

Abstract
This study examined the effects of the COX inhibitors, ketorolac and ibuprofen, and the NOS inhibitor L-NAME for their potential to both inhibit the development and reverse tolerance to the antinociceptive action of morphine.
Repeated administration of intrathecal morphine (15 μg), once daily, resulted in a progressive decline of antinociceptive effect and an increase in the ED50 value in the tailflick and paw pressure tests. Co-administration of ketorolac (30 and 45 μg) or S(+) ibuprofen (10 μg) with morphine (15 μg) prevented the decline of antinociceptive effect and increase in ED50 value. Similar treatment with L-NAME (100 μg) exerted weaker effects. Administration of S(+) but not R(−) ibuprofen (10 mg kg−1) had similar effects on systemic administration of morphine (15 mg kg−1).
Intrathecal or systemic administration of the COX or NOS inhibitors did not alter the baseline responses in either tests. Acute keterolac or S(+) ibuprofen also did not potentiate the acute actions of spinal or systemic morphine, but chronic intrathecal administration of these agents increased the potency of acute morphine.
In animals already tolerant to intrathecal morphine, subsequent administration of ketorolac (30 μg) with morphine (15 μg) partially restored the antinociceptive effect and ED50 value of acute morphine, reflecting the reversal of tolerance. Intrathecal L-NAME (100 μg) exerted a weaker effect.
These data suggest that spinal COX activity, and to a lesser extent NOS activity, contributes to the development and expression of opioid tolerance. Inhibition of COX may represent a useful approach for the prevention as well as reversal of opioid tolerance.
Keywords: Morphine tolerance, cyclo-oxygenase, prostaglandins, nitric oxide, spinal cord, antinociception
Introduction
Morphine and related opioid drugs produce potent analgesia by activating specific receptors associated with spinal and brain neurons involved in nociceptive signalling. Chronic administration of these drugs, however, produces a state of tolerance, indicated by loss of drug potency, and physical dependence, indicated by the appearance of a withdrawal syndrome. In clinical situations, opioid tolerance can result in escalation of drug dose and it can limit the usefulness of opioids in the management of severe pain syndromes. The mechanisms underlying the development of tolerance are complex but they involve neural adaptations that reduce opioid potency and compromise the analgesic response. Currently, this phenomenon is viewed as a cellular adaptation that is mediated by activity of the neuronal N-methyl-D-asparate (NMDA) receptor, a class of excitatory amino acid receptor, since its development can be effectively inhibited by competitive or non-competitive antagonists for this receptor (Trujillo & Akil, 1991; 1994). The central loci at which the NMDA receptor activity mediates this adaptation to chronic opioids have not been fully identified, but the dorsal region of the spinal cord is an important site in this respect. The involvement of spinal NMDA receptor activity in opioid tolerance is suggested by studies showing that co-administration of a non-competitive NMDA receptor antagonist, MK 801, blocks the development of tolerance to the analgesic effect of intrathecal morphine (Dunbar & Yaksh, 1996a; Mao et al., 1994). Since the activation of spinal NMDA receptors in the dorsal horn elicits hyperalgesia (Malmberg & Yaksh, 1992b), an adaptive increase in the activity of these receptors under the chronic opioid exposure would produce a physiological antagonism of opioid analgesia. Thus, agents that interfere with NMDA receptor-mediated hyperalgesia would be expected to prevent the loss of opioid analgesic response.
Several studies have demonstrated that nitric oxide (NO) is an intermediary in the function of NMDA receptors in the brain and spinal cord (Meller & Gebhart, 1993; Garthwaite, 1991). Studies using nitric oxide synthase (NOS) inhibitors such as NG-nitro-L-arginine methyl ester (L-NAME) have demonstrated that NO mediates hyperalgesia induced by the activation of spinal NMDA receptors (Malmberg & Yaksh, 1993b). Thus, the ability of NMDA receptor antagonists to inhibit the development of tolerance may be due, in part, to their ability to prevent the formation of NO. Indeed, recent studies have demonstrated that the development of tolerance to the analgesic effects of systemic morphine can be attenuated by co-administration of L-NAME or NG-nitro-L-arginine (L-NOARG), agents which inhibit NOS activity (Kolesnikov et al., 1992; Majeed et al., 1994). Additionally, repeated administration of L-NOARG has been shown to gradually reverse established tolerance to systemic morphine (Kolesnikov et al., 1993). In contrast, spinal administration of L-NAME has a poor effect on the development of tolerance produced by continuous infusions of intrathecal morphine (Dunbar & Yaksh, 1996b), a finding which argues against the role of spinal NO in the development of spinal tolerance. Thus, alternative messengers of the NMDA receptor might mediate the development of tolerance at the spinal level.
