

Abstract
Protease-activated receptors (PARs) are activated by an irreversible proteolytic mechanism which renders cleaved receptors unresponsive to subsequent challenges with activating enzymes. Non-specific proteolysis of PARs downstream of the activation site also prevents subsequent enzymic activation. Therefore, we investigated the effects of non-activating amino-terminal proteolysis with the bacterial protease thermolysin on PAR-mediated relaxation of porcine coronary artery ring preparations contracted with the thromboxane A2 mimetic U46619 (1–10 nM).
Treatment of contracted artery ring segments with thermolysin (0.01–1 u ml−1, 20 min) caused no response, but abolished endothelium-dependent relaxations induced by the enzymic activators of PAR-1 and PAR-2, thrombin (0.01–0.3 u ml−1) and trypsin (0.003–0.1 u ml−1) respectively. The same treatment, however, did not affect similar responses to the proteolysis-independent PAR-1 and PAR-2 activating peptides, SFLLRN-NH2 and SLIGRL-NH2 respectively (0.1–10 μM).
The inhibition of responsiveness to trypsin after thermolysin treatment recovered in a time-dependent manner, with maximal recovery (77.3±8.0% of time controls) occurring 150 min after thermolysin treatment. No recovery of responsiveness to thrombin after thermolysin treatment was observed within this time, however, the thrombin response returned to control levels after 20 h.
The recovery of responsiveness to trypsin was inhibited by the translation inhibitor cycloheximide (100 μM; 17.3±4.7%) and the protein trafficking inhibitor brefeldin A (10 μM; 12.1±4.8%) but was unaffected by the transcription inhibitor actinomycin D (2 μM; 65.1±3.6%), which did, however, abolish upregulation of B1-kinin receptors in this preparation.
In conclusion, our findings indicate that activation-independent amino-terminal proteolysis of PARs stimulates selective recovery of endothelial cell PAR-2 responsiveness, which appears to be regulated by translation. Such a novel mechanism for the maintenance of responsiveness to enzymic PAR-2 activators may imply that these receptors play important roles in vascular homeostasis.
Keywords: Endothelium, porcine coronary artery, protease-activated receptors, PAR-2, trypsin, thrombin, receptor recycling
Introduction
The cloning of the first thrombin receptor, protease-activated receptor-1 (PAR-1; Vu et al., 1991a) revealed a unique mechanism of receptor activation which explained the established observation that the cellular effects of proteases such as thrombin are dependent on their proteolytic activity (Davey & Luscher, 1967). The subsequent cloning of the trypsin-sensitive PAR-2 (Nystedt et al., 1994; Böhm et al., 1996b), thrombin-activated PAR-3 (Ishihara et al., 1997), and PAR-4 (Xu et al., 1998), which can be activated by both thrombin and trypsin, indicated the emergence of a family of seven transmembrane domain, G protein-coupled receptors which detect particular extracellular enzymes (Coughlin, 1994). PAR activation occurs via enzymic cleavage of the amino-terminal exodomain at a site specific for each receptor subtype (Vu et al., 1991a; Nystedt et al., 1994; Ishihara et al., 1997; Xu et al., 1998). The newly exposed amino-terminal sequence than acts as a ‘tethered ligand', binding intramolecularly to initiate cellular signaling (Vu et al., 1991a,1991b; Gerszten et al., 1994; Blackhart et al., 1996). For PAR-1, PAR-2 and PAR-4, but surprisingly not PAR-3, receptor activation can be mimicked by synthetic peptides corresponding to their respective tethered ligand sequences (Vu et al., 1991a; Nystedt et al., 1994; Blackhart et al., 1996; Ishihara et al., 1997; Xu et al., 1998).
