

Abstract
The aims of this study were to compare, in the rat isolated perfused lung preparation, the antagonist effects of a nonselective β-adrenoceptor agonist (isoprenaline), a selective β2-adrenoceptor agonist (salbutamol) and a selective β3-adrenoceptor agonist (SR 59104A) on the hypoxic pulmonary pressure response, and to investigate the role of K+ channels, endothelium derived relaxing factor and prostaglandins in these effects. K+ channels were inhibited by glibenclamide, charybdotoxin or apamin, NO synthase and cyclo-oxygenase were inhibited by NG-nitro-L-arginine methyl ester (L-NAME) and indomethacin, respectively.
Hypoxic ventilation produced a significant increase in perfusion pressure (+65%, P<0.001) and L-NAME significantly increased this response further (+123%, P<0.01). After apamin, L-NAME, indomethacin, post-hypoxic basal pressure did not return to baseline values (P<0.001).
Glibenclamide partially inhibited the relaxant effects of isoprenaline (P<0.05) and salbutamol (P<0.001) but not that of SR 59104A. In contrast, charybdotoxin and apamin partially inhibited the relaxant effects of SR 59104A (P=0.053 and <0.01, respectively) but did not modify the effects of isoprenaline and salbutamol. L-NAME partially inhibited the dilator response of salbutamol (P<0.01) and SR 59104A (P<0.05) but not that of isoprenaline.
We conclude that (a) EDRF exerts a significant inhibition of the hypoxic pulmonary response, (b) SKCa channel activation, EDRF and prostaglandins contribute to the reversal of the hypoxic pressure response, (c) the vasodilation induced by isoprenaline is mediated in part by activation of KATP channels, that of salbutamol by activation of KATP channels and EDRF. In contrast, SR 59104A partly operates through BKCa, SKCa channels and EDRF activation, differing in this from the β1 and β2-adrenoceptor agonists.
Keywords: Hypoxic pulmonary vasoconstriction, isoprenaline, salbutamol, SR 59104A, nitric oxide/endothelium derived relaxing factor, ATP-sensitive K+ channels, Ca2+-activated K+ channels
Introduction
Pulmonary hypertension may be secondary to hypoxic vasoconstriction resulting in complex changes in the pulmonary vascular bed. Despite extensive research, the mechanism underlying hypoxic pulmonary vasoconstriction is yet unknown. Suppression of endogenous vasodilator substances such as endothelium derived relaxing factor (EDRF/NO) may be one mechanism mediating the hypoxic vasoconstriction (Liu et al., 1991). The ability of potassium channels to modulate hypoxic vasoconstriction is suspected because of the excitatory effects of K+ channel blockers such as tetraethylammonium or 4-aminopyridine (Hasunuma et al., 1991a; Dumas et al., 1994; Reeve et al., 1997).
Currently, available therapies remain limited to correction of hypoxaemia and generalized non-specific pulmonary vasodilation (Leach & Treacher, 1995). The recent development of inhaled therapy (NO, prostacyclin and analogues) represents a significant advance in the management of hypoxic pulmonary vasoconstriction (Olschewski et al., 1996; Manktelow et al., 1997). However the therapeutic effects of this approach are limited by the short half-life of these compounds. A new pharmacological approach consists in prolonging the effects of prostacyclin by the addition of subtherapeutic doses of another aerosolized dilator agent limiting thereby the intrapulmonary shunt and systemic side effects (Ghofrani et al., 1998). In this context, the development of selective compounds may be of interest. In the adrenergic system, there are indications that atypical β-adrenoceptors, which are sensitive to preferential pharmacological agonists that have little effects on β1- and β2- adrenoceptors (e.g., BRL 37344 and SR 58611), but relatively insensitive to the conventional β-adrenoceptor antagonists are present in the vascular smooth muscle (Berlan et al., 1994; Oriowo, 1994; 1995). In a recent study the relaxant effects of three β3- adrenoceptor agonists (SR 58611A, SR 59104A and SR 59119A) have been investigated in the pulmonary circulation (Dumas et al., 1998).
β-adrenoceptor agonists are usually regarded as agents mediating their effects through specific receptors coupled to adenylate cyclase, inducing smooth muscle relaxation by increasing the intracellular concentration of adenosine 3′, 5′-cyclic monophosphate (cyclic AMP) (Bylund et al., 1994; Torphy, 1994). Recent evidence however suggests that β-adrenoceptor agonists can activate multiple signalling pathways. It has been shown that ATP-sensitive K+ (KATP) channels (Randall & McCulloch, 1995; Nakashima & Vanhoutte, 1995; Chang, 1997) or Ca2+-activated K+ channels (Satake et al., 1996; Huang & Kwok, 1997) or EDRF (Wang et al., 1993; Hamdad et al., 1996) are involved in the non selective- or the β2-selective adrenoceptor agonists-induced relaxation of vascular smooth muscle. To our knowledge, these various mechanisms of action have not yet been investigated in the pulmonary circulation.
The purpose of this study was to investigate (1) the contribution of ATP sensitive K+ channels, large conductance Ca2+-activated K+ (BKCa) channels, small conductance Ca2+-activated K+ (SKCa) channels, EDRF, and prostaglandins to hypoxic vasoconstriction in rat lungs perfused with physiological solutions, and (2) the role of these mechanisms in the vasodilating actions exerted by the nonselective β-adrenoceptor agonist (isoprenaline), a selective β2-adrenoceptor agonist (salbutamol) and a selective β3-adrenoceptor agonist (SR 59104A).
