoal, were measured. For each animal, GIT was calculated as the percentage (%) of distance travelled by the charcoal, relative to the total length of the small intestine (% of GIT).
Intestinal permeability (PER)
Permeability of the small intestine was assessed by measuring the passage of a radioactive marker (51Cr-EDTA) from blood to lumen, using a technique adapted from Miller et al. (1991). Prior to the study, mice were fasted for 18 h except for free access to water. A laparotomy was performed under light ether anaesthesia, and both renal pedicles ligated to prevent rapid excretion of the radioactive marker into the urine. Animals were allowed to recover for a period of 50 min, and at this time 4 μCi of 51Cr-EDTA were injected intravenously in a vein of the tail. Forty minutes later, the small intestine was removed and the intestinal lumen washed with 1 ml of saline; the 51Cr-EDTA present in the fluid was then measured with a gamma counter (LKB-Wallac 1282 CompuGamma). Results are expressed as d.p.m. g−1 of wet weight tissue. In these experiments, opioid agonists were given 15 min before the intravenous administration of 51Cr-EDTA.
Intracerebroventricular injections
Intracerebroventricular injections were carried out into the left lateral ventricle of ether-anaesthetized mice in a final volume of 5–10 μl. Control animals received the same volume of saline. Injections were performed using a Hamilton microlitre syringe (Microdispenser Socorex, PANREAC S.A.) fitted with a 26-gauge needle, according to the method of Haley & McCormick (1957). The site of injection was 2 mm caudal and 2 mm lateral to bregma, and 3 mm in depth from the skull surface.
Antibodies to μ-OR
The ABs used in the study were: the polyclonal antisera MAS/2 which was generated in rabbits against the amino-terminal portion 1–16 (MDSSTGPGNTSDCSDP) of the cloned μ-OR (Garzón et al., 1995) and the anti μ-receptor antiserum MU/2EL raised against the peptide sequence 208–216 (TKYRQGSID) of the second extracellular loop of this receptor (Garzón & Sánchez-Blázquez, 1995). Each AB was tested in three dilutions: 1 : 100, 1 : 1000 and 1 : 10.000. A single dose of diluted immunized sera was injected i.c.v., 24 h prior to the experiment, in a final volume of 5 μl. Preimmune sera or immune sera heated at 100°C for 10 min, were used as a control.
Synthesis of oligodeoxynucleotides (ODN)
End-capped phosphorothiodate ODN were synthesized on a CODER 300 DNA synthesizer by phosphoramidite chemistry (Matteucci & Caruthers, 1981). The introduction of phosphorothioate linkages was achieved by tetraethylthiuram disulphide sulphurization (Vu & Hischbein, 1991). Crude ODN were purified by reverse-phase chromatography with COP cartridges (Cruachem, Glasgow, U.K.). The eluded ODN in 50% acetonitrile-water were then lyophilized (Rouan RC 1009/RCT 90 France). Sequences were as follows: (a) ODN μ16–32 5′ C*T*GATGTTCCCTGGG*C*C-3 a 17-base oligo corresponding to nucleotides 16–32 of the murine μ OR gene sequence (Min et al., 1994; Sánchez-Blázquez et al., 1997) and (b) a random ODN with the sequence 5′- G*C*CTTATTTACTACTTTC*G*C-3′ served as control (Gillardon et al., 1994; Sánchez-Blázquez et al., 1995).
Oligodeoxynucleotides solutions were reconstructed in sterile saline immediately before use. To assess the specificity of the treatments with ODN three separate control groups of mice (6–8 per group) were used: noninjected animals, animals receiving i.c.v. saline and mice injected with a random-sequence ODN (ODN-random). In all experiments, treatments were administered according to the following schedule: 1 nmol on days 1 and 2; 2 nmol on days 3 and 4 and 3 nmol on day 5. On day 6, the inhibitory effects of opioids on GIT or PER were evaluated.
Groups of experiments
The effects of μ and δ opioid agonists administered by i.c.v. or s.c. routes, were evaluated on GIT and PER. We determined: (a) dose response curves to i.c.v. or s.c. morphine, fentanyl and DPDPE in control animals (non-treated); (b) the effect/s of i.c.v. morphine, fentanyl and DPDPE in animals treated with i.c.v. μ-OR ABs or ODN and (c) the effects of s.c. PL017 (a peripherally acting μ agonist) and morphine, after the i.c.v. administration of μ-OR ABs.