Since activation of PARs by specific endogenous proteases is an irreversible process, precise desensitization and resensitization mechanisms most likely control cellular responsiveness to subsequent enzymic activation. For example, biochemical and molecular studies on PAR-1 (Hoxie et al., 1993; Hein et al., 1994) and PAR-2 (Böhm et al., 1996a) in isolated cells have shown that once cleaved, activated receptors are rapidly internalized and replenished from a pool of pre-formed receptors located in the Golgi. However, amino-terminal proteolysis by non-activating proteases which cleave distal to the receptor's specific activation site have also been shown to disable the receptor from subsequent challenges with activating enzymes (Molino et al., 1995; 1997; Renesto et al., 1997; Déry et al., 1998). Inflammatory cells release many proteases (Davey & Luscher, 1967; Mitchinson & Ball, 1987; Kovanen et al., 1995; Welle, 1997), including elastase, cathepsin G and proteinase 3–all of which are capable of PAR inactivation (Renesto et al., 1997). Since PARs are proposed to modulate vascular tone in inflammatory conditions (Tesfamariam et al., 1993; Saifeddine et al., 1996), we investigated whether endothelial PARs maintain responsiveness to PAR activators after non-activating amino-terminal cleavage with thermolysin, an inflammatory bacterial protease which increases vascular permeability (Molle et al., 1987). Our data demonstrate that rapid recovery of endothelial cell PAR-2 responsiveness in porcine isolated coronary arteries is stimulated by cleavage of the amino-terminal exodomain without concomitant receptor activation. Our observation of a novel and efficient mechanism for the replenishment of PAR-2 in native endothelial cells provides further evidence for the importance of maintained function of these receptors in the vasculature.
Methods
Preparation of porcine coronary arteries
Right coronary arteries were dissected from hearts of Large White pigs (either sex, 30–40 kg) freshly slaughtered at a local abattoir. Three mm long artery ring preparations were mounted between two parallel wire hooks in 30 ml organ baths containing Krebs solution (composition in mM: Na+ 144, Cl− 128.7, HCO3− 25, K+ 5.9, Ca2+ 2.5, Mg2+ 1.2, H2PO4− 1.2, SO42− 1.2 and glucose 11) maintained at 37°C and continuously bubbled with 95% O2, 5% CO2 to keep the pH at 7.4. One hook was attached to a micrometer-adjustable support leg and the other to an isometric force transducer (Grass Instruments, model FT03C) to record changes in isometric force which were amplified and displayed on flat bed chart recorders (W&W Scientific Instruments).
Following a 60 min equilibration period, tissues were twice stretched to 5 g passive force and allowed to recover for 30 min before being exposed to an isotonic, high potassium Krebs solution (KPSS; composition in mM: K+ 124.9, Cl− 128.7, Na+ 25.0, HCO3− 25.0, Ca2+ 2.5, Mg2+ 1.2, SO42− 1.2, H2PO4− 1.2 and glucose 6.1) to obtain a maximum contraction for each artery ring (KPSSmax; Drummond & Cocks, 1996). The KPSS was then replaced with normal Krebs solution and the tissues allowed to return to their optimal passive force level over 30–60 min.
Effect of thermolysin on PAR-mediated responses
Artery ring segments were contracted to approximately 50% KPSSmax with the thromboxane A2 mimetic, U46619 (1–10 nM) and were either untreated or treated cumulatively with thermolysin (0.01–1 u ml−1). Treated tissues were left exposed to the maximum concentration of thermolysin (1 u ml−1) for 20 min and then either washed with Krebs solution (containing the appropriate concentration of U46619 to maintain the level of precontraction) or had thermolysin left in the organ bath. Responses to cumulative additions of thrombin and trypsin (0.0001–0.3 u ml−1) and the PAR-1 and PAR-2 tethered ligand sequences, SFLLRN-NH2 and SLIGRL-NH2 respectively (0.01–10 μM), were then obtained. Following the maximum relaxation to each PAR activator, the maximum endothelium-dependent and -independent relaxation capabilities of each ring preparation were determined with the addition of bradykinin (0.3 μm) and isoprenaline (1 μm) respectively. To determine the effect of thermolysin on responses to other, non-PAR endothelium-dependent dilators, a similar protocol to that described for the PAR activators was carried out for bradykinin (0.1–300 nM) and substance P (0.01–30 nM).
Recovery of PAR-mediated responses after thermolysin treatment
Tissues were treated with thermolysin (0.01–1 u ml−1; 20 min) in a similar manner to that described above except at resting levels of active force. After thorough washout of thermolysin, ring segments were contracted to approximately 50% KPSSmax with U46619 in a timed manner to allow responses to thrombin or trypsin (0.1 u ml−1) to be recorded at one of 50, 100, 150 or 200 min time points after thermolysin washout. This protocol was repeated in preparations similarly contracted to 50% KPSSmax with KCl (10–18 mM).
Inhibition of recovery of PAR-mediated responses after thermolysin treatment
In this series of experiments the protocol described above for investigating the recovery of responses to thrombin and trypsin was repeated, at only the 150 min recovery time, in the absence and presence of one of the transcription inhibitor, actinomycin D (2 μM), the translation inhibitor, cycloheximide (100 μM), or the protein trafficking inhibitor, brefeldin A (10 μM).