Methods
Rat perfused isolated lung preparation
Twenty-eight groups (n=4–9 per group) of male Wistar rats (Dépré, St Doulchard, France), weighing 280–340 g, were anaesthetized with sodium pentobarbitone (100 mg kg−1) and tracheotomized. After thoracotomy, a polyethylene cannula was inserted into the main pulmonary artery and the lungs were removed quickly and allowed to equilibrate in the perfusion circuit maintained at 38°C by a surrounding water bath and consisting of a perfusion reservoir, a roller pump (Harvard 77, Ealing, Les Ullis, France), connecting tubing and bubble trap. Mean perfusion pressure which was measured from a side-arm of the arterial line (Harvard transducer, −50 to 300 mmHg), was recorded continuously (Ankersmit WR 3701 recorder, Graphtec Corp., Japan) and reflected pulmonary vascular resistances because the flow rate was constant (0.025 ml g−1 min−1). The lungs were perfused with a salt solution containing (mM): NaCl 116, KCl 5.4, NaH2PO4 1.04, MgSO4 0.83, CaCl2 1.8, NaHCO3 19 and D glucose 5.5. Ficoll (1 g 100 ml−1, type 70, Sigma) was included as a colloïd (Hasunuma et al, 1991b). The lungs were ventilated with a Harvard rodent ventilator at a tidal volume of 10 ml kg−1 body weight and a frequency of 55 breaths min−1. The end expiratory pressure was set at 2.5 cm H2O. The pressure of airways was measured with a Validyne DP45 (0–88 cm H2O) differential pressure transducer. A 20–30 min equilibration period was allowed to establish a stable baseline for pulmonary airway and vascular pressures before experiments were started. During this period the lungs were ventilated with a humid mixture of 21% O2, 5% CO2, 74% N2 (normoxia). Lungs of which the weight had increased in excess of 10% (indicative of oedema) at the end of the experiments were discarded.
Experimental protocols
Vasoconstrictor responses to hypoxia
After the equilibration period, the pulmonary vasculature was precontracted twice, using a bolus of 0.25–0.5 μg angiotensin II to prime the otherwise low vascular reactivity seen in salt solution-perfused lungs. The lungs were then challenged with a hypoxic gas mixture (5% CO2, 95% N2) as described previously (Dumas et al., 1996). Each hypoxic challenge (4 min) was followed by the addition of 0.25 μg angiotensin II in normoxic ventilation (4 min) and the pressure was allowed to return to baseline before the initiation of hypoxic ventilation. The perfusate gas tensions were measured during hypoxic challenges, by collecting perfusate anaerobically from the arterial cannula and analysing it immediately (Corning 170 pH/blood gas analyzer). During hypoxic periods, PO2 was maintained below 35 mmHg and the pH was between 7.3 and 7.4. After three or four hypoxic pulmonary vasoconstrictions the responses became reproducible (Dumas et al., 1996). Drugs were tested after a stable response to hypoxia was reached.
Effects of isoprenaline, salbutamol and SR 59104A on pulmonary vasoreactivity: influence of K+ channels, nitric oxide synthase and cyclo-oxygenase inhibitors
Non-cumulative concentration-response curves to the three agonist compounds were obtained by perfusing lungs during six to seven hypoxic periods with a salt solution containing isoprenaline (0.001–1 μM), salbutamol (0.01–10 μM) or SR 59104A (0.3–100 μM). In another series of experiments, concentration-response curves were obtained in the presence of one of the following inhibitors: glibenclamide (1 μM), a KATP channel blocker. Charybdotoxin (0.1 μM), a BKCa channel blocker, apamin (0.05 μM), a SKCa channel blocker, NG-nitro-L-arginine methyl ester (L-NAME 100 μM), a nitric oxide synthase inhibitor, or indomethacin (10 μM), a cyclo-oxygenase inhibitor were administered during the fifth, sixth and seventh hypoxic periods whereas the agonists were used during the sixth and seventh periods (Dumas et al., 1997), at concentrations inducing about 75% inhibition of the pressure response to hypoxia (isoprenaline 0.03 μM, salbutamol 0.6 μM, SR 59104A 20 μM).
Chemicals/drugs
The drugs used were: SR 59104A (N- [6 hydroxy-1,2,3,4-tetrahydronaphtalen-(2R)-2yl) methyl]-(2R)-2hydroxy-2-(3-chlorophenyl) ethanamine hydrochloride (Sanofi-Midy, Research Centre, Milan, Italy), glibenclamide (Laboratoire Hoechst, Paris la Défense, France), isoprenaline, salbutamol, angiotensin II, NG-nitro-L-arginine methyl ester, indomethacin (Sigma Chimie, La Verpillère, France) and charybdotoxin and apamin (Latoxan, Rosans, France). Isoprenaline, salbutamol, SR 59104A, apamin, angiotensin II and NG-nitro-L-arginine methyl ester were dissolved in distilled water, charybdotoxin in saline, glibenclamide in a mixture of dimethylsulphoxide-distilled water (1 : 1) and indomethacin in a carbonate salt solution ( 4 :100). All the stock solutions were kept frozen until use and all the diluted solutions were prepared just before administration. The maximal concentrations of dimethylsulphoxide (2%) added to the bath did not by themselves exert any effect and did not modify the reactivity of the preparation.
Data analysis
Hypoxic pressure response was measured at the time of the peak increase and expressed as absolute changes from baseline values. For agonists, the one-half effective maximum concentration values expressed in the negative logarithm to base 10 form (pD2) were determined from individual concentration-response curves. For the experiments performed with an inhibitor, the level of hypoxic pressure response being different in the presence of inhibitors, the effects of the latter were calculated as:
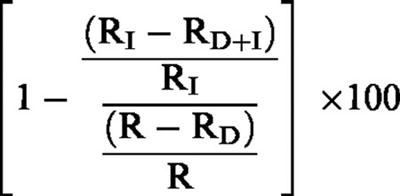 |
where (a) RD+I and RI are the hypoxic pressure responses observed with the inhibitor with and without the agonist, respectively, and (b) RD and R are the corresponding hypoxic pressure responses observed in control experiments with and without the agonist, respectively.