Drugs
The drugs used were: morphine sulphate (Alcaiber S.A., Madrid, Spain), fentanyl (Syntex Latino S.A., Madrid, Spain), PL017 [N-MePhe3, D-Pro4–morphiceptin] (Peninsula Laboratories, Belmont, CA, U.S.A.), and DPDPE [Enkephalin, [D-Pen2,5] (Research Biochemicals Incorporated, U.S.A.). Drugs were dissolved in sterile pyrogen-free 0.9% sodium chloride just before use, and injected s.c. (10 ml kg−1) or i.c.v. (5 μl).
Data analysis
The inhibitory effects of opioid agonists are expressed as the percentage of inhibition of the GIT or PER in a drug treated animal (test) when compared with the mean GIT or PER measured in the corresponding group of control mice (n=6–8):
Data are expressed as a group mean±s.e.mean All statistical calculations were performed as described by Tallarida & Murray (1986). ED50±s.e. (dose which produced a 50% effect) values were determined by linear regression analysis of dose-response relations based on at least six to eight mice per dose. Statistical analysis for significant differences between two groups were obtained by Student's t-test; when multiple groups were compared, one way analysis of variance (ANOVA) followed by a Student-Newman-Keuls test, was used whenever applicable. A value of P<0.05 was considered as significant.
Results
Dose-response curves to morphine, fentanyl and DPDPE on gastrointestinal transit and intestinal permeability
In each preparation, opioid agonists were evaluated after administration by the i.c.v. or s.c. routes. Dose-response curves were performed for morphine, fentanyl and DPDPE (Figures 1 and 2), and their ED50 values calculated as a measure of potency (Table 1). For each route, the curves to morphine and fentanyl were parallel and reached Emax values approximately 80%. The i.c.v. administration of DPDPE also generated dose-response curves, with an Emax for PER of 80% while that on GIT was 52.2%. When DPDPE was administered by the s.c. route, a dose response curve was obtained on PER, that showed a significantly different slope than morphine or fentanyl, and an Emax of 46.2%. No dose-response curve could be obtained after the s.c. administration of DPDPE and a maximal inhibitory effect was obtained with the administration of 38.5 nmol. The results show the DPDPE has a lower potency than morphine or fentanyl, in all experimental conditions. On the basis of the dose-response curves obtained with DPDPE, propose that δ-OR could participate in the inhibitory control of intestinal PER in mice.
Figure 1.
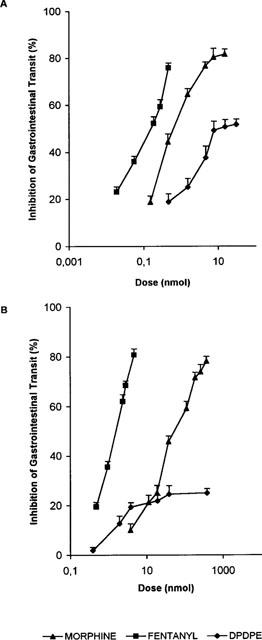
Dose response relationships for the i.c.v. (A) and s.c. (B) administration of morphine, fentanyl and DPDPE on the inhibition of GIT in mice. Each point represents the mean±s.e.mean of six or more animals.
Figure 2.
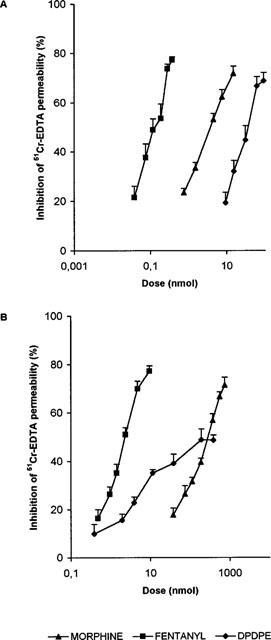
Dose response relationships for the i.c.v. (A) and s.c. (B) administration of morphine, fentanyl and DPDPE on the inhibition of intestinal PER in mice. Each point represents the mean±s.e.mean of six or more animals.
Table 1.