To verify that actinomycin D blocked transcription in porcine coronary endothelial cells, artery ring preparations were left untreated or were treated with actinomycin D (2 μM) immediately after they were initially placed at 37°C. Endothelium-dependent relaxations to the selective B1- or B2-kinin receptor agonists, des-arg9-bradykinin (3 μM) and bradykinin (3 μM) respectively, were then recorded in U46619-contracted preparations after 3 or 13 h of incubation at 37°C. In addition, to determine if prior exposure of tissues to thermolysin inhibited the ability of actinomycin D to block transcription, some tissues were treated with thermolysin (0.01–1 u ml−1, 20 min) following the initial exposure to des-arg9-bradykinin (3 μM) and bradykinin (3 μM). These tissues were then washed thoroughly, again treated with actinomycin D (2 μM) and responses to the kinin receptor agonists re-examined after 13 h.
Analysis and statistics
All responses are expressed as a percentage of the tissue's response to isoprenaline (1 μM) and data is presented as mean±s.e.mean. Mean concentration-response curves were computer fitted to a sigmoidal regression curve (Graphpad Prism, Graphpad Software Inc.) to generate values for sensitivity (pEC50). Differences in mean pEC50 and maximum response (Rmax) were tested for significance either by unpaired Student's t-test or one-way analysis of variance (ANOVA) with a Tukey-Kramer modified t statistic for multiple comparisons. In all cases, differences were considered significant at P<0.05.
Materials
Actinomycin D, bradykinin (acetate salt), brefeldin A, cycloheximide, des-arg9-bradykinin (acetate salt), (−)-isoprenaline, substance P (acetate salt), thermolysin (type X, from Bacillus polymyca) and α-thrombin (bovine serum) were obtained from Sigma (MO, U.S.A.). 9, 11-dideoxy-9α,11α-methanoepoxy-Prostaglandin F2α (U46619) was from Sapphire Bioscience (NSW, Australia). Trypsin (bovine pancreas) was from Worthington Biochem (NJ, U.S.A.) while the peptides SLIGRL-NH2 and SFLLRN-NH2 were obtained from Auspep (Vic, Australia). Stock solutions of U46619 (1 mM) were in absolute ethanol with further dilutions in distilled water. Solutions of all other drugs were in distilled water.
Results
Effect of thermolysin on PAR-mediated responses
Trypsin (0.003–0.1 u ml−1) and thrombin (0.01–0.3 u ml−1) each caused concentration-dependent relaxation of U46619-contracted pig coronary artery ring preparations (Figure 1). The sensitivity (pEC50, −log u ml−1) and maximum response (Rmax, percentage response to 1 μM isoprenaline) values for trypsin were 2.1±0.1 and 92.3±3.9% respectively, while those for thrombin were 1.6±0.1 and 77.1±4.0%. Responses to both enzymes were abolished by prior removal of the endothelium (data not shown). Treatment of contracted artery preparations with cumulative additions of thermolysin (0.01–1 u ml−1, 20 min) caused no response (not shown) but abolished relaxations to both trypsin and thrombin whether it was left in contact with, or washed out of, the tissue (Figure 1).
Figure 1.
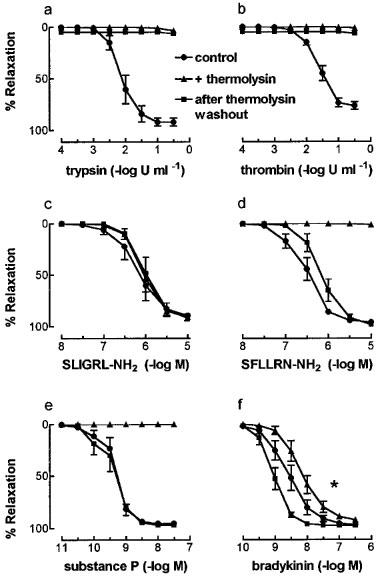
The effect of thermolysin (0.01–1 u ml−1, 20 min) on relaxations to (a) trypsin, (b) thrombin, (c) SLIGRL-NH2, (d) SFLLRN-NH2, (e) substance P and (f) bradykinin in endothelium-intact, U46619-contracted pig coronary artery ring segments. Cumulative concentration-response curves were generated in the absence (control), presence, and after washout of thermolysin. Data are mean±s.e.mean from 4–8 experiments and are expressed as a percentage of the maximum relaxation to isoprenaline (1 μM). *P<0.05 (t-test) for pEC50 values for bradykinin in the presence of thermolysin vs after thermolysin washout.