Data are shown as mean±s.e.mean. Statistical significance was assessed with the Mann-Whitney U-test for simple comparisons and the ANOVA-Bonferroni multiple t-test for multiple comparisons; P values <0.05 were considered significant.
Results
In lung preparations, the mean baseline inflation pressure was 10.32±0.09 cm H2O (n=192) and was not significantly modified by hypoxic ventilation or addition of the various drugs. After equilibration of the preparation, the baseline perfusion pressure in normoxic ventilation was similar in all series of rats (6.12±0.09 mmHg, n=192). Ventilation with a hypoxic mixture of gas produced a significant increase of the perfusion pressure (+3.95±0.05 mmHg, n=160, +65% from baseline values, P<0.001) which, starting from the fourth period of hypoxia, was reproducible for at least nine subsequent periods.
Effects of K+ channel, EDRF and cyclo-oxygenase inhibitors on pulmonary reactivity
Glibenclamide, charybdotoxin, apamin, L-NAME and indomethacin did not affect significantly the baseline values of perfusion pressure (+0.05±0.01, +0.08±0.17, −0.15±0.15, +0.05±0.13 and +0.12±0.24 mmHg respectively). As shown in Figure 1a, glibenclamide at 1 μM, charybdotoxin at 0.1 μM, apamin at 0.05 μM and indomethacin at 10 μM did not by themselves affect the hypoxic pressure response. In contrast the hypoxic pressure response was significantly increased by L-NAME (+9.17±1.60 versus 3.92±0.40 mmHg, P<0.01). As shown in Figure 1b, post-hypoxic basal pressure did not return to baseline values after apamin (+0.9±0.17 mmHg, P<0.001), L-NAME (+0.79±0.18 mmHg, P<0.001) and indomethacin (+0.69±0.34 mmHg, P<0.001).
Figure 1.
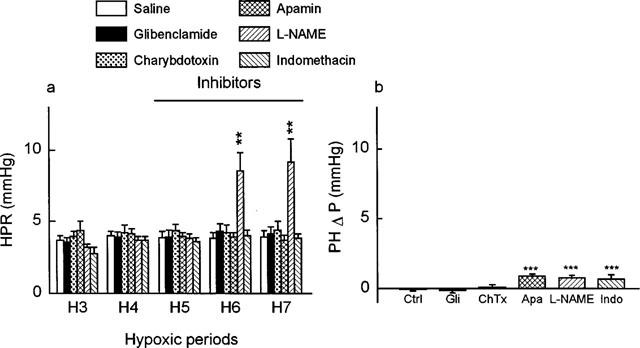
Influence of normal saline and inhibitors: glibenclamide (1 μM) (Gli), charybdotoxin (0.1 μM) (ChTx), apamin (0.05 μM) (Apa), L-NAME (100 μM) or indomethacin (10 μM) (Indo) on (a) hypoxic pressure response (n=7, 5, 5, 7 and 4, respectively), and (b) the post-hypoxic relaxation (n= 6, 7, 5, 5, 7 and 4, respectively) in the isolated rat perfused lung. Values shown are increases of perfusion pressure above basal values (a) from the third to the seventh hypoxic period with infusion of the inhibitors from the fifth to the seventh hypoxic period (hypoxic pressure response: HPR), or (b) after the seventh hypoxic period (post hypoxic Δ pressure: PHΔP). Data represent mean±s.e.mean. Asterisks indicate responses that were significantly different from corresponding responses in control experiments (**P<0.01, ***P<0.001).
Effects of isoprenaline, salbutamol and SR 59104A on pulmonary reactivity; influence of K+ channel, EDRF and cyclo-oxygenase inhibitors
As shown in Figure 2, isoprenaline, salbutamol and SR 59104A concentration-dependently decreased the hypoxic pressure response (P<0.001). Per cent inhibition of this response was 73% with isoprenaline 0.03 μM, 75% with salbutamol 0.6 μM and 75% with SR 59104A 20 μM. Table 1 shows that isoprenaline was significantly (P<0.001) more potent than salbutamol and SR 59104A at decreasing the hypoxic pressure response, the decreasing order of potency being: isoprenaline>salbutamol>SR 59104A. During the hypoxic pressure response, the KATP channel inhibitor, glibenclamide produced a rightward shift of the concentration-response curves to isoprenaline and salbutamol (P<0.05 and P<0.001 respectively, Figure 2a and b), resulting in a significantly lower pD2 value of salbutamol than in the control conditions (P<0.01, Table 1). Glibenclamide inhibited the response to 0.03 μM isoprenaline and 1 μM salbutamol by approximately 43 and 40% respectively. In contrast, the antagonist effects of SR 59104A against the hypoxic vasoconstriction remained unaffected by glibenclamide (Figure 2c and Table 1). The BKCa channel inhibitor, charybdotoxin, was devoid of any effect on the response to isoprenaline and salbutamol but reduced that to SR 59104A, there being in this respect a difference between the selective β2- and β3-adrenoceptor agonists (P<0.05, Figure 3a). The antagonist effects of SR 59104A in presence of charybdotoxin were reduced by 33% as compared to those observed in control conditions (P=0.053, Table 2). Similar results were observed with the SKCa channel inhibitor, apamin (Figure 3b) which showed no effect on the responses to isoprenaline and salbutamol but significantly decreased that to SR 59104A (−32%, P<0.001, Table 2). As shown in Figure 4, L-NAME increased 2 fold the hypoxic pressure responses (+8.53±1.30 and +9.17±1.60 mmHg during the sixth and the seventh hypoxic periods as compared to +3.71±0.26 mmHg and +3.92±0.40 mmHg during the same hypoxic periods with saline only, P<0.01). L-NAME (Table 2) did not significantly affect the relaxant effects of isoprenaline but significantly reduced those of salbutamol (−38%) and SR 59104A (−25%) (P<0.01 and P<0.05, respectively, Table 2).