ED50 values (nmol), slopes and Emax of i.c.v. and s.c. opioid agonists on % inhibition of GIT and intestinal PER in mice
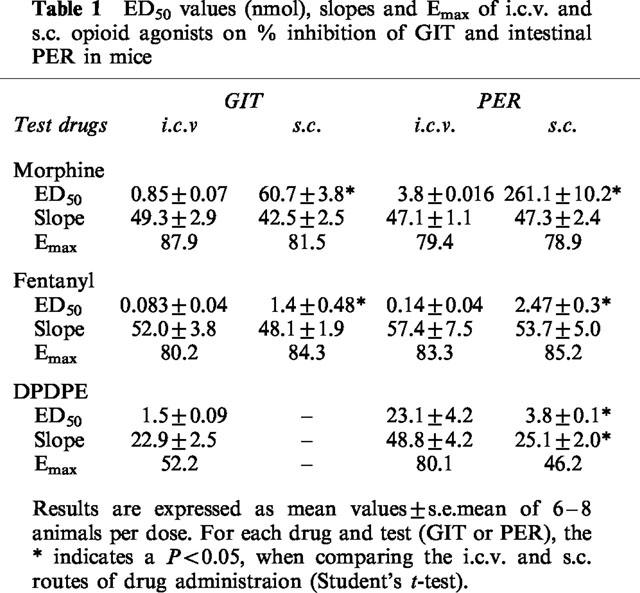
In order to assess the most effective route of administration, ratios of the ED50 values of s.c. and i.c.v. opioids were calculated (Table 2). Morphine and fentanyl were approximately 70 and 17 times more potent respectively by the i.c.v. route, both on GIT and PER. Thus, in all instances, the potency of μ opioids inhibiting intestinal function is greater when drugs are given centrally. Ratios between the ED50 values on PER and GIT were obtained as an indication of the potency of μ opioids inhibiting a certain intestinal function. Regardless of the route, morphine and fentanyl were 4.3 and 1.7 times, respectively more potent inhibiting GIT than PER. Potency ratios for DPDPE could not be obtained due to the characteristics (Emax, slope) of the dose-response curves obtained.
Table 2.
ED50 ratios of morphine and fenanyl according to the route of administration (s.c./i.c.v.) and experimenal preparation (PER/GIT)

Effects of i.c.v. anti-μ-OR ABs (MAS/2 and MU/2EL) on the inhibition of GIT and PER induced by i.c.v. opioid agonists
The i.c.v. administration of the polyclonal ABs MAS/2 and MU/2EL, to the cloned μ-OR, induced 11.7 and 8.5% decreases in GIT and PER respectively in basal conditions; however the differences were not statistically significative (GIT: basal 52.0±3.1 vs treated 45.9±1.1%) (PER: 2587±326.5 vs 2366.5±306.3 d.p.m. g−1). Similar results were obtained with MAS/2 and MU/2EL. The effects of a single dose of i.c.v. morphine, fentanyl or DPDPE on GIT and PER, were evaluated in animals treated (24 h before) with each antiserum at three different dilutions (1 : 100, 1 : 1000 and 1 : 10000). Doses of opioids that produced approximately a 60–70% inhibition of GIT or PER were utilized in all experiments, except when testing the DPDPE on GIT, where the dose that produced the maximal effect was used (7.7 nmol). Control animals received i.c.v. preimmune sera instead of the ABs. In all experiments, opioid agonists produced the same inhibitory effect in noninjected control conditions and in animals receiving i.c.v. immune sera. Treatment with anti-μ-OR ABs, significantly decreased the effects of morphine and fentanyl on GIT and PER. Figure 3A shows the inhibition of GIT induced by a fixed dose of i.c.v. morphine (1.5 nmol) in control conditions and in animals pretreated with immune sera, MAS/2 or MU/2EL antiserum. When comparing the GIT effects of morphine in the different experimental conditions, ANOVA revealed a significant effect of both ABs at dilutions of 1 : 100 and 1 : 1000 (P<0.01); similar results were obtained with 0.28 nmol of i.c.v. fentanyl (ANOVA P<0.01, Figure 3B). In contrast, the antitransit effects of i.c.v. DPDPE a δ-selective agonist (7.7 nmol), was unaltered in the presence of any of the ABs dilutions tested (Figure 3C).
Figure 3.
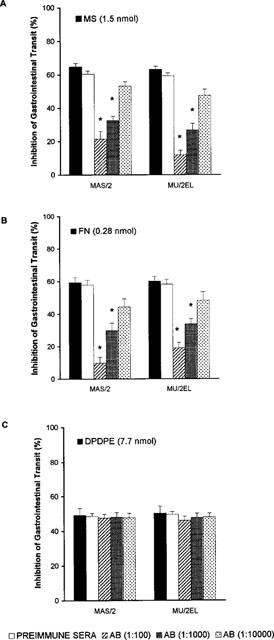
Inhibition of GIT induced by the i.c.v. administration of 1.5 nmol morphine (A), 0.28 nmol fentanyl (B) and 7.7 nmol DPDPE (C). Animals were pretreated with preimmune sera or ABs to μ-OR (MAS/2 or MU/2EL) at three dilutions (1 : 100; 1 : 1000 and 1 : 10000). Solid bars show the effects of morphine (MS), fentanyl (FN) or DPDPE in noninjected mice. Each column represents the mean±s.e.mean of six or more animals. *P<0.05, when compared to control (Student-Newman-Keuls test).