Similar responses as those for trypsin and thrombin were obtained with the synthetic PAR-2 and PAR-1 tethered ligand sequences, SLIGRL-NH2 and SFLLRN-NH2 respectively (Figure 1). The pEC50 (−log M) and Rmax values for SLIGRL-NH2 were 6.2±0.2 and 89.4±4.7% respectively, while those for SFLLRN-NH2 were 6.5±0.1 and 95.9±0.7%. As observed with trypsin and thrombin, relaxations to both peptides were abolished by endothelium denudation (data not shown). In contrast to the enzyme-induced responses, thermolysin treatment had no effect on responses to SLIGRL-NH2 (Figure 1). Also, relaxations to SFLLRN-NH2 were abolished in the presence of thermolysin, but these relaxations returned to control levels after thermolysin washout (Figure 1).
The effect of thermolysin on responses to non-PAR endothelium-dependent peptide vasodilators was examined using bradykinin and substance P. Relaxations to substance P (pEC50 9.3±0.1; Rmax 94.4±0.6%) were abolished in the presence of thermolysin but returned to control values after washout (pEC50 9.4±0.2; Rmax 96.9±1.0%) (Figure 1). By contrast, relaxations to bradykinin (pEC50 8.6±0.2; Rmax 96.4±0.8%) were unaffected by thermolysin treatment (Figure 1), except for a significant (P<0.05) increase in sensitivity after thermolysin washout (pEC50 9.1±0.1) compared with that in its presence (pEC50 8.2±0.1).
Mechanism of recovery of PAR-mediated responses after thermolysin treatment
Whilst thermolysin treatment initially abolished trypsin-induced relaxations in U46619-contracted preparations, the trypsin response recovered with time after thermolysin washout (Figure 2). After 50 min, trypsin (0.1 u ml−1) caused 17.8±9.1% of the time control response in thermolysin-treated artery preparations. This increased to 44.9±10.3% of the time control response after 100 min. Maximum recovery occurred 150 min after thermolysin washout (77.3±8.0% of time controls) since no further significant recovery was observed after 200 min (83.3±8.8% of time controls). Similar responses were observed in artery ring preparations contracted with KCl (data not shown) indicating that recovery of the trypsin response was not specific for U46619-contracted tissues.
Figure 2.
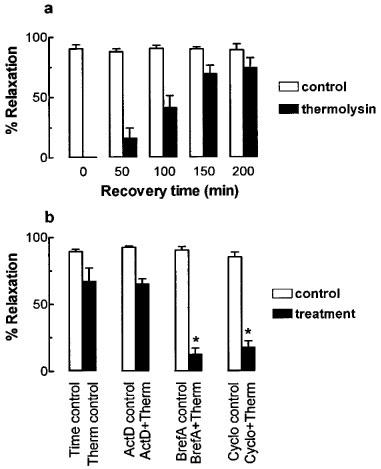
The time course and mechanism of recovery of responsiveness to trypsin (0.1 u ml−1) following desensitization with thermolysin (0.01–1 u ml−1, 20 min) in U46619-contracted rings of pig coronary artery. (a) Endothelium-dependent relaxations to trypsin (0.1 u ml−1) were determined in matched time control and thermolysin-treated arteries at the times indicated after thermolysin washout. (b) Relaxations to trypsin (0.1 u ml−1) 150 min after treatment washout, in artery preparations treated with actinomycin D (ActD; 2 μM), brefeldin A (BrefA; 10 μM) or cycloheximide (Cyclo; 100 μM) alone or after prior treatment with thermolysin (Therm; 0.01–1 u ml−1, 20 min). Data are mean±s.e.mean from 4–6 experiments and are expressed as percentages of the maximum relaxation to isoprenaline (1 μM). *P<0.05 (ANOVA) vs thermolysin control group.
Recovery of the relaxation to trypsin (0.1 u ml−1) 150 min after thermolysin treatment (74.9±10.1% of time controls) was significantly inhibited by either the protein trafficking inhibitor, brefeldin A (10 μM, 12.1±4.8%; P<0.05 ANOVA) or the translation inhibitor, cycloheximide (100 μM, 17.3±4.7%; P<0.05 ANOVA) (Figure 2). In the absence of thermolysin treatment none of these inhibitors affected the time control response to trypsin (Figure 2). The recovery of the trypsin (0.1 u ml−1) response was unaffected by the transcription inhibitor, actinomycin D (2 μM, 65.1±3.6%) (Figure 2). The same concentration of actinomycin D (2 μM), however, abolished the time-dependent increase in endothelium-dependent relaxation to the selective B1-kinin receptor agonist des-arg9-bradykinin without affecting similar relaxations to the selective B2-kinin receptor agonist bradykinin (Figure 3). Also, inhibition of the increased responsiveness to des-arg9-bradykinin by actinomycin D was unaffected by prior treatment of the tissue with thermolysin (0.01–1 u ml−1, 20 min) (Figure 3).