Recent studies demonstrate that prostaglandins mediate the pain behaviours elicited by direct (Malmberg & Yaksh, 1992b) or indirect (Malmberg & Yaksh, 1993a) activation of spinal NMDA receptors. In addition, intrathecal injection of non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit cyclo-oxygenase (COX) and block prostaglandin synthesis, can inhibit hyperalgesia produced by spinal injection of NMDA (Malmberg & Yaksh, 1992b). This suggests that prostaglandins mediate pain behaviours elicited by NMDA receptor activity and COX inhibitors can inhibit these behaviours by functionally antagonizing the NMDA receptor (Malmberg & Yaksh, 1992b). Since spinal NMDA receptors are implicated in tolerance, prostaglandins may mediate the neural adaptation that is expressed through these receptors and results in tolerance. Thus, agents that inhibit prostaglandins production would be expected to block the behavioural manifestations of tolerance. The effects of COX inhibitors in this context have not been examined. As these inhibitors have often been used in combination with opioid drugs (Foley, 1985; Twycross, 1988), a synergy between the acute analgesic effects of COX inhibitors and opioids might be a factor that offsets the development of tolerance. However, the COX inhibitors have the potential to influence opioid tolerance by a mechanism that is more specific than the acute synergy of analgesic action. This possibility is suggested by previous studies demonstrating that classical NMDA receptor antagonists such as MK 801 can impair morphine tolerance in tests of acute thermal nociception without producing an analgesia on their own or demonstrating additivity/synergism with acutely administered morphine (Dunbar & Yaksh, 1996a; Trujillo & Akil, 1994). Thus, as functional antagonists of the spinal NMDA receptors, COX inhibitors would be expected to share this activity profile.
The present study was conducted to investigate the effects of spinally or systemically administered COX inhibitors on analgesic tolerance produced by repeated administration of morphine. An important objective was to compare the effects of COX and NOS inhibition on the development of spinal morphine tolerance and to investigate whether this inhibition has the potential to reverse established tolerance. The effects of ketorolac and S(+) ibuprofen, non-selective COX inhibitors, and L-NAME, a NOS inhibitor, were examined in a model of morphine tolerance.
Methods
Intrathecal catheter implantation and drug injection
Adult male Sprague-Dawley rats (200–250 g) (Charles River, Quebec, Canada) were used in this study. Animals were given free access to food and water and were maintained according to the guidelines of the Canadian Council on Animal Care. Under halothane anaesthesia (4%), animals were implanted with indwelling polyethylene (PE10) catheters (Yaksh & Rudy, 1976). The atlanto-occipital membrane of the cisterna magna was exposed and catheters (7.5 cm) were inserted through a small puncture into the subarachnoid space such that the caudal tip rested on the lumbar enlargement of the spinal cord. The rostral end of the catheter was exteriorized through the skin on the head and the would closed with sutures. Animals were allowed 4 days to recover from surgery and those showing signs of motor dysfunction (e.g. hind limb paralysis) were excluded from experiments. All drugs were injected into the exteriorized portion of the catheter in a volume of 10 μl followed by 10 μl of 0.9% saline to flush the catheter.
Behavioural assessment of nociception
Nociceptive testing was evaluated using two spinal reflex tests; the tailflick and paw pressure tests. Testing was performed before and 30 min after drug administration to determine baseline and drug induced analgesic effects, respectively.
Tailflick test
The tailflick test (D'Amour & Smith, 1941) was used to evaluate the response to a thermal nociceptive stimulus. Radiant heat was applied to the base of the tail using an analgesic meter (Owen et al., 1981) and the time latency for removal of the tail from the stimulus was recorded. The heat source was adjusted to yield a baseline response of 2–3 s and a maximum cut-off time of 10 s was used to prevent tissue damage.
Paw pressure test
The paw pressure test (Loomis et al., 1987) was used to evaluate the response to a mechanical nociceptive stimulus. Pressure was applied to the dorsal surface of the hindpaw using an inverted air-filled syringe connected to a pressure gauge. The pressure was gradually increased until a paw withdrawal response was observed and the value (mmHg) at which this occurred was recorded. A maximum cut-off pressure of 300 mmHg was used. The measurements of tailflick responses preceded the paw pressure responses in each animal and previous experiments have demonstrated no interactions between responses in these two tests (Loomis et al., 1985).