Figure 2.
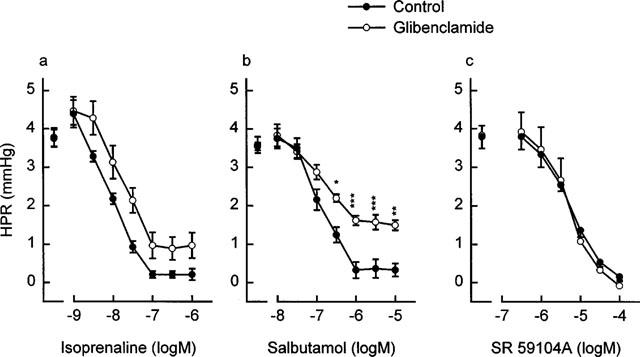
Effects of infusions of (a) isoprenaline (0.001–1 μM), (b) salbutamol (0.01–10 μM) or (c) SR 59104A (0.3–100 μM) on hypoxic pressor responses (HPR) in the absence (n=7, 6 and 6, respectively) (•) and in the presence of glibenclamide (1 μM) (n=9, 6 and 6, respectively) (○) in the isolated rat perfused lung. Values shown are increases of perfusion pressure above basal values. Data represent mean±s.e.mean. Asterisks indicate responses with glibenclamide that were significantly different from corresponding responses obtained without glibenclamide (*P<0.05, **P<0.01, ***P<0.001).
Table 1.
Potencies expressed as pD2 values of isoprenaline, salbutamol and SR 59104A in pulmonary vessels contracted by hypoxia: Influence of glibenclamide

Figure 3.
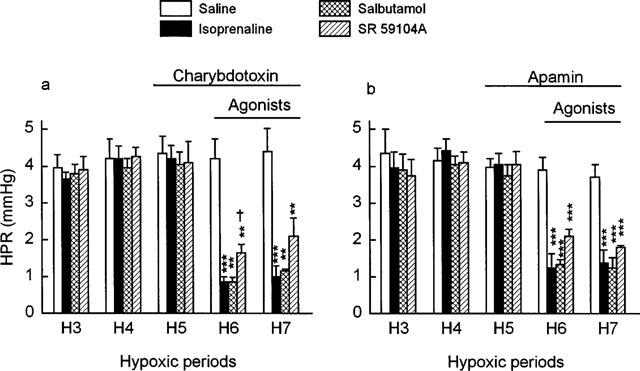
Influence of (a) charybdotoxin (0.1 μM) and (b) apamin (0.05 μM) on the effects of infusions of normal saline (n=5 and 5, respectively), isoprenaline (0.03 μM) (n=5 and 6, respectively), salbutamol (0.6 μM) (n=5 and 6, respectively) or SR 59104A (20 μM) (n=5 and 5, respectively) on hypoxic pressure responses (HPR) in the rat isolated perfused lung. Charybdotoxin and apamin were infused from the fifth hypoxic period and agonists were infused from the sixth hypoxic period. Values shown are increases of perfusion pressure above basal values. Data represent mean±s.e.mean. Asterisks indicate responses to the agonists that were significantly different from corresponding responses obtained in the control period (**P<0.01, ***P<0.001). † indicates a response to SR 59104A that was significantly different (P<0.05) from the corresponding response to salbutamol.
Table 2.
Percentage of the maximal relaxation obtained with β-adrenoceptor agonists on hypoxic pulmonary vasoconstriction: Influence of charybdotoxin, apamin or L-NAME

Figure 4.

Influence of L-NAME (100 μM) on the effects of infusions of normal saline (n=7), isoprenaline (0.03 μM) (n=5), salbutamol (0.6 μM) (n=5) and SR 59104A (20 μM) (n=5) on hypoxic pressure responses (HPR) in the isolated rat perfused lung. Inhibitors were infused from the fifth hypoxic period and agonists were infused from the sixth hypoxic period. Values are increases of perfusion pressure above basal values. Data represent mean±s.e.mean. Asterisks indicate responses to the agonists that were significantly different from corresponding responses obtained in the control period (*P<0.05, **P<0.01). † indicates responses to L-NAME that were significantly different (P<0.01) from corresponding responses obtained during the fourth hypoxic period.
Discussion
In this study, we investigated the effects of various β-adrenoceptor agonists on the pulmonary pressor response to hypoxia and the mechanisms involved, in a rat model of isolated perfused lung according to a method based on that described by McMurtry (1984). This model allows the investigation of the vasomotor tone in all pulmonary vessels and particularly in the small arteries and veins which are known to account for the greatest part of pulmonary vascular resistances (Madden et al., 1985; Zhao et al., 1993).
