

Abstract
The putative inhibitory effects of verapamil and diltiazem on neuronal non-L-type Ca2+ channels were studied by investigating their effects on either K+- or veratridine-evoked [3H]-dopamine ([3H]-DA) release in rat striatal slices. Involvement of N-, P- and Q-type channels was identified by sensitivity of [3H]-DA release to ω-conotoxin GVIA (ω-CTx-GVIA), ω-agatoxin IVA (ω-Aga-IVA) and ω-conotoxin MVIIC (ω-CTx-MVIIC), respectively.
KCl (50 mM)-evoked [3H]-DA release was abolished in the absence of Ca2+, and was insensitive to dihydropyridines (up to 30 μM). It was significantly blocked by ω-CTx-GVIA (1 μM), ω-Aga-IVA (30 nM) and was confirmed to be abolished by ω-CTx-MVIIC (3 μM), indicating involvement of N-, P- and Q-type channel subtypes.
Verapamil and diltiazem inhibited K+-evoked [3H]-DA release in a concentration-dependent manner. The inhibitory effects of verapamil or diltiazem (each 30 μM) were fully additive to the effect of ω-CTx-GVIA (1 μM), whereas co-application with ω-Aga-IVA (30 nM) produced similar effects to those of ω-Aga-IVA alone.
As shown previously, veratridine-evoked [3H]-DA release in Ca2+ containing medium exclusively involves Q-type Ca2+ channels. Here, diltiazem (30 μM) did not inhibit veratridine-evoked [3H]-DA release, whereas verapamil (30 μM) partially inhibited it, indicating possible involvement of Q-type channels in verapamil-induced inhibition. However, verapamil (30 μM) inhibited this release even in the absence of extracellular Ca2+, suggesting that Na+ rather than Q-type Ca2+ channels are involved.
Taken together, our results suggest that verapamil can block P- and at higher concentrations possibly N- and Q-type Ca2+ channels linked to [3H]-DA release, whereas diltiazem appears to block P-type Ca2+ channels only.
Keywords: Dopamine release, striatum, dihydropyridines, verapamil, diltiazem, ω-conotoxins, ω-agatoxins, veratridine
Introduction
Synaptic transmission is dependent upon the entry of Ca2+ through presynaptic voltage-activated Ca2+ channels (VACCs). In peripheral and central mammalian neurons, one low (T-type) and at least six high VACCs (L-, N-, O-, P-, Q-, and R-type) have been identified on the basis of their electrophysiological and pharmacological properties (Olivera et al., 1994; Varadi et al., 1995; Randall & Tsien, 1995). Striatal dopamine (DA) transmission is mediated mainly by N-type Ca2+ channels which are blocked selectively by ω-conotoxin-GVIA (ω-CTx-GVIA) (Tsien et al., 1988), whereas L-type Ca2+ channels are not involved (Harvey et al., 1996; Prince et al., 1996; Dobrev et al., 1998). Recently, it has been demonstrated that P-type Ca2+-channels which are blocked by ω-agatoxin-IVA (ω-Aga-IVA) (Randall & Tsien, 1995) and Q-type Ca2+ channels (Wheeler et al., 1994) which are blocked by ω-conotoxin-MVIIC (ω-CTx-MVIIC) also modulate dopamine synaptic transmission (Turner et al., 1993; Kimura et al., 1995; Soliakov & Wonnacott, 1996; Harvey et al., 1996; Dobrev & Andreas, 1997; Okada et al., 1998). The role of R- and O-type Ca2+ channels in synaptic transmissions is still unclear, because selective ligands of these channels have not yet been discovered (Olivera et al., 1994; Randall & Tsien, 1995).
Agents that selectively modulate VACCs are valuable tools in basic research. For example, low concentrations (⩽1 μM) of dihydropyridines (DHPs), inhibit Ca2+ currents in a frequency- and voltage-dependent manner due to selective blockade of L-type VACCs. However, higher concentrations of these drugs (⩾30 μM) are no longer selective, but also block other ion channels as for instance Na+ channels (Yatani & Brown, 1985). Together with the peptide neurotoxins that block other specific classes of VACCs (Olivera et al., 1994), the DHPs have been used to delineate the contributions of calcium channel subtypes to specific biological functions; however the Ca2+ channel blockers of phenylalkylamine and benzothiazepine structure have not been sufficiently investigated.