Figure 4 shows that pretreatment with antisera to μ-OR similarly blocks the effects of i.c.v. morphine and fentanyl (but not DPDPE) on PER. In the upper panel (Figure 4A) we show the inhibition of PER induced by morphine (7.5 nmol); ANOVA revealed a significant effect of both ABs (P<0.05) at 1 : 100 and 1 : 1000; similar results were observed with 0.28 nmol of fentanyl (Figure 4B, P<0.05). However, the effects of DPDPE (61.6 nmol) were not significantly altered by any of the concentrations of the ABs (Figure 4C).
Figure 4.
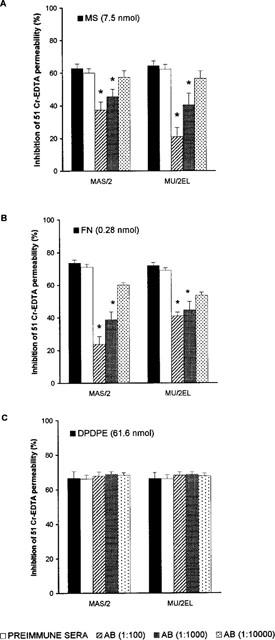
Inhibition of intestinal PER induced by the i.c.v. administration of 7.5 nmol morphine (A), 0.28 nmol fentanyl (B) and 61.6 nmol DPDPE (C). Animals were pretreated with preimmune sera or ABs to μ-OR (MAS/2 or MU/2EL) at three different dilutions (1 : 100; 1 : 1000 and 1 : 10000). Solid bars show the effects of morphine (MS), fentanyl (FN) or DPDPE in noninjected mice. Each column represents the mean±s.e.mean of six or more animals. *P<0.05, when compared to control (Student-Newman-Keuls test).
The differences observed regarding the degree of antagonism obtained with each AB cannot be accurately quantified. Grossly, when we evaluate the dilution 1 : 100, the MU/2EL AB seems to be more effective antagonizing the antitransit effects of morphine (81.7% inhibition), while MAS/2 is more active blocking the antitransit effects of fentanyl (83.5%). In addition, the inhibition of the effects of opioids by the ABs are more apparent on GIT.
Effects of antisense ODN to μ-OR mRNA on the inhibition of GIT and PER induced by the i.c.v. administration of opioids
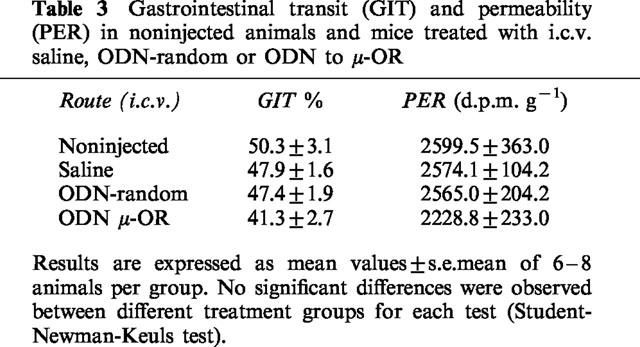
The effects of opioid-agonists were evaluated after reducing the density of μ-OR in the CNS by the i.c.v. administration of antisense ODN directed to μ-OR mRNA. Initial experiments were performed in order to determine the effects of the i.c.v. administration of ODN to μ-OR on basal GIT and PER. The results show that the ODN decreased GIT and PER by 18 and 14% respectively (Table 3). However, when the results were compared with the different controls (noninjected, saline, ODN-random), no significant differences were observed (one way ANOVA). In each preparation, we tested the same doses of i.c.v. opioids used in the experiments with ABs.
Table 3.
Gastrointestinal transit (GIT) and permeability (PER) in noninjected animals and mice treated with i.c.v. saline, ODN-random or ODN to μ-OR
The results show (Figure 5) that treatment with ODN to μ-OR, significantly decreased the effects of morphine (P<0.05) and fentanyl (P<0.05) on GIT and PER, while those of DPDPE were unaltered. The ODN blocked 63% of the effect of morphine on GIT and 74% of PER; the effects of fentanyl were similarly antagonized (52% on GIT and 60% on PER).
Figure 5.