The baseline perfusion pressure under normoxic conditions was not influenced in our study by L-NAME, indomethacin and K+ channel inhibitors suggesting that the low pulmonary vascular tone is not dependent upon EDRF or prostacyclin release, or upon KATP, BKCa channels (Leeman et al., 1994; Dumas et al., 1996; Nossaman et al., 1997) and SKCa channels. It should however be stressed that other K+ channels with a non-inactivating, voltaged gated potassium current identified as IK(N) in pulmonary artery rings (Osipenko et al., 1998) or novel ATP dependent delayed-rectifier K+ channels in pulmonary artery myocytes (Patel et al., 1997) have been claimed to be involved in the maintenance of the pulmonary circulation resting tone. In our study, K+ channels or cyclo-oxygenase inhibitors did not affect significantly the hypoxic pulmonary vasoconstriction, thus suggesting the absence of any modulating effect of KATP, BKCa channels (Hasunuma et al., 1991a; Dumas et al., 1996) and SKCa channels or of prostacyclin release. Although Nossaman et al. (1997) described a small increase in the pressor response to hypoxia after charybdotoxin, recent studies suggest that oxygen-sensitive K+ channels involved in the modulation of vessel tone during hypoxia are not KCa channels but other K+ channels identified as Kv1.2 and Kv1.5 channels (Wang et al., 1997), novel Kv2.1/ Kv9.3 ATP dependent delayed-rectifier K+ channels (Patel et al., 1997) or K+ channels with IK(N) currents (Osipenko et al., 1998). In contrast to the lack of modulation of the hypoxic vasoconstriction observed in our study with glibenclamide, charybdotoxin, apamin or indomethacin, L-NAME enhanced the hypoxic vasoconstriction thus confirming that EDRF modulates the hypoxic pulmonary vasoconstriction as described previously (Liu et al., 1991; Dumas et al., 1994). A striking observation in our study was the partial inhibition of the post-hypoxic return to baseline pulmonary pressure induced by apamin, indomethacin and L-NAME which suggests that SKCa channels, prostacyclin and EDRF modulate post-hypoxic relaxation. These results support the view that the mechanisms involved in initiating the hypoxic smooth muscle contraction are different from those involved in initiating post hypoxic relaxation. The role of potassium channels and EDRF in the latter phenomenon deserve further investigations.
In this study, we investigated the effects of a non selective β-adrenoceptor agonist (isoprenaline), a selective β2-adrenoceptor agonist (salbutamol) and a selective β3-adrenoceptor agonist (SR 59104A, Croci et al., 1995) on the pulmonary vascular response to hypoxia, a physiopathological condition that produces vasoconstriction, to study potential dilator agents for the treatment of diseases such as pulmonary hypertension (Dumas et al., 1994). Isoprenaline, salbutamol and SR 59104A shared the ability to relax the pulmonary circulation during hypoxia (O'Donnell et al., 1991; Dumas et al., 1998) and in doing so, the relaxant potencies of isoprenaline and salbutamol were similar to or higher than those observed in pulmonary artery and in various other normoxic tissues (Jones et al., 1990; Oriowo, 1994; Sooch & Marshall, 1996; Huang & Kwok, 1997). Our findings suggest that hypoxia does not influence the relaxant effects of β-adrenoceptor agonists, thus contrasting with other investigations which reported a negative influence of hypoxia on the relaxant effects of these drugs in pulmonary arteries precontrated with adrenaline or U46619 (McIntyre et al., 1994; Wagner et al., 1997). These discrepancies might be related to the different agents used to contract the preparations.
Our experiments with glibenclamide clearly show that KATP channels contribute to the vasodilation induced by isoprenaline and salbutamol but not to that produced by SR 59104A. As regards isoprenaline and salbutamol, our results are in agreement with previous studies that suggested that the relaxant effects of β1- and β2-adrenoceptor agonists in various smooth vascular tissues were mediated in part through activation of KATP channels (Nakashima & Vanhoutte, 1995; Randall & McCulloch, 1995; Ming et al., 1997; Chang, 1997). As regards SR 59104A, glibenclamide failed to oppose its pulmonary relaxant effects. To our knowledge the role of KATP channels in the effects of β3-adrenoceptor agonists have not yet been investigated and this study clearly shows these drugs exhibit a pharmacological profile different from that of β1/β2-adrenoceptor agonists in the pulmonary circulation. These data are consistent with our previous demonstration that in the same vascular bed propranolol antagonizes the effects of isoprenaline but not those of the β3-adrenoceptor agonists SR 59104A, SR 59119A and SR 58611A (Dumas et al., 1998). In another study investigating the influence of glibenclamide on the responses elicited by agents acting through direct activation of adenylate cyclase such as forskolin, or through specific receptors such as iloprost and prostaglandin E1, we showed that only forskolin and iloprost, but not prostaglandin E1, were partially inhibited by glibenclamide indicating that the role of cyclic AMP in these effects is not clear (Dumas et al, 1997). This may account for the discrepancies observed among drugs activating the cyclic AMP pathway through different β-adrenoceptors. Differential activation of protein kinases A and G may also be suggested as is the case in airway smooth muscle (Torphy, 1994). Other studies are consistent with the hypothesis of an interaction of β3-adrenoceptor agonists with both Gs and Gi proteins (Chaudry et al., 1993; Gauthier et al., 1996; Roberts & Summers, 1998). Finally, it has been suggested that β-adrenergic activation could be linked via a cyclic AMP-independent and pertussis toxin-sensitive process to an activation of K+ channels (Khac et al., 1996), a hypothesis that might explain the effects observed with SR 59104A. Thus, at the high concentrations used, a mechanism of action other than the stimulation of β-adrenoceptors cannot be ruled out and further investigations are necessary to establish the pharmacological properties of β3-adrenoceptor agonists according to tissues and/or species.
As regards KCa channels, charybdotoxin and apamin failed to block the relaxant effects of isoprenaline and salbutamol but inhibited partly those of SR 59104A, demonstrating that β3-, but not β2- and/or β1-adrenoceptor agonists, act in part through BKCa and SKCa channels. Regarding isoprenaline, Nossaman et al. (1997) also failed to inhibit its relaxant effects with charybdotoxin in the rat isolated perfused lung. However other studies have suggested that activation of BKCa channels is one of the underlying mechanisms responsible for isoprenaline or β2-adrenoceptor agonist-induced relaxation in various tissues and species as rat aorta (Satake et al., 1996), mesenteric artery (Huang & Kwok, 1997) or guinea-pig mesenteric lymphatic vessels (Von der Weid et al., 1996). However, these data were obtained in isolated vessels rings whereas our study was performed in the isolated whole lung preparation.