Although phenylalkylamines and benzothiazepines in low concentrations are also considered to be selective for L-type Ca2+ channels, there is now evidence that they may also affect non-L-type VACCs at higher concentrations (for review see McDonald et al., 1994). Recently, Ishibashi et al. (1995) demonstrated that micromolar concentrations of verapamil and diltiazem block P-type Ca2+ currents in freshly dissociated cerebellar Purkinje neurons and Diochot et al. (1995) showed that these drugs also have inhibitory effects on other types of neuronal non-L-type Ca2+ currents (see also Cai et al., 1997). Little data is currently available concerning the effects of verapamil and diltiazem on dihydropyridine-resistant Ca2+ channels involved in striatal dopamine transmission. There is evidence from one recent study that verapamil strongly inhibits K+-evoked [3H]-DA release from striatal synaptosomes in a concentration-dependent manner (Prince et al., 1996). Since K+-evoked [3H]-DA release is known to be predominantly mediated by N- and P-type but not by L-type Ca2+ channels, and also to be tetrodotoxin-resistant (Turner et al., 1993; Prince et al., 1996), its block by verapamil and diltiazem would suggest involvement of non-L-type Ca2+ channels.
Here, we investigated the putative inhibitory effects of verapamil and diltiazem on neuronal non-L-type VACCs. Since direct electrophysiological investigation of the Ca2+ currents of most nerve terminals in central neurons is generally limited by their small size (see Dunlap et al., 1995), we chose to study the effects of verapamil and diltiazem on K+-evoked [3H]-DA release from rat striatal slices in order to establish the contribution of N- and P-type Ca2+ channels to [3H]-DA release. In addition, we also investigated the drugs' effects on veratridine-evoked [3H]-DA release because this release mechanism involves predominantly Q-type VACCs (Dobrev et al., 1998). Thus, the involvement of N-, P- and Q-type channels was identified pharmacologically by sensitivity of evoked [3H]-DA release to ω-CTx-GVIA, ω-Aga-IVA and ω-CTx-MVIIC, respectively (see Dobrev & Andreas, 1997; Dobrev et al., 1998). Some of the results reported here have appeared previously in abstract form (Dobrev & Andreas, 1998).
Methods
All studies complied with the German home office regulations governing the care and use of laboratory animals.
[3H]-DA loading of rat striatal slices
The release experiments were performed as previously described (Dobrev & Andreas, 1997; Dobrev et al., 1998). In brief, the rats were anaesthetized with CO2, and after decapitation the brain was immersed in chilled (4°C) with atmospheric oxygen equilibrated Krebs-phosphate buffer (KPB) of the following composition (in mM): NaCl 118, KCl 4.7, CaCl2 1.8, MgSO4 7H2O 1.2, Na2HPO42H2O 15.9, ascorbic acid 0.6 and glucose 5.6 (pH 7.4). Subsequently 200-μm-thick coronal slices of the striatum were prepared and pre-incubated for 20 min at room temperature (22–24°C). The slices were then incubated with 0.1 μM [3H]-DA for additional 30 min at 37°C. After loading and washing of the slices, 10 μM nomifensine was applied to KPB to block [3H]-DA re-uptake, 10 μM pargyline to prevent its metabolism and 1.3 mM Na2-EDTA to prevent auto-oxidation of [3H]-DA. The slices (∼7 mg of tissue wet weight) were then transferred into a superfusion chamber (37°C) and were superfused with KPB using a peristaltic pump (flow rat 0.6 ml min−1, Desaga PLG-Peristaltic pump, Heidelberg, Germany).
Release experiments
Base-line release became stable within 20 min of wash-out, 30 or 40 successive 1 min fractions were collected. The release of [3H]-DA from the slices was stimulated either once with 25 μM veratridine (S1) or twice at fraction 7 (S1) and 27 (S2) (i.e. 27 and 47 min after the onset of superfusion) with 50 mM KCl (NaCl was replaced by KCl to maintain an isosmotic condition, see inset in Figure 1). Unless otherwise stated, the test drugs were applied to the medium 12 min before S2 in the experiments with K+ stimulation, and 6 min before starting fraction collection in those with veratridine stimulation. At the end of the experiment, the slices were sonicated and after adding of scintillation cocktail to the probes, total radioactivity (3H) in the collected fractions and in 500 μl aliquots of the homogenized slice preparation was determined by liquid scintillation-spectrometry (LKB 1219 Rackbeta scintillation counter).
Figure 1.