Figure 3.
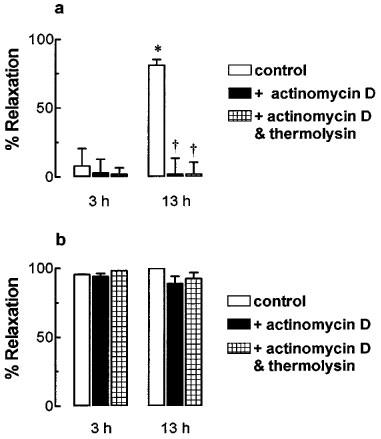
The effect of actinomycin D on endothelium-dependent relaxations to (a) des-arg9-bradykinin (3 μM) and (b) bradykinin (3 μM) in U46619-contracted pig coronary artery ring preparations. Tissues were either untreated (control), treated with actinomycin D (2 μM) or treated with actinomycin D (2 μM) and thermolysin (0.01–1 u ml−1, 20 min). Responses were examined after 3 or 13 h of incubation at 37°C. Data are mean±s.e.mean from four experiments and are expressed as percentages of the maximum relaxation to isoprenaline (1 μM). *P<0.05 (t-test) vs 3 h response. †P<0.05 (t-test) vs untreated control.
In contrast to trypsin, the loss of responsiveness to thrombin after thermolysin treatment failed to recover up to 200 min after washout, but eventually returned to control levels within 20 h (Figure 4).
Figure 4.
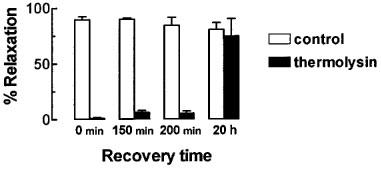
The effect of thermolysin (0.01–1 u ml−1, 20 min) on the recovery of responsiveness to thrombin (0.1 u ml−1) in U46619-contracted rings of pig coronary artery. Endothelium-dependent relaxations to thrombin (0.1 u ml−1) were examined in time control and matched artery preparations treated with thermolysin (0.01–1 u ml−1, 20 min) at the times indicated after thermolysin washout. Data are mean±s.e.mean from five experiments and are expressed as a percentage of the maximum relaxation to isoprenaline (1 μM).
Discussion
The main finding of this study was that vascular endothelial cell PAR-2 signal rapid replenishment of new receptors to the cell surface following non-activating amino-terminal proteolysis. The enzyme used to stimulate this PAR-2 turnover, bacterial thermolysin, cleaves peptide bonds amino-terminally to the hydrophobic residues leucine (L) and isoleucine (I), with some specificity for phenylalanine (F) and valine (V). Therefore, thermolysin would be expected to remove the exodomain of PAR-1 and PAR-2 – including their tethered ligand sequences. Also, with no specificity for the cleavage site required for activation of either PAR-1 or PAR-2, thermolysin was not expected to initiate receptor activation. This was confirmed functionally by the findings that thermolysin failed to cause any response in contracted artery preparations, yet it abolished the endothelium-dependent relaxation to thrombin and trypsin. Any non-specific effects of thermolysin were unlikely to explain its effect on the relaxations to thrombin and trypsin since the same treatment did not affect activation of PAR-1 or PAR-2 by their respective synthetic tethered ligand sequences after removal of thermolysin from the bathing solution. The predicted thermolysin cleavage site(s) within SF/L/LRN-NH2 (indicated by /) most likely explains the lack of activity of SFLLRN-NH2 in the presence of thermolysin, since the residues at positions 2 (F) and 5 (R) are both critical for PAR-1 activation (Hollenberg et al., 1992; Scarborough et al., 1992; Vassallo et al., 1992). Interestingly, full activity of SLIGRL-NH2 was observed in the presence of thermolysin despite three predicted cleavage sites within this sequence (S/L/IGR/L). The remaining trimer (IGR), however, caused no relaxation in this preparation (Hamilton & Cocks, unpublished data). It is possible that thermolysin failed to cleave SLIGRL-NH2 due to the proximity of the predicted cleavage sites. Regardless of the reason for the resistance of SLIGRL-NH2 to be cleaved by thermolysin, our results indicate that thermolysin prevented proteolytic, but not proteolysis-independent activation of both PAR-1 and PAR-2, presumably due to amino-terminal cleavage of these receptors at a position distal to their respective activation sites.