There are only few studies investigating the influence of the nitric oxide pathway on the pharmacological activity of β-adrenoceptor agonists in the pulmonary circulation (Priest et al., 1997). In our study, L-NAME partly inhibited the relaxant effects of salbutamol and SR 59104A but not those of isoprenaline. These results are in agreement with previous reports where EDRF was involved in the β2-adrenergic dilatation but not in that induced by β1-adrenoceptor stimulation (Hamdad et al., 1996) or isoprenaline (Bea et al., 1994). These results suggest that the modulation of β-adrenoceptor mediated relaxation may be dependent upon the β-adrenoceptor subtypes. In contrast, in another report, it was shown that the relaxations induced by a non selective agonist, isoprenaline, and a β2-adrenoceptor selective agonist, salbutamol, were both antagonized by NO-synthase inhibitors mainly in large pulmonary arteries and to a lesser extent in small pulmonary arteries suggesting differences according to the conduits rather than to the β-adrenoceptor subtypes (Priest et al., 1997). Thus, the influence of EDRF in the modulation of the relaxant effects of β-adrenoceptor agonists in the pulmonary circulation deserves further investigations.
In conclusion, it appears from our data that in the pulmonary circulation: (a) the low resting tone is not modulated by KATP, BKCa, SKCa channels or the release of nitric oxide or prostaglandins, (b) EDRF exerts a significant inhibition of the hypoxic pulmonary vasoconstriction, (c) SKCa channels, EDRF and prostaglandins partially contribute to the reversal of the hypoxic pulmonary vasoconstriction, (d) isoprenaline, salbutamol and SR 59104A share the ability to oppose the hypoxic pulmonary vasoconstriction, (e) the vasodilation induced by isoprenaline is mediated in part by KATP channels, that of salbutamol by KATP channels and EDRF, and (f) SR 59104A also vasodilates the pulmonary vascular bed, partly through BKCa and SKCa channels activation and EDRF release, differing in this from β1 and/or β2-adrenoceptor agonists.
Abbreviations
- BKCa
large conductance Ca2+-activated K+
- EDRF
endothelium derived relaxing factor
- KATP
ATP-sensitive K+
- L-NAME
NG-nitro-L-arginine methyl ester
- NO
nitric oxide
- SKCa
small conductance Ca2+-activated K+
References
- BEA M.L., GHALEH B., GIUDICELLI J.F., BERDEAUX A. Lack of importance of NO in β-adrenoceptor mediated relaxation of large epicardial canine coronary arteries. Br. J. Pharmacol. 1994;111:981–982. doi: 10.1111/j.1476-5381.1994.tb14839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERLAN M., GALITZKY J., BOUSQUET-MELOU A., LAFONTAN M., MONTASTRUC J.L. β3-adrenoceptor-mediated increase in cutaneous blood flow in the dog. J. Pharmacol. Exp. Ther. 1994;268:1444–1451. [PubMed] [Google Scholar]
- BYLUND D.B., EIKENBERG D.C., HIEBLE J.P., LANGER S.Z., LEFKOWITZ R.J., MINNEMAN K.P., MOLINOFF P.B., RUFFOLO R.R., TRENDELENBURG U. International union of pharmacology nomenclature of adrenoceptors. Pharmacol. Rev. 1994;46:121–136. [PubMed] [Google Scholar]
- CHANG H-Y. The involvement of ATP-sensitive potassium channels in β2-adrenoceptor agonist-induced vasodilatation on rat diaphragmatic microcirculation. Br. J. Pharmacol. 1997;121:1024–1030. doi: 10.1038/sj.bjp.0701192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAUDRY A., MACKENZIE R.G., GEORGIC L.M., GRANNEMAN J. Differential interaction of β1-and β3-adrenergic receptors with Gi in rat adipocytes. Cell. Signal. 1993;6:457–465. doi: 10.1016/0898-6568(94)90093-0. [DOI] [PubMed] [Google Scholar]
- CROCI T., CECCHI R., GUZZI M., LANDI T., MENINI T., PAROTTI L., LE FUR G., MANARA L. The novel β3-adrenoceptor agonists SR 59119A and SR 59104A are stereospecifically antagonized by SR 59230A. Br. J. Pharmacol. 1995;116:204P. [Google Scholar]
- DUMAS M., DUMAS J.P., BARDOU M., ROCHETTE L., ADVENIER C., GIUDICELLI J.F. Influence of β-adrenoceptor agonists on the pulmonary circulation. Effects of a β3-adrenoceptor antagonist, SR 59104A. Eur. J. Pharmacol. 1998;348:223–228. doi: 10.1016/s0014-2999(98)00146-0. [DOI] [PubMed] [Google Scholar]
- DUMAS M., DUMAS J.P., ROCHETTE L., ADVENIER C., GIUDICELLI J.F. Comparison of the effects of nicorandil, pinacidil and nitroglycerin on hypoxic and hypercapnic pulmonary vasoconstriction in the isolated perfused lung of rat. Br. J. Pharmacol. 1996;117:633–638. doi: 10.1111/j.1476-5381.1996.tb15237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMAS M., DUMAS J.P., ROCHETTE L., ADVENIER C., GIUDICELLI J.F. Role of potassium channels and nitric oxide in the effects of iloprost and prostaglandin E1 on hypoxic vasoconstriction in the isolated perfused lung of the rat. Br. J. Pharmacol. 1997;120:405–410. doi: 10.1038/sj.bjp.0700912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUMAS J.P., DUMAS M., SGRO C., ADVENIER C., GIUDICELLI J.F. Effects of two K+ channel openers, aprikalim and pinacidil, on hypoxic pulmonary vasoconstriction. Eur. J. Pharmacol. 1994;263:17–23. doi: 10.1016/0014-2999(94)90518-5. [DOI] [PubMed] [Google Scholar]