Effects of tetrodotoxin, Ca2+ and ω-CTx-MVIIC on K+-evoked [3H]-DA release. After 20 min washout period, 1 min fractions of superfusate were collected, and [3H]-DA release was evoked twice (S1, S2, see inset) for 2 min with 50 mM KCl containing buffer. Tetrodotoxin was added to the superfusion medium or CaCl2 was replaced by equimolar MgCl2, 12 min before S2, ω-CTx-MVIIC was applied 10 min before S2. Values are means±s.e.mean (bars) of the S2/S1 ratios in per cent of control, n indicates number of experiments. Statistical analysis was performed on untransformed data (S2/S1) by one-way ANOVA and LSD multiple comparison test. *P<0.05 compared with the controls.
Data presentation and statistics
The stimulation-evoked [3H]-DA release was calculated from the total release during the 2 min stimulation period and the subsequent 9 min (in the case of KCl) or 15 min (in the case of veratridine), corrected for basal release during this time. The evoked release during S1 and S2 was then expressed as per cent of the total radioactivity present in the slices just before the respective stimulation (S1 and S2%). To quantify drug effects on [3H]-DA release evoked by K+ stimulation, the ratio S2/S1 was calculated and expressed as per cent of that in the control. When stimulation was performed with veratridine, S1 values obtained with the test drugs were expressed as per cent of those in the control group. If drug effects were expressed in per cent of the mean S2/S1 ratio or S1 values of the corresponding control, the law of error propagation was considered for calculating the final s.e. mean values.
Data are presented as means±s.e.mean of the S2/S1 ratios or S1 values for the indicated number of experiments. In experiments on inhibition of release by various drugs, all data were statistically analysed as S2/S1 ratios or S1 values before transformation of data into percentage of control release.
Statistical significance was evaluated by one-way analysis of variance (ANOVA), followed by LSD multiple comparisons test when P<0.05 by using the computer program SPSS for Windows (SPSS software GmbH, version 6.0.1, München, Germany).
Drugs
[3H]-Dopamine (3,4-[7,8-3H]-dihydroxyphenylethylamine; specific activity 21.5 Ci mmol−1) was from American Radiolabeled Chemicals Inc., St. Louis, MO, U.S.A. Diltiazem, nifedipine, nitrendipine, nicardipine, (±)-Bay K 8644, nomifensine maleate, pargyline hydrochloride were from RBI, Natick, MA, U.S.A. L-ascorbic acid, (±)-verapamil, veratridine, tetrodotoxin, ω-conotoxin-GVIA, ω-agatoxin-IVA, and ω-conotoxin-MVIIC were from Sigma, Deisenhofen, Germany. Nominally Ca2+ free solution was prepared by replacing CaCl2 with equimolar MgCl2 (1.8 mM).
Results
Pharmacological classification of VACCs involved in K+-evoked [3H]-DA release
Under the conditions of the present study, stimulation of the slices with 50 mM K+ for 2 min (S1) caused a [3H]-DA release of 38.3 ± 1.8% (n=24) of total radioactivity in excess of basal release. When two 2 min pulses of KCl (50 mM) were applied 20 min apart, the S2/S1 ratio in the controls averaged 0.82±0.02 (n=24) (see inset in Figure 1). The nominal absence of Ca2+ with Ca2+ replaced by equimolar MgCl2 completely abolished K+-evoked [3H]-DA release. In contrast, the Na+ channel blocker TTX (1 μM) did not affect K+-evoked [3H]-DA release (Figure 1).
Moreover, the non-selective N-, P/Q-type Ca2+ channel blocker ω-CTx-MVIIC (3 μM), reduced K+-evoked [3H]-DA release to 12% of control, this value being similar to the inhibition of release to 7% of control in the absence of external Ca2+ (Figure 1, difference not statistically different). Finally, K+ evoked [3H]-DA release was not sensitive either to nifedipine (10–30 μM, Figure 2), nitrendipine (30 μM), nicardipine (30 μM) or to the activator of L-type VACCs (±)-Bay K 8644 (1 μM) (Table 1).
Figure 2.

Concentration-response curves for the inhibition produced by nifedipine, verapamil and diltiazem on K+-evoked [3H]-DA release. The drugs were added 12 min before S2 and were kept at the indicated final concentrations until the end of the experiments. The maximum inhibition was to 8.9±1.3% and 49.4±7.7% of control, for 100 μM verapamil and 100 μM diltiazem, respectively. Each point represents the mean S2/S1±s.e.mean for the given concentration from 4–7 independent experiments, with error bars when bigger than symbols. The control S2/S1 ratios were 0.79±0.02 (n=8), 0.79±0.04 (n=7) and 0.81±0.03 (n=6) for nifedipine, verapamil and diltiazem, respectively, and are defined as 100%. Statistical analysis was performed on untransformed data (S2/S1) by one-way ANOVA and LSD multiple comparison test. *P<0.05 compared with the respective controls.