Further confirmation of the activity and specificity of thermolysin was obtained by examining its actions on the two non-PAR endothelium-dependent peptide vasodilators, substance P and bradykinin. Thermolysin is predicted to remove the C-terminal methionyl (M) and leucyl (L) residues of substance P (RPLPNNFFG/LM) which are essential for the peptide's peripheral vasodilator activity (Chipkin et al., 1979; Djokic et al., 1989) and hence relaxations to substance P were abolished in the presence of thermolysin. The complete recovery of relaxations to substance P after thermolysin washout is important since it indicates that thermolysin did not affect the function of the neurokinin-1 receptor. By contrast, responses to bradykinin (RPPGFSPFR), which should not be cleaved by thermolysin, were not inhibited either in the presence or after washout of thermolysin. The cause of the small increase in sensitivity to bradykinin after thermolysin washout compared with that in its presence is unknown.
Recovery of responsiveness to trypsin after thermolysin treatment most likely represents trafficking of new, amino-terminal intact receptors to the cell surface since it was abolished by either cycloheximide or brefeldin A. The finding that this recovery process was resistant to actinomycin D may indicate that replenishment of intact PAR-2 was dependent on translation rather than transcription. Also, any spontaneous turnover of PAR-2 was unlikely since none of the inhibitors affected the recovery to trypsin when given alone.
Since actinomycin D failed to block the recovery of the trypsin-induced relaxation, it was necessary to establish that actinomycin D effectively inhibited transcription in porcine coronary artery endothelial cells. The B1-kinin receptor was chosen as an appropriate marker since it mediates endothelium-dependent relaxation and has been reported to be upregulated in human (Drummond & Cocks, 1995a) and cow (Drummond & Cocks, 1995b) coronary arteries and in the cow this upregulation was inhibited by actinomycin D. The present study established that a similar actinomycin D-sensitive upregulation of endothelial B1-kinin receptors coupled to endothelium-dependent relaxation mechanisms occurs in porcine coronary arteries. This inhibition of B1-kinin receptor upregulation by actinomycin D was unlikely to be due to a non-specific decrease in responsiveness to endothelium-dependent dilators, since endothelium-dependent relaxation to the B2-kinin receptor agonist bradykinin was unaffected by similar treatment. In addition, it was unlikely that prior treatment of tissues with thermolysin prevented actinomycin D from blocking transcription since actinomycin D also inhibited the upregulation of responses to des-arg9-bradykinin in tissues previous exposed to thermolysin. Therefore, the rapid recovery of responsiveness of endothelial cell surface PAR-2 following disabling (non-activating) amino-terminal proteolysis with thermolysin, which was not blocked by actinomycin D, was most likely regulated by translation.
Such a mechanism of PAR-2 resensitization differs from that reported by Böhm et al. (1996a) who showed that in transfected kidney epithelial cells, rapid resensitization to PAR-2 was entirely dependent on intracellular stores of pre-formed receptors. Mobilization of intracellular PAR stores is dependent on receptor activation (Shapiro et al., 1996). However, in contrast to the method of Böhm et al. (1996a) we have examined PAR-2 turnover independently of receptor activation. Consequently, we may have uncovered a new mechanism for PAR-2 turnover whereby translation is stimulated by proteolysis of the amino-terminal exodomain alone. Evidence suggests that PAR activation is dependent on the rate of receptor cleavage (Ishii et al., 1993). Therefore, whilst it is possible that the thermolysin-induced PAR turnover is similarly dependent on receptor cleavage rate (and therefore enzyme concentration), this was not investigated in this study.
This activation-independent stimulation of PAR turnover appeared to be specific for PAR-2 since no short-term recovery of responsiveness to thrombin, the enzymic activator of PAR-1, was observed following thermolysin treatment. The eventual return of the thrombin response within 20 h most likely represented the tonic cycling of these receptors described by Shapiro et al. (1996).