- GAUTHIER C., TAVERNIER G., CHARPENTIER F., LANGIN D., LE MAREC H. Functional β3-adrenoceptor in the human heart. J. Clin. Invest. 1996;98:556–562. doi: 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHOFRANI H.A., SCHERMULY R., ROHL A., WEISSMANN N., WALMRATH D., SEEGER W., GRIMMINGER F. Synergism of theophyllin and inhaled prostacyclin (PGI2) in a model of acute pulmonary hypertension. Am. J. Resp. Crit. Care Med. 1998;157:A592. [Google Scholar]
- HAMDAD N., MING Z., PARENT R., LAVALLEE M. β2-adrenergic dilation of conductance coronary arteries involves flow-dependent NO formation in conscious dogs. Am. J. Physiol. 1996;271:H1926–H1937. doi: 10.1152/ajpheart.1996.271.5.H1926. [DOI] [PubMed] [Google Scholar]
- HASUNUMA K., RODMAN D.M., McMURTRY I.F. Effects of K+ channel blockers on vascular tone in the perfused rat lung. Am. Rev. Resp. Dis. 1991a;144:884–887. doi: 10.1164/ajrccm/144.4.884. [DOI] [PubMed] [Google Scholar]
- HASUNUMA K., YAMAGUCHI T., RODMAN D.M., O'BRIEN R.F., McMURTRY I.F. Effects of inhibitors of EDRF and EDHF on vasoreactivity of perfused rat lungs. Am. J. Physiol. 1991b;260:L97–L104. doi: 10.1152/ajplung.1991.260.2.L97. [DOI] [PubMed] [Google Scholar]
- HUANG Y., KWOK K.H. Effects of putative K+ channel blockers on β-adrenoceptor-mediated vasorelaxation of rat mesenteric artery. J. Cardiovasc. Pharmacol. 1997;29:515–519. doi: 10.1097/00005344-199704000-00013. [DOI] [PubMed] [Google Scholar]
- JONES T.R., CHARETTE L., GARCIA M.L., KACZOROWSKI G.J. Selective inhibition of relaxation of guinea-pig trachea by charybdotoxin, a potent Ca++-activated K+ channel inhibitor. J. Pharmacol. Exp. Ther. 1990;255:697–706. [PubMed] [Google Scholar]
- KHAC L.D., ARNEAUDEAU S., LEPRETRE N., MIRONNEAU J., HARBON S. Beta adrenergic receptor activation attenuates the generation of inositol phosphates in the pregnant rat myometrium. Correlation with inhibition of Ca++ influx, a cAMP-independent mechanism. J. Pharmacol. Exp. Ther. 1996;276:130–136. [PubMed] [Google Scholar]
- LEACH R.M., TREACHER D.F. Clinical aspects of hypoxic pulmonary vasoconstriction. Exp. Physiol. 1995;80:865–875. doi: 10.1113/expphysiol.1995.sp003894. [DOI] [PubMed] [Google Scholar]
- LEEMAN M., ZEGERS DE BEYL V., DELCROIX M., NAEIJE R. Effects of endogenous nitric oxide on pulmonary vascular tone in intact dogs. Am. J. Physiol. 1994;266:H2343–H2347. doi: 10.1152/ajpheart.1994.266.6.H2343. [DOI] [PubMed] [Google Scholar]
- LIU S., CRAWLEY D.E., BARNES P.J., EVANS T.W. Endothelium-derived relaxing factor inhibits hypoxic pulmonary vasoconstriction in rats. Am. Rev. Resp. Dis. 1991;143:32–37. doi: 10.1164/ajrccm/143.1.32. [DOI] [PubMed] [Google Scholar]
- MADDEN J.A., DAWSON C.A., HARDER D.A. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J. Appl. Physiol. 1985;59:113–118. doi: 10.1152/jappl.1985.59.1.113. [DOI] [PubMed] [Google Scholar]
- MANKTELOW C., BIGATELLO L.M., HESS D., HURFORD W.E. Physiologic determinants of the response to inhaled nitric oxide in patients with acute respiratory distress syndrome. Anesth. 1997;87:297–307. doi: 10.1097/00000542-199708000-00017. [DOI] [PubMed] [Google Scholar]
- McINTYRE R.C., BANERJEE A., BENSARD D.D., BREW E.C., HAHN A.R., FULLERTON D.A. Adenosine A1-receptor mechanisms antagonize β-adrenergic pulmonary vasodilation in hypoxia. Am. J. Physiol. 1994;267:H2179–H2185. doi: 10.1152/ajpheart.1994.267.6.H2179. [DOI] [PubMed] [Google Scholar]
- McMURTRY I.F. Angiotensin is not required for hypoxic constriction in salt-solution-perfused lungs. J. Appl. Physiol. 1984;56:375–380. doi: 10.1152/jappl.1984.56.2.375. [DOI] [PubMed] [Google Scholar]
- MING Z., PARENT R., LAVALLEE M. β2-adrenergic dilation of resistance coronary vessels involves KATP channels and nitric oxide in conscious dogs. Circ. 1997;95:1568–1576. doi: 10.1161/01.cir.95.6.1568. [DOI] [PubMed] [Google Scholar]
- NAKASHIMA M., VANHOUTTE P.M. Isoproterenol causes hyperpolarisation through opening of ATP-sensitive potassium channels in vascular smooth muscle of the canine saphenous vein. J. Pharmacol. Exp. Ther. 1995;272:379–384. [PubMed] [Google Scholar]