Table 1.
Effects of L-type Ca2+ channel blockers on K+-evoked [3H]-DA release from rat striatal slices
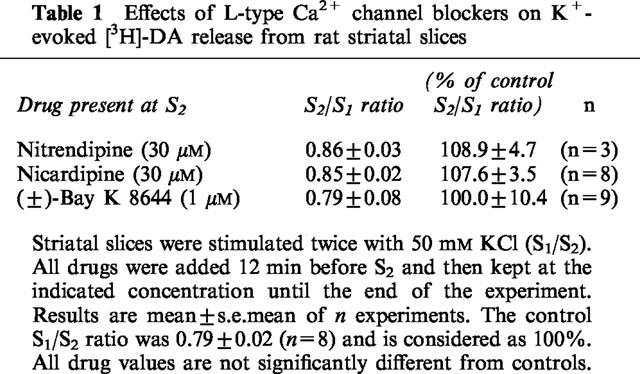
In contrast to the lack of effects to the selective L-type VACC blockers of the DHP group (±)-verapamil and diltiazem inhibited K+-evoked [3H]-DA release in a concentration-dependent manner (Figure 2), with profound quantitative differences between verapamil and diltiazem at 100 μM (see below).
Verapamil- and diltiazem-induced inhibition of multiple classes of VACCs
In order to investigate whether (±)-verapamil and diltiazem were acting on N- or P-type VACCs in our preparation, we tested the effects of co-application of selective neurotoxins and (±)-verapamil or diltiazem. The selective N-type Ca2+ channel blocker ω-CTx-GVIA (1 μM) inhibited [3H]-DA release to 78.5±11.6% (n=4) and the selective P-type Ca2+ channel blocker ω-Aga-IVA (30 nM) to 40.5±3.9% (n=4) of control, indicating involvement of both channel subtypes (Figure 3). Applicating of 30 μM (±)-verapamil alone produced an inhibition of K+-evoked [3H]-DA release to 55.7±6.5% (n=7) of control (compare Figure 2), and simultaneous application of 30 μM (±)-verapamil and 1 μM ω-CTx-GVIA decreased the release to 35.4±1.6% (n=4) of control (Figure 3). In contrast, 30 μM (±)-verapamil could not enhance inhibition of release caused by P-type VACC blocker ω-Aga-IVA (30 nM) alone (Figure 3). The inhibitory effects of 30 μM diltiazem and 1 μM ω-CTx-GVIA were also fully additive, whereas the inhibition produced by co-application with 30 nM ω-Aga-IVA was the same as with ω-Aga-IVA alone (Figure 3).
Figure 3.

Inhibitory effects of ω-CTx-GVIA alone and in combination with verapamil or diltiazem and respective effects of ω-Aga-IVA on K+-evoked [3H]-DA release. Verapamil (or diltiazem) and ω-CTx-GVIA were simultaneously applied 12 min before S2, whereas verapamil (or diltiazem) and ω-Aga-IVA were applied sequentially. In these experiments verapamil or diltiazem were added 12 min before S2, 6 min later the superfusion buffer was switched to medium containing both verapamil or diltiazem and ω-Aga-IVA. All drugs were then present until the end of the experiments. The control S2/S1 ratio was 0.82±0.02 (n=29), and is considered as 100%. Each column represents the mean±s.e.mean (bars) values from n independent experiments. Statistical analysis was performed on untransformed data (S2/S1) by one-way ANOVA and LSD multiple comparison test. All drug effects are significantly different from controls. *P<0.05 compared with ω-CTx-GVIA alone.
Effects of verapamil and diltiazem on veratridine-evoked [3H]-DA release
From the almost complete inhibition of K+-evoked [3H]-DA release by 100 μM (±)-verapamil (see Figure 2) we hypothesized that Q-type Ca2+ channels might be involved as well. This was tested by employing veratridine-evoked [3H]-DA release, of which we have shown prevously that Ca2+-dependent release is mediated solely by Q-type VACCs (Dobrev et al., 1998).
Like DHPs at the concentration of 30 μM, diltiazem did not inhibit veratridine-evoked [3H]-DA release (Table 2), whereas 30 μM (±)-verapamil significantly inhibited veratridine-evoked [3H]-DA release. With 100 μM diltiazem applied the release was inhibited and with 100 μM (±)-verapamil it was blocked further (Table 2). In order to determine whether the inhibition of release by 30 μM (±)-verapamil was exclusively mediated through blocking of Q-type VACCs, we investigated the effect of this drug on the component of veratridine-evoked [3H]-DA release which was independent of external Ca2+. (±)-Verapamil (30 μM) partially inhibited the veratridine-evoked [3H]-DA release in the absence of external Ca2+ (Table 2).