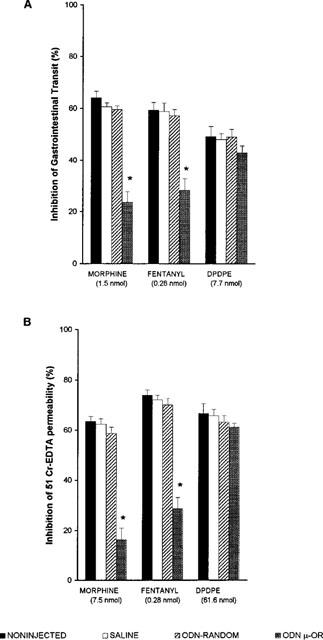
Inhibition of GIT (A) and intestinal PER (B) induced by the i.c.v. administration of morphine, fentanyl and DPDPE, in animals pretreated with: saline, ODN-random or ODN to μ-OR mRNA. Each column represents the mean±s.e.mean of six or more animals. *P<0.05, when compared to control (Student-Newman-Keuls test).
Effects of s.c. PL017 (a peripheral μ-opioid agonist) and morphine on GIT and PER, after the i.c.v. administration of μ-OR ABs
These experiments were designed to evaluate the intestinal (peripheral) effects of s.c. opioids, in animals treated with i.c.v. ABs. We evaluated: (a) the effects of s.c. PL017, a μ opioid agonist that has limited access to the brain and (b) two doses of s.c. morphine (high and low), an opioid that binds to central and/or intestinal OR according to the dose administered (Shook et al., 1987). In our experiments we used doses of morphine that produced approximately a 20 and 70% inhibition of GIT or PER. Figure 6 shows that the ABs (1 : 100) did not alter the effects of PL017 or low doses of morphine on GIT or PER. However, the inhibitory effects of high doses of s.c. morphine were partially antagonized in animals treated with i.c.v. ABs.
Figure 6.
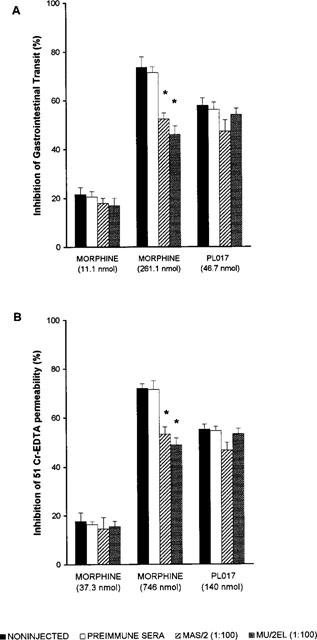
Inhibition of GIT (A) and intestinal PER (B) induced by the s.c. administration of morphine (low and high doses) and PL017, in animals pretreated with preimmune sera or ABs to μ-OR (MAS/2 or MU/2EL) at a 1 : 100 dilution. Each column represents the mean±s.e.mean of six or more animals. *P<0.05, when compared to control (Student-Newman-Keuls test).
The results show that the effects of high doses of morphine are mediated by binding to central as well as intestinal OR, while the effects of low doses of morphine and PL017 are mostly peripheral.
Discussion
The present study demonstrates that morphine and fentanyl, administered i.c.v. or s.c., produce dose-related inhibitions of GIT and PER. Both agonists were more potent on the inhibition of GIT than PER, suggesting that transit was more susceptible to the inhibitory effects of μ opioids. Probably, when measuring the effects of opioids on transit, the simultaneous inhibition of fluid transfer to the lumen (PER), contributes to the antitransit effects measured. However, we were able to show for the first time that DPDPE a δ-OR agonist, is more effective decreasing PER than GIT. Interestingly, i.c.v. DPDPE produced a maximal inhibitory effect on GIT of 52%, while on PER the Emax was over 80%, suggesting that δ-OR play an important role in the inhibitory control of intestinal PER.
Morphine and fentanyl were both more potent when administered by the i.c.v. route, a finding previously reported in the literature (Kromer, 1989; Pol & Puig, 1997). In our experimental conditions, morphine was about 70 times more potent by the i.c.v. than the s.c. route, and fentanyl only 17 times; this could be related to the lipophilic properties of fentanyl, which when injected i.c.v., binds to OR located on the site of injection, while morphine (a hydrophilic drug) migrates into the CSF and reach OR located in other brain regions. Unfortunately the anatomical sites that regulate intestinal function in the CNS, are very little known at present.
The i.c.v. administration of ABs (MAS/2 and MU/2EL) or ODN to cloned μ-OR did not significantly alter basal GIT or PER; however, basal GIT has been reported to be approximately 30% lower in μ-OR knockout mice when compared to heterozygous and wild type animals (Roy et al., 1998). This discrepancy could be explained by the presence of residual functional μ-OR that remain active after treatment with ODN (Sánchez-Blázquez et al., 1997), suggesting that ODN preferentially affect a small pool of receptors of recent synthesis or with rapid turnover. The results indicate that eliminating but not decreasing the density of functional OR has a significant effect on intestinal function.