- NOSSAMAN B.D., KAYE A.D., FENG C.J., KADOWITZ P.J. Effects of charybdotoxin on responses to nitrovasodilators and hypoxia in the rat lung. Am. J. Physiol. 1997;272:L787–L791. doi: 10.1152/ajplung.1997.272.4.L787. [DOI] [PubMed] [Google Scholar]
- O'DONNELL S.R., WANSTALL J.C., KAY C.S., ZENG X. Tissue selectivity and spasmogen selectivity of relaxant drugs in airway and pulmonary vascular smooth muscle contracted by PGF2α or endothelin. Br. J. Pharmacol. 1991;102:311–316. doi: 10.1111/j.1476-5381.1991.tb12171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLSCHEWSKI H., WALMRATH D., SCHERMULY R., GHOFRANI A., GRIMMINGER F., SEEGER W. Aerosolized prostacyclin and iloprost in severe pulmonary hypertension. Ann. Int. Med. 1996;124:820–824. doi: 10.7326/0003-4819-124-9-199605010-00006. [DOI] [PubMed] [Google Scholar]
- ORIOWO M.A. Atypical β-adrenoceptors in the rat isolated common carotid artery. Br. J. Pharmacol. 1994;113:699–702. doi: 10.1111/j.1476-5381.1994.tb17049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORIOWO M.A. Different atypical β3-adrenoceptors mediate isoprenaline-induced relaxation in vascular and non vascular smooth muscles. Pharmacol. Lett. 1995;56:269–275. doi: 10.1016/0024-3205(95)00076-3. [DOI] [PubMed] [Google Scholar]
- OSIPENKO O.N., ALEXANDER D., MACLEAN M.R., GURNEY A.M. Influence of chronic hypoxia on the contributions of non-activating and delayed rectifier K currents to the resting potential and tone of rat pulmonary artery smooth muscle. Br. J. Pharmacol. 1998;124:1335–1337. doi: 10.1038/sj.bjp.0702006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATEL A.J., LAZDUNSKI M., HONORE E. Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ Channel in oxygen-sensitive pulmonary artery myocytes. EMBO J. 1997;16:6615–6625. doi: 10.1093/emboj/16.22.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRIEST R.M., HUCKS D., WARD J.P.T. Noradrenaline, β-adrenoceptor mediated vasorelaxation and nitric oxide in large and small pulmonary arteries of the rat. Br. J. Pharmacol. 1997;122:1375–1384. doi: 10.1038/sj.bjp.0701528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANDALL M.D., McCULLOCH A.J. The involvement of ATP-sensitive potassium channels in β-adrenoceptor-mediated vasorelaxation in the rat isolated mesenteric arterial bed. Br. J. Pharmacol. 1995;115:607–612. doi: 10.1111/j.1476-5381.1995.tb14975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REEVE H.L., ARCHER S.L., WEIR K. Ion channels in the pulmonary vasculature. Pulm. Pharmacol. Ther. 1997;10:243–252. doi: 10.1006/pupt.1998.0107. [DOI] [PubMed] [Google Scholar]
- ROBERTS S.J., SUMMERS R.J. Cyclic AMP accumulation in rat soleus: stimulation by β2- but not β3-adrenoceptors. Eur. J. Pharmacol. 1998;348:53–60. doi: 10.1016/s0014-2999(98)00021-1. [DOI] [PubMed] [Google Scholar]
- SATAKE N., SHIBATA M., SHIBATA S. The inhibitory effects of iberiotoxin and 4-aminopyridine on the relaxation induced by β1- and β2-adrenoceptor activation in rat aortic rings. Br. J. Pharmacol. 1996;119:505–510. doi: 10.1111/j.1476-5381.1996.tb15700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOOCH S., MARSHALL I. Evidence for atypical β-adrenoceptor in the rat vasculature. Br. J. Pharmacol. 1996;117:261P. [Google Scholar]
- TORPHY T.J. β-adrenoceptors, cAMP and airway smooth muscle relaxation: challenges to the dogma. Trends Pharmacol. Sci. 1994;15:370–374. doi: 10.1016/0165-6147(94)90157-0. [DOI] [PubMed] [Google Scholar]
- VON DER WEID P.-Y., VAN HELDEN D.F. β-adrenoceptor-mediated hyperpolarization in lymphatic smooth muscle of guinea-pig mesentery. Am. J. Physiol. 1996;270:H1687–H1695. doi: 10.1152/ajpheart.1996.270.5.H1687. [DOI] [PubMed] [Google Scholar]
- WAGNER R.S., SMITH C.J., TAYLOR A.M., RHOADES R.A. Phosphodiesterase inhibition improves agonist-induced relaxation of hypertensive pulmonary arteries. J. Pharmacol. Exp. Ther. 1997;282:1650–1657. [PubMed] [Google Scholar]
- WANG J., JUHASZOVA M., RUBIN L.J., YUAN X.-J. Hypoxia inhibits gene expression of voltage-gated K+ channel α subunits in pulmonary artery smooth muscle cells. J. Clin. Invest. 1997;100:2347–2353. doi: 10.1172/JCI119774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Y-X, , POON K.S., RANDALL D.J., PANG C.C.Y. Endothelium-derived nitric oxide partially mediates salbutamol-induced vasodilatations. Eur. J. Pharmacol. 1993;250:335–340. doi: 10.1016/0014-2999(93)90018-d. [DOI] [PubMed] [Google Scholar]
- ZHAO L., PACKER S., RHOADES R.A. Pulmonary veins contract in response to hypoxia. Am. J. Physiol. 1993;265:L87–L92. doi: 10.1152/ajplung.1993.265.1.L87. [DOI] [PubMed] [Google Scholar]