Table 2.
Effects of L-type Ca2+ channel blockers on veratridine-evoked [3H]-DA release from rat striatal slices

Discussion
We provide evidence that in rat striatal slices verapamil inhibits P- and at higher concentrations possibly also N- and Q-type VACCs, whereas comparable concentrations of diltiazem block P-type Ca2+ channels only. In our experimental models of K+- or veratridine-evoked [3H]-DA release, L-type VACCs were not involved, since DHPs (up to 30 μM) were without effect. The latter conclusion is in agreement with a large body of evidence demonstrating that neurotransmitter release in central neurons is insensitive to modulations of L-type VACCs (Miller, 1987; Takahashi & Momiyama, 1993). Further evidence against the involvement of L-type channels comes from the lack of stimulatory effect of Bay K 8644, as this activator of L-type VACCs would have to enhance DA release if these channel subtypes played a significant role in the release process (Herdon & Nahorski, 1989).
In contrast to dihydropyridines, the L-type Ca2+ channel blockers (±)-verapamil (1–100 μM) and diltiazem (10–100 μM) led to a concentration-dependent inhibition of K+-evoked [3H]-DA release (Figure 2). Similar inhibitory effects of verapamil and diltiazem have been demonstrated in other neurotransmitter release systems such as K+-evoked release of endogenous glutamate from cerebellar slices (Barnes & Davies, 1988), of [3H]-D-aspartate from hippocampal slices (Mangano et al., 1991), and of [3H]-GABA from cortical synaptosomes (Carvalho et al., 1986) as well as in striatal dopamine release measured by microdialysis in freely moving rats (Kato et al., 1992). Since we have demonstrated that block of N-, P- and possibly Q-type VACCs entirely abolishes K+-evoked [3H]-DA release, it is difficult to envision a scheme that could also accommodate inhibition of release by (±)-verapamil and diltiazem via block of L-type VACCs. The finding that DHPs were ineffective but (±)-verapamil and diltiazem inhibited K+-evoked [3H]-DA release suggests the involvement of presynaptic non-L-type Ca2+ channels or other ion channels as for instance Na+ channels. Although (±)-verapamil and diltiazem selectively block L-type VACCs in concentrations up to 1 μM, they may have additional effects on neuronal Na+ channels at higher concentrations (Bustamante, 1985; Yatani & Brown, 1985; Yokoo et al., 1998). However, the finding that the K+-evoked [3H]-DA release was not modified by tetrodotoxin in the first place, excludes involvement of Na+ channel block by (±)-verapamil and diltiazem in this release system. Therefore, (±)-verapamil and diltiazem must block non-L-type VACCs. In other neuronal systems, both verapamil and diltiazem block P-type Ca2+ currents (rat cerebellar Purkinje neurons, Ishibashi et al., 1995) or inhibit N- and P/Q-type Ca2+ currents (mouse sensory neurons, rat motoneurons: Diochot et al., 1995). Even recombinant Ca2+ channels of P/Q- and R-type were inhibited by verapamil and diltiazem (Cai et al., 1997) and many of those effects occurred in the same concentration range as reported here for inhibition of dopamine release. Therefore, it is tempting to explain our findings by interactions of (±)-verapamil and diltiazem with striatal N-, P- or Q-type Ca2+ channels.
These possibilities were tested by employing ω-CTx-GVIA at 1 μM (saturating concentration for N-type VACCs, see Olivera et al., 1994) and ω-Aga-IVA at 30 nM (saturating concentration for P-type VACCs, see Mintz et al., 1992). Each of the two peptides inhibited a fraction of K+-evoked [3H]-DA release which was completely blocked by the non-selective N-, P-, Q-type VACC blocker ω-CTx-MVIIC (3 μM), suggesting also involvement of Q-type VACCs.
The inhibiting effects of 1 μM ω-CTx-GVIA and of 30 μM (±)-verapamil on K+-evoked [3H]-DA release were additive, indicating that both compounds acted at different channel subtypes. Since no additional (±)-verapamil-induced inhibition of release was observed on top of 30 nM ω-Aga-IVA, we conclude that (±)-verapamil acts on P-type Ca2+ channels in striatal neurons. Similar conclusions hold true for diltiazem. Interaction of diltiazem with P-type Ca2+ channels is indirectly supported by the lack of effect of the drug in a neurotransmitter release model which does not involve any P-type VACCs in the first place, i.e. the veratridine-evoked [3H]-DA release (Dobrev et al., 1998).