The probes (ABs and ODN) significantly reduced the supraspinal inhibitory effects (GIT and PER) of μ opioid agonists, while those of the δ specific agonist DPDPE remained unaltered, confirming the specificity of the ABs and ODN. Using the same ABs, Garzón & Sánchez-Blázquez (1995) had previously reported a similar antagonistic effect on antinociception in the tail flick test. However, Roy et al. (1998) have shown that in μ-OR knockout animals, the i.c.v. administration of δ or κ agonists had no effect GIT, suggesting a crossover of these agonists to μ-OR, an assumption that is not supported by our results.
In our experimental model, the antagonistic effect of the AB MU/2EL, was more prominent blocking the effects of morphine than fentanyl, suggesting that the 2nd extracellular loop of the μ-OR, could participate in the binding of morphine, while the N-terminal could influence that of fentanyl. However, on the basis of the small differences observed between the effects of the ABs, no conclusive evidence can be derived.
Repeated i.c.v. injections of an ODN complementary to the bases 16–32 of the murine μ-OR mRNA, significantly reduced the central GIT and PER effects of morphine and fentanyl. The same ODN (Sánchez-Blázquez et al., 1997) as well an another ODN targeted to the first 18 nucleotides of the rat μ-OR (Chen et al., 1995), have been shown to significantly decrease the antinociceptive effects of morphine. However, our results disagree with those of Rossi et al. (1995) who showed that only ODNs targeting exon 4, were active blocking the effects of μ opioids on the GIT; this author also showed that none of the probes directed against exon 1 could decreased the antitransit effect of i.c.v. morphine. The discrepancies with our results could be explained by the different sequence of the ODN used, and/or dissimilar protocol of administration.
In mammals, the effects of systemic opioids on gastrointestinal function are mediated by interaction with OR located in the brain and in the gastrointestinal tract, where they are present with particular abundance in the myenteric and submucosal plexuses (Fickel et al., 1997). OR are also present on villus and crypts of enterocytes where they are a possible target for opioids inhibiting intestinal secretion (Lang et al., 1996). In our study, the administration of i.c.v. ABs did not alter the peripheral effects of opioids. In these experiments we used the systemic administration of a peripherally acting μ agonist (PL017), as well as two doses of morphine, based on the assumption that low doses of morphine act exclusively at peripheral sites (Shook et al., 1987; Pol et al., 1996a), while high doses would have a central and a peripheral effect. Our results support this possibility, since the ABs partially blocked the effects of high doses of morphine, while it did not alter the effects of lower doses, or those induced by PL017. Moreover, when morphine is administered systemically seems to reach the gut in a larger proportion than the brain (Manara et al., 1986).
In conclusion the study shows, that μ opioid agonists administered i.c.v. or s.c. produce dose-related inhibitions of GIT and PER. Both agonists are more potent by the i.c.v. route and produce a greater inhibition of GIT than PER. Delta (δ) agonists were also more effective by the i.c.v. route, but had a greater potency inhibiting intestinal PER than GIT. The i.c.v. administration of the μ-OR ABs (MAS/2 and MU/2EL), or antisense ODN 16–32 (to exon 1) to μ-OR , specifically blocked the central effects of μ (but not δ) agonists on GIT and PER. The antagonistic effect of the AB MU/2EL, was greater for morphine than fentanyl, a pure μ-OR agonist.
Acknowledgments
The authors wish to thank Mr Sergi Leánez for his excellent technical assistance. The work was partially supported by grants from CICYT, PM96-0039, Madrid and Fundació La Marató de TV3, 2032/97, Barcelona, Spain.
Abbreviations
- ABs
antibodies
- CNS
central nervous system
- DPDPE
[D-Pen2,5]- enkephalin
- i.c.v.
intracerebroventricular
- GIT
gastrointestinal transit
- ODN
oligodeoxynucleotides
- OR
opioid receptor
- s.c.