In conclusion, the present study provides functional evidence that cleavage of the PAR-2 amino-terminal exodomain, without concomitant receptor activation, triggers rapid recovery of receptor responsiveness, most likely regulated by translation. We propose that such a novel pathway would provide endothelial cells with a remarkable capacity to maintain responsiveness to trypsin and other trypsin-like enzymes, such as the PAR-2-activating mast cell-derived tryptase (Molino et al., 1997), even if they lose their amino-terminal exodomain via non-selective proteolysis.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council (Australia). The authors thank Vitina Sozzi for expert technical assistance.
Abbreviations
- KPSS
high potassium Krebs solution
- PAR
protease-activated receptor
- Rmax
maximum response
- u
unit for enzyme activity
References
- BLACKHART B.D., EMILSSON K., NGUYEN D., TENG W., MARTELLI A.J., NYSTEDT S., SUNDELIN J., SCARBOROUGH R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- BÖHM S.K., KHITIN L.M., GRADY E.F., APONTE G., PAYAN D.G., BUNNETT N.W. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 1996a;271:22003–22016. doi: 10.1074/jbc.271.36.22003. [DOI] [PubMed] [Google Scholar]
- BÖHM S.K., KONG W., BROMME D., SMEEKENS S.P., ANDERSON D.C., CONNOLLY A., KAHN M., NELKEN N.A., COUGHLIN S.R., PAYAN D.G., BUNNETT N.W. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996b;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIPKIN R.E., STEWART J.M., SWEENEY V.E., HARRIS K., WILLIAMS R. In vitro activities of some synthetic substance P analogs. Arch. Int. Pharmacodyn. 1979;240:193–202. [PubMed] [Google Scholar]
- COUGHLIN S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9200–9202. doi: 10.1073/pnas.91.20.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVEY M.G., LUSCHER E.F. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature. 1967;216:857–858. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- DÉRY O., CORVERA C.U., STEINHOFF M., BUNNET N.W. Proteinase-activated receptor: novel mechanisms of signaling by serine proteases. Am. J. Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- DJOKIC T.D., SEKIZAWA K., BORSON D.B., NADEL J.A. Neutral endopeptidase inhibitors potentiate substance P-induced contraction in gut smooth muscle. Am. J. Physiol. 1989;256:G39–G43. doi: 10.1152/ajpgi.1989.256.1.G39. [DOI] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Endothelium-dependent relaxation to the B1 kinin receptor agonist des-Arg9-bradykinin in human coronary arteries. Brit. J. Pharmacol. 1995a;116:3083–3085. doi: 10.1111/j.1476-5381.1995.tb15108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRUMMOND G.R., COCKS T.M. Endothelium-dependent relaxations mediated by inducible B1 and constitutive B2 kinin receptors in the bovine isolated coronary artery. Brit. J. Pharmacol. 1995b;116:2473–2481. doi: 10.1111/j.1476-5381.1995.tb15098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRUMMOMD G.R., COCKS T.M. Evidence for mediation by endothelium-derived hyperpolarizing factor of relaxation to bradykinin in the bovine isolated coronary artery independently of voltage-operated Ca2+ channels. Br. J. Pharmacol. 1996;117:1035–1040. doi: 10.1111/j.1476-5381.1996.tb16693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERSZTEN R.E., CHEN J., ISHII M., ISHII K., WANG L., NANEVICZ T., TURCK C.W., VU T.-K.H., COUGHLIN S.R. Specificity of the thrombin receptor for agonist peptide is defined by its extracellular surface. Nature. 1994;368:648–651. doi: 10.1038/368648a0. [DOI] [PubMed] [Google Scholar]
- HEIN L., ISHII K., COUGHLIN S.R., KOBILKA B.K. Intracellular targetting and trafficking of thrombin receptors. J. Biol., Chem. 1994;269:27719–27726. [PubMed] [Google Scholar]
- HOLLENBERG M.D., YANG S.-G., LANIYONU A.A., SAIFEDDINE M., MOORE G.J. Action of the thrombin receptor polypeptide in gastric smooth muscle: identification of a core pentapeptide retaining full thrombin-mimetic intrinsic activity. Mol. Pharmacol. 1992;42:186–191. [PubMed] [Google Scholar]