Increasing the (±)-verapamil concentration to 100 μM completely abolished the K+-evoked [3H]-DA release, indicating that at this high concentration the drug may also affect N- and Q-type VACCs in addition to P-type. Although we did not test the additive effects of 100 μM diltiazem and 30 nM ω-Aga-IVA, 100 μM diltiazem inhibited K+-evoked [3H]-DA release to the same degree as ω-Aga-IVA (compare Figures 2 and 3), suggesting no significant interaction of diltiazem with N- or Q-type VACCs.
Veratridine-evoked [3H]-DA release was used as an additional probe for involvement of Q-type VACCs (Dobrev et al., 1998). In the present study, the application of 30 μM diltiazem had no effect on veratridine-evoked [3H]-DA release, excluding interaction with Q-type VACCs at this concentration. In contrast, 30 μM (±)-verapamil and very high concentrations of either (±)-verapamil or diltiazem (100 μM) inhibited veratridine-evoked [3H]-DA release, suggesting possible interaction with Q-type VACCs (see Table 2). In order to test whether low concentrations of (±)-verapamil (30 μM) interact with Q-type VACCs, veratridine-evoked [3H]-DA release in the absence of external Ca2+ was measured. (±)-Verapamil (30 μM) reduced the release even in the absence of external Ca2+ and this inhibitory effect of (±)-verapamil could be explained by an antagonistic effect on VACCs only, if we assume that in the absence of external Ca2+, Na+ enters into the neurons through VACCs. However, we have recently demonstrated that the Ca2+-independent component of veratridine-evoked [3H]-DA release is abolished by tetrodotoxin (Dobrev et al., 1998). Moreover, the (±)-verapamil- and diltiazem-induced inhibition of release at 100 μM was much greater both than that which could be obtained with ω-CTx-MVIIC (∼35%, see Dobrev et al., 1998) and than the Ca2+-dependent component of release (Table 2). Inhibitory effects of L-type channel blockers on Na+ channels when used at high concentrations were reported in various cell types (Callanan & Keenan, 1984; Yatani & Brown, 1985; Yokoo et al., 1998; for review see McDonald et al., 1994). Thus, although we cannot exclude an interaction of (±)-verapamil and diltiazem with Q-type VACCs, it is reasonable to suggest that in the case of veratridine-evoked [3H]-DA release they inhibit the release via block of Na+ channels.
Finally, because of its strong lipophilicity, (±)-verapamil–especially at high concentrations–could impair channel function indirectly by perturbation of membrane fluidity. At this time, we cannot exclude such an effect of (±)-verapamil from our results. If non-specific membrane perturbation was the operative mechanism, verapamil would also be expected to inhibit the release of other neurotransmitters. However, the verapamil effects reported in other systems were highly variable including inhibition (Barnes & Davies, 1988; Mangano et al., 1991; Prince et al., 1996), enhancement (Ebstein & Daly, 1982) or even no effect at all (Bentue-Ferrer et al., 1993). In addition, verapamil did not inhibit ionomycin-evoked [3H]-D-aspartate release from hippocampal slices (Mangano et al., 1991).
In summary, we have demonstrated that at low μM concentrations (⩽30 μM), (±)-verapamil and diltiazem block P-type VACCs in rat striatum. The inhibition of P-type Ca2+ channels by these drugs occurs at concentrations which overlap with those required for complete block of cardiac L-type Ca2+ channels (Diochot et al., 1995). At concentrations >30 μM, (±)-verapamil, but not diltiazem, also appears to interact with N- and Q-type Ca2+ channels. The significance of these findings awaits further study, but has practical consequences, since P-type VACCs are found in many brain regions besides the striatum and in peripheral neurons. Therefore, (±)-verapamil and diltiazem are expected to modify the function of those neuronal cells through interaction with P-type VACCs. On the other hand, at higher concentrations both drugs also appear to have inhibitory effects on Na+ channels, so that lack of specificity of these drugs should be taken into account when probing for presence of L-type channels in central neurons. Finally, these effects may contribute to their therapeutic efficacy.
Acknowledgments
The authors wish to thank Ms Martina Rossmann for her excellent technical assistance in a part of the neurotransmitter release studies.