subcutaneous
References
- BILSKY E.J., BERNSTEIN R.N., HRUBY V.J., ROTHMAN R.B., LAI J., PORRECA F. Characterization of antinociception to opioid receptor selective agonists after antisense oligodeoxynucleotide-mediated ‘knock-down' of opioid receptors in vivo. J. Pharmacol. Exp. Ther. 1996;277:491–501. [PubMed] [Google Scholar]
- CHEN X.H., ADAMS J.U., GELLER E.B., DERIEL J.K., ADLER M.W., LIU CHEN L.Y. An antisense oligodeoxynucleotides to mu-opioid receptors inhibits mu-opioid receptor agonist-induced analgesia in rats. Eur. J. Pharmacol. 1995;275:105–108. doi: 10.1016/0014-2999(95)00012-a. [DOI] [PubMed] [Google Scholar]
- CHEN Y., MESTEK A., LIU J., YU L. Molecular cloning of a rat kappa-opioid receptor reveals sequence similarities to the mu and delta opioid receptors. Biochem. J. 1993;295:625–628. doi: 10.1042/bj2950625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS C.J., KEITH D.E., MORRISON H., MAGENDZO K., EDWARDS R.H. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- FICKEL J., BAGNOL D., WATSON S.J., AKIL H. Opioid receptor expression in the rat gastrointestinal tract: a quantitative study with comparison to the brain. Mol. Brain Res. 1997;46:1–8. doi: 10.1016/s0169-328x(96)00266-5. [DOI] [PubMed] [Google Scholar]
- GARZON J., CASTRO M.A., JUARROS J.L., SANCHEZ-BLAZQUEZ P. Antibodies raised against the N-terminal sequence of the delta opioid receptor blocked delta-mediated supraspinal antinociception in mice. Life Sci. Pharmacol. Lett. 1994;54:PL191–PL196. doi: 10.1016/0024-3205(94)90167-8. [DOI] [PubMed] [Google Scholar]
- GARZON J., JUARROS J.L., CASTRO M.A., SANCHEZ-BLAZQUEZ P. Antibodies to the cloned mu-opioid receptor detect various molecular weight forms in areas of mouse brain. Mol. Pharmacol. 1995;47:738–744. [PubMed] [Google Scholar]
- GARZON J., SANCHEZ-BLAZQUEZ P. In vivo injection of antibodies directed against the cloned mu opioid receptor blocked supraspinal analgesia induced by mu-agonist in mice. Life Sci. Pharmacol. Lett. 1995;56:PL237–PL242. doi: 10.1016/0024-3205(95)00064-d. [DOI] [PubMed] [Google Scholar]
- GILLARDON F., BECK H., UHLMANN E., HERDEGEN T., SANDKUHLER J., PEYMAN A., ZIMMERMANN M. Inhibition of c-fos protein expression in rat spinal cord by antisense oligodeoxynucleotide superfusion. Eur. J. Neurosci. 1994;6:880–884. doi: 10.1111/j.1460-9568.1994.tb00999.x. [DOI] [PubMed] [Google Scholar]
- HALEY T.J., MCCORMICK W.G. Pharmacological effects produced by intracerebral injection of drug in the conscious mouse. Br. J. Pharmacol. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KROMER W. The current status of opioid research on gastrointestinal motility. Life Sciences. 1989;44:579–589. doi: 10.1016/0024-3205(89)90190-2. [DOI] [PubMed] [Google Scholar]
- LAI J., BILSKY E.J., ROTHMAN R.B., PORRECA F. Treatment with antisense oligodeoxynucleotide to the opioid delta receptor selectively inhibits delta2-agonist antinociception. NeuroReport. 1994;5:1049–1052. doi: 10.1097/00001756-199405000-00008. [DOI] [PubMed] [Google Scholar]
- LANG M.E., DAVISON J.S., BATES S.L., MEDDINGS J.B. Opioid receptors on guinea-pig intestinal crypt epithelial cells. J. Physiol. 1996;497:161–174. doi: 10.1113/jphysiol.1996.sp021757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANARA L., GIANCARLO B., FERRETTI P., TAVANI A. Inhibition of gastrointestinal transit by morphine in rats results primarily from direct drug action on gut opioid sites. J. Pharmacol. Exp. Ther. 1986;237:945–949. [PubMed] [Google Scholar]
- MANSOUR A., FOX C.A., AKIL H., WATSON S.J. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- MATTEUCCI M.D., CARUTHERS M.H. Synthesis of deoxyoligo nucleotides on a polymer support. J. Am. Chem. Soc. 1981;103:3185–3191. [Google Scholar]
- MENG F., XIE G.X., THOMPSON R.C., MANSOUR A., GOLDSTEIN A., WATSON S.J., AKIL H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9954–9958. doi: 10.1073/pnas.90.21.9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MILLER M.J.S., ZHANG X.J., GU X., CLARK D.A. Acute intestinal injury induced by acetic acid and casein: prevention by intraluminal misoprostol. Gastroenterol. 1991;101:22–30. doi: 10.1016/0016-5085(91)90455-t. [DOI] [PubMed] [Google Scholar]