- HOXIE J.A., AHUJA M., BELMONTE E., PIZARRO S., PARTON R., BRASS L.F. Internalization and recycling of activated thrombin receptors. J. Biol. Chem. 1993;268:13756–13763. [PubMed] [Google Scholar]
- ISHIHARA H., CONNOLLY A.J., ZENG D., KAHN M.L., ZHENG Y.W., TIMMONS C., TRAM T., COUGHLIN S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- ISHII K., HEIN L., KOBILKA B., COUGHLIN S.R. Kinetics of thrombin receptor cleavage on intact cells: relation to signalling. J. Biol. Chem. 1993;268:9780–9786. [PubMed] [Google Scholar]
- KOVANEN P.T., KAARTINEN M., PAAVONEN T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion of rupture in myocardial infarction. Circulation. 1995;92:1084–1088. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- MITCHINSON M.J., BALL R.Y. Macrophages and atherogenesis. Lancet. 1987;18:146–149. doi: 10.1016/s0140-6736(87)92341-5. [DOI] [PubMed] [Google Scholar]
- MOLINO M., BARNATHAN E.S., NUMEROF R., CLARK J., DREYER M., CUMASHI A., HOXIE J.A., SCHECHTER N., WOOLKALIS M., BRASS L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- MOLINO M., BLANCHARD N., BELMONTE E., TARVER A.P., ABRAMS C., HOXIE J.A., CERLETTI C., BRASS L.F. Proteolysis of the human platelet and endothelial cell thrombin receptor by neutrophil-derived cathepsin G. J. Biol. Chem. 1995;270:11168–11175. doi: 10.1074/jbc.270.19.11168. [DOI] [PubMed] [Google Scholar]
- MOLLE A., MATSUMURA Y., YAMAMOTO T., OKAMURA R., MAEDA H. Pathogenic capacity of proteases from Serratia marcescens and Pseudomonas aeruginosa and their suppression by chicken egg white ovomacroglobulin. Infect. Immun. 1987;55:2509–2517. doi: 10.1128/iai.55.10.2509-2517.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYSTEDT S., EMILSSON K., WAHLESTEDT C., SUNDELIN J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RENESTO P., SI-TAHAR M., MONIATTE M., BALLOY V., VAN DORSSELAER A., PIDARD D., CHIGNARD M. Specific inhibition of thrombin-induced cell activation by the neutrophil proteinases elastase, cathepsin G, and proteinase 3: evidence for distinct cleavage sites within the amino-terminal domain of the thrombin receptor. Blood. 1997;89:1944–1953. [PubMed] [Google Scholar]
- SAIFEDDINE M., AL-ANI B., CHENG C.-H., WANG L., HOLLENBERG M.D. Rat proteinase-activated receptor-2 (PAR-2): cDNA sequence and activity of receptor-derived peptides in gastric and vascular tissue. Br. J. Pharmacol. 1996;118:521–530. doi: 10.1111/j.1476-5381.1996.tb15433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCARBOROUGH R.M., NAUGHTON M.-A., TENG W., HUNG D.T., ROSE J., VU T.-K.H., WHEATON V.I., TUREK C.W., COUGHLIN S.R. Tethered ligand agonist peptides: structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J. Biol. Chem. 1992;267:13146–13149. [PubMed] [Google Scholar]
- SHAPIRO M.J., TREJO J., ZENG D., COUGHLIN S.R. Role of thrombin receptors's cytoplasmic tail in intracellular trafficking. J. Biol. Chem. 1996;271:32874–32860. doi: 10.1074/jbc.271.51.32874. [DOI] [PubMed] [Google Scholar]
- TESFAMARIAM B., ALLEN G.T., NORMANDIN D., ANTONACCIO M.J. Involvement of the ‘tethered ligand' receptor in thrombin-induced endothelium-mediated relaxations. Am. J. Physiol. 1993;265:H1744–H1749. doi: 10.1152/ajpheart.1993.265.5.H1744. [DOI] [PubMed] [Google Scholar]
- VASSALLO R.R. , JR, KIEBER-EMMONS T., CICHOWSKI K., BRASS L.F. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J. Biol. Chem. 1992;267:6081–6085. [PubMed] [Google Scholar]
- VU T.-K.H., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991a;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- VU T.-K.H., WHEATON V.I., HUNG D.T., ISRAEL C., COUGHLIN S.R. Domains specifying thrombin-receptor interaction. Nature. 1991b;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- WELLE M. Development, significance, and heterogeneity of mast cells with particular regard to the mast cell-specific proteases chymase and tryptase. J. Leuk. Biol. 1997;61:233–245. doi: 10.1002/jlb.61.3.233. [DOI] [PubMed] [Google Scholar]
- XU W., ANDERSEN H., WHITMORE T.E., PRESNELL S.R., YEE D.P., CHING A., GILBERT T., DAVIE E.W., FOSTER D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Nat. Acad. Sci. U.S.A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]