Abbreviations
- ω-Aga-IVA
ω-agatoxin IVA
- ANOVA
analysis of variance
- DA
dopamine
- ω-CTx-GVIA
ω-conotoxin GVIA
- ω-CTx-MVIIC
ω-conotoxin MVIIC
- KPB
Krebs-phosphate buffer
- LSD-test
least significant difference test
- VACCs
voltage-activated calcium channels
References
- BARNES S., DAVIES J.A. The effects of calcium channel agonists and antagonists on the release of endogenous glutamate from cerebellar slices. Neurosci. Lett. 1988;92:58–63. doi: 10.1016/0304-3940(88)90742-2. [DOI] [PubMed] [Google Scholar]
- BENTUE-FERRER D., DECOMBE R., SAIAG B., ALLAIN H., VAN DEN DRISSCHE J. L-type voltage-dependent calcium channels do not modulate aminergic neurotransmitter release induced by transient global cerebral ischemia: an in vivo microdialysis study in rat. Exp. Brain Res. 1993;93:288–292. doi: 10.1007/BF00228396. [DOI] [PubMed] [Google Scholar]
- BUSTAMANTE J.O. Block of Na+-currents by the Ca2+-antagonist D-600 in human heart cell segments. Pflügers Arch. Eur. J. Physiol. 1985;403:225–227. doi: 10.1007/BF00584106. [DOI] [PubMed] [Google Scholar]
- CAI D., MULLE J.G., YUE D.T. Inhibition of recombinant Ca2+ channels by benzothiazepines and phenylalkylamines: class-specific pharmacology and underlying molecular determinants. Mol. Pharmacol. 1997;51:872–881. [PubMed] [Google Scholar]
- CALLANAN K.M., KEENAN A.K. Differential effects of D600, nifedipine and dantrolene sodium on excitation-secretion coupling and presynaptic β-adrenoceptor responses in rat atria. Br. J. Pharmacol. 1984;83:1440–1447. doi: 10.1111/j.1476-5381.1984.tb16240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARVALHO C.M., SANTOS S.V., CARVALHO A.P. γ-Aminobutyric acid release from synaptosomes as influenced by Ca2+ and Ca2+ channel blockers. Eur. J. Pharmacol. 1986;131:1–12. doi: 10.1016/0014-2999(86)90509-1. [DOI] [PubMed] [Google Scholar]
- DIOCHOT S., RICHARD S., BALDY-MOULINIER M., NARGEOT J., VALMIER J. Dihydropyridines, phenylalkylamines and benzothiazepines block N-, P/Q- and R-type calcium currents. Pflügers Arch. Eur. J. Physiol. 1995;431:10–19. doi: 10.1007/BF00374372. [DOI] [PubMed] [Google Scholar]
- DOBREV D., ANDREAS K. Modulation of potassium-evoked [3H]dopamine release from rat striatal slices by voltage-activated calcium channel ligands: effects of ω-conotoxin-MVIIC. Neurochem. Res. 1997;22:1085–1093. doi: 10.1023/a:1027305016440. [DOI] [PubMed] [Google Scholar]
- DOBREV D., ANDREAS K. Verapamil and diltiazem affect N- and P-type Ca2+ channels coupled to K+-evoked [3H]dopamine release in rat striatal slices. Naunyn Schmiedeberg's Arch. Pharmacol. 1998;357 suppl. 4:R73. [Google Scholar]
- DOBREV D., MILDE A.S., ANDREAS K., RAVENS U. Voltage-activated Ca2+ channels involved in veratridine-evoked [3H]dopamine release from rat striatal slices. Neuropharmacology. 1998;37:973–982. doi: 10.1016/s0028-3908(98)00103-8. [DOI] [PubMed] [Google Scholar]
- DUNLAP K., LUEBKE J.I., TURNER T.J. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- EBSTEIN R.P., DALY J.W. Release of norepinephrine and dopamine from brain vesicular preparations: effects of calcium antagonists. Cell. Mol. Neurobiol. 1982;2:205–213. doi: 10.1007/BF00711148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARVEY J., WEDLEY S., FINDLAY J.D., SIDELL M.R., PULLAR I.A. ω-Agatoxin identifies a single calcium channel subtype which contributes to the potassium-induced release of acetylcholine, 5-hydroxytryptamine, dopamine, γ-aminobutyric acid and glutamate from rat brain slices. Neuropharmacology. 1996;35:385–392. doi: 10.1016/0028-3908(96)00010-x. [DOI] [PubMed] [Google Scholar]
- HERDON H., NAHORSKI S.R. Investigations of the roles of dihydropyridine and ω-conotoxin-sensitive calcium channels in mediating depolarization-evoked endogenous dopamine release from striatal slices. Naunyn Schmiedeberg's Arch. Pharmacol. 1989;340:36–40. doi: 10.1007/BF00169204. [DOI] [PubMed] [Google Scholar]