- MIN B.H., AUGUSTIN L.B., FELSHEIM R.F., FUCHS J.A., LOH H.H. Genomic structure and analysis of promoter sequence of a mu opioid receptor gene. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9081–9085. doi: 10.1073/pnas.91.19.9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POL O., FERRER I., PUIG M.M. Diarrhoea associated with intestinal inflammation increases the potency of mu and delta opioids on the inhibition of gastrointestinal transit in mice. J. Pharmacol. Exp. Ther. 1994;270:386–391. [PubMed] [Google Scholar]
- POL O., PUIG M.M. Reversal of tolerance to the antitransit effects of morphine during acute intestinal inflammation in mice. Br. J. Pharmacol. 1997;122:1216–1222. doi: 10.1038/sj.bjp.0701472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POL O., SANCHEZ B., PUIG M.M. Peripheral effects of opioids in a model of intestinal inflammation in mice. Pharmacol. 1996a;53:340–350. doi: 10.1159/000139449. [DOI] [PubMed] [Google Scholar]
- POL O., VALLE L., FERRER I., PUIG M.M. The inhibitory effects of α2-adrenoceptor agonists on gastrointestinal transit during croton oil-induced intestinal inflammation. Br. J. Pharmacol. 1996b;119:1649–1655. doi: 10.1111/j.1476-5381.1996.tb16085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PORRECA F., MOSBERG H.I., HRUST R., HRUBY V.J., BURKS T.F. Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J. Pharmacol. Exp. Ther. 1984;230:341–348. [PubMed] [Google Scholar]
- PUIG M.M., POL O., WARNER W. Interaction of morphine and clonidine on gastrointestinal transit in mice. Anesthesiology. 1996;85:1403–1412. doi: 10.1097/00000542-199612000-00022. [DOI] [PubMed] [Google Scholar]
- ROSSI G.C., PAN Y-X., BROWN G.P., PASTERNAK G.W. Antisense mapping the MOR-1 opioid receptor: evidence for alternative splicing and a novel morphine-6β-glucuronide receptor. FEBS Lett. 1995;369:192–196. doi: 10.1016/0014-5793(95)00757-z. [DOI] [PubMed] [Google Scholar]
- ROSSI G.C., PAN YX., CHENG J., PASTERNAK G.W. Blockade of morphine analgesia by an antisense oligodeoxynucleotide against the mu receptor. Life. Sci. Pharmacol. Sci. 1994;54:PL375–PL379. doi: 10.1016/0024-3205(94)90038-8. [DOI] [PubMed] [Google Scholar]
- ROY S., LIU H-C., LOH H.H. μ-Opioid receptor-knockout mice: the role of μ-opioid receptor in gastrointestinal transit. Mol. Brain. Res. 1998;56:281–283. doi: 10.1016/s0169-328x(98)00051-5. [DOI] [PubMed] [Google Scholar]
- SANCHEZ-BLAZQUEZ P., GARCIA-ESPAÑA A., GARZON J. In vivo injection of oligodeoxynucleotides to Gα subunits and supraspinal analgesia evoked by mu and delta opioid agonists. J. Pharmacol. Exp. Ther. 1995;275:1590–1596. [PubMed] [Google Scholar]
- SANCHEZ-BLAZQUEZ P., GARCIA-ESPAÑA A., GARZON J. Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: role of delta-2 opioid receptors. J. Pharmacol. Exp. Ther. 1997;280:1423–1431. [PubMed] [Google Scholar]
- SHOOK J.E., PELTON J.T., HRUBY V.J., BURKS T.F. Peptide opioid antagonist separates peripheral and central opioid antitransit effects. J. Pharmacol. Exp. Ther. 1987;243:492–500. [PubMed] [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of Pharmacologic Calculations with Computer Programs. New York: Springer-Verlag; 1986. [Google Scholar]
- THOMPSON R.C., MANSOUR A., AKIL H., WATSON S.J. Cloning and pharmacological characterization of a rat μ opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- VU A., HIRSCHBEIN B.L. Internucleotide phosphite sulfurization with tetraethylthiuram disulfide phosphorothioate oligonucleotide synthesis via phosphoramidite chemistry. Tetrahedron Lett. 1991;32:3005–3008. [Google Scholar]
- WAHLESTEDT C., PICH E.M., KOOB G.F., YEE F., HELLING M. Modulation of anxiety and neuropeptide Y-Y1 receptors by antisense oligodeoxynucleotides. Science. 1993;259:528–531. doi: 10.1126/science.8380941. [DOI] [PubMed] [Google Scholar]