- ISHIBASHI H., YATANI A., AKAIKE N. Block of P-type Ca2+ channels in freshly dissociated rat cerebellar Purkinje neurons by diltiazem and verapamil. Brain Res. 1995;695:88–91. doi: 10.1016/0006-8993(95)00815-8. [DOI] [PubMed] [Google Scholar]
- KATO T., OTSU Y., FURUNE Y, YAMAMOTO T. Different effects of L-, N- and T-type channel blockers on striatal dopamine release measured by microdialysis in freely moving rats. Neurochem. Int. 1992;21:99–107. doi: 10.1016/0197-0186(92)90072-y. [DOI] [PubMed] [Google Scholar]
- KIMURA M., YAMANISHI Y., HANADA T., KAGAYA T., KUWADA M., WATANABE T., KATAYAMA K, NISHIZAWA Y. Involvement of P-type calcium channels in high potassium-elicited release of neurotransmitters from rat brain slices. Neuroscience. 1995;66:609–615. doi: 10.1016/0306-4522(95)00023-c. [DOI] [PubMed] [Google Scholar]
- MANGANO T.J., PATEL J., SALAMA A.I., KEITH R.A. Inhibition of K+-evoked [3H]D-aspartate release and neuronal calcium influx by verapamil, diltiazem and dextromethorphan: evidence for non-L/non-N voltage-sensitive calcium channels. Eur. J. Pharmacol. 1991;192:9–17. doi: 10.1016/0014-2999(91)90062-u. [DOI] [PubMed] [Google Scholar]
- MCDONALD T.F., PELZER S., TRAUTWEIN W., PELZER D.J. Regulation and modulation of calcium channels in cardiac skeletal and smooth muscle cells. Physiol. Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MILLER R.J. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- MINTZ I.M., VENEMA V.J., SWIDEREK K.M., LEE T.D., BEAN B.P., ADAMS M.E. P-type calcium channels blocked by the spider toxin ω-aga-IVA. Nature. 1992;335:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- OKADA M., WADA K., KIRYU K., KAWATA Y., MIZUNO K., KONDO T., TASAKI H., KANEKO S. Effects of Ca2+ channel antagonists on striatal dopamine and DOPA release, studied by in vivo microdialysis. Br. J. Pharmacol. 1998;123:805–814. doi: 10.1038/sj.bjp.0701675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLIVERA B.M., MILJANICH G.P., RAMACHANDRAN J., ADAMS M.E. Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. A. Rev. Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- PRINCE R.J., FERNANDES K.G., GREGORY J.C., MARTYN I.D., LIPPIELLO P.M. Modulation of nicotine-evoked [3H]dopamine release from rat striatal synaptosomes by voltage-sensitive calcium channel ligands. Biochem. Pharmacol. 1996;52:613–618. doi: 10.1016/0006-2952(96)00313-9. [DOI] [PubMed] [Google Scholar]
- RANDALL A., TSIEN R.W. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J. Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOLIAKOV L., WONNACOTT S. Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat striatal synaptosomes. J. Neurochem. 1996;67:163–170. doi: 10.1046/j.1471-4159.1996.67010163.x. [DOI] [PubMed] [Google Scholar]
- TAKAHASHI T., MOMIYAMA A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- TSIEN R.W., LIPSCOMBE D., MADISON D.V., BLEY K.R., FOX A.P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- TURNER T.J., ADAMS M.E., DUNLAP K. Multiple Ca2+ channel types coexist to regulate synaptosomal neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9518–9522. doi: 10.1073/pnas.90.20.9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VARADI G., MORI Y., MIKALA G., SCHWARTZ A. Molecular determinants of Ca2+ channel function and drug action. Trends Pharmacol. Sci. 1995;16:43–49. doi: 10.1016/s0165-6147(00)88977-4. [DOI] [PubMed] [Google Scholar]
- WHEELER D.B., RANDALL A., TSIEN R.W. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- YATANI A., BROWN A.M. The Ca2+ channel blocker nitrendipine blocks sodium channels in neonatal rat cardiac myocytes. Circulation Res. 1985;56:868–875. doi: 10.1161/01.res.56.6.868. [DOI] [PubMed] [Google Scholar]
- YOKOO H, , SHIRAISHI S., KOBAYASHI H., YANAGITA T., YAMAMOTO R., WADA A. Selective inhibition by riluzole of voltage-dependent sodium channels and catecholamine secretion in adrenal chromaffin cells. Naunyn Schmiedeberg's Arch. Pharmacol. 1998;357:526–531. doi: 10.1007/pl00005203. [DOI] [PubMed] [Google Scholar]