Experimental paradigm
Induction of tolerance to intrathecal morphine
In order to induce spinal morphine tolerance, animals were given 15 μg of morphine intrathecally once daily for 7 days. The injections of morphine were given between 10.00 h and 12.00 h, and nociception tests were performed both before and 30 min after the drug injection, as described above. On the eighth day, cumulative morphine dose-response curves were generated to examine the extent of reduction in morphine potency. To determine these curves, animals were given increasing doses of intrathecal morphine every 30 min and nociceptive testing was performed 25 min after each morphine injection. This procedure was continued until a maximal response was obtained in each nociceptive test. The morphine ED50 value, an indicator of morphine potency, was derived from the constructed dose-response curve for each animal. A state of tolerance was indicated by a progressive decline in antinociception produced by morphine, a rightward shift in the acute morphine dose-response curve, and an increase in the ED50 value of the agonist.
Induction of tolerance to systemic morphine
Tolerance to systemic morphine was induced by administration of a single intraperitoneal dose (15 mg kg−1) of the agonist once daily for 7 days. The effects of ibuprofen isomers on tolerance were investigated by co-administration with morphine. On day 8 cumulative dose-response curves for the acute action of systemic morphine were generated as described above. Morphine ED50 values were calculated from these curves.
Study 1: The effect of cyclo-oxygenase and nitric oxide synthase inhibition on the development of morphine tolerance
To examine the effect of COX and NOS inhibition on the development of morphine tolerance, the COX inhibitor (ketorolac; S(+) and R(−) ibuprofen) or the NOS inhibitor (L-NAME) was co-administered with intrathecal morphine according to the paradigm described above. The ability of drug treatments to influence the development of tolerance was assessed by examining their effect on (a) the decline of morphine-induced antinociception and (b) the potency of morphine as indicated by a shift in the dose-response curves and a change in the ED50 values.
Study 2: The effect of cyclo-oxygenase inhibition on acute intrathecal and systemic morphine
To determine if COX inhibitors influenced the antinociceptive response to acute intrathecal morphine, the agents under study were co-injected with morphine in drug naïve animals. In the case of intrathecal morphine, the action of the COX inhibitors was examined on a single dose of morphine producing a submaximal antinociceptive effect. In the case of systemic morphine, the drug under study or vehicle was administered 20 min prior to determination of a cumulative dose response curve for the opioid agonist. The ED50 values in animals given morphine with and without the COX inhibitor were calculated from these curves.
Study 3: The effect of cyclo-oxygenase and nitric oxide synthase inhibition on established morphine tolerance
To examine the effect of COX and NOS inhibition in animals with established morphine tolerance, ketorolac (30 μg) or L-NAME (100 μg) was administered intrathecally after tolerance to morphine (15 μg) was produced by daily administration over a 5 day period. On day 6, ketorolac or L-NAME was administered either alone or in combination with morphine once daily for 5 days. The cumulative dose-response curves for the acute effects of morphine in each group of animals were derived on day 11. The ability of drug treatments to reverse tolerance was indicated by (a) a recovery of morphine-induced antinociception and (b) a recovery of morphine potency (see above).
Drugs
Morphine sulphate was obtained from BDH Pharmaceuticals (Toronto, Ontario, Canada). Ketorolac tromethamine was a gift from Syntex Ireland Ltd. (Palo Alto, California, U.S.A.), and NG-nitro-L-arginine methyl ester hydrochloride (L-NAME) was obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). S(+) and R(−) ibuprofen were obtained from Sigma Chemical Co (St. Louis, MO, U.S.A.). Ketorolac and L-NAME were dissolved in physiological saline (0.9%) and ibuprofen isomers were dissolved in 5% cyclodextrin.
Data analysis
Tailflick and paw pressure values were converted to a maximum per cent effect (M.P.E.): M.P.E.=100×[post-drug response–baseline response]/[cut-off value–baseline response]. Data are expressed as mean (±s.e.mean) in the figures. The ED50 values were determined using a non-linear regressional analysis (Prizm 2, GraphPad Software Inc.). Statistical significance (P<0.05) was determined using a one-way ANOVA followed by a student Newman-Keuls post hoc test for multiple comparisons between groups. Relative potency values are a ratio of saline ED50 values to drug treatment ED50 values.
Results
Study 1: The effect of cyclo-oxygenase and nitric oxide synthase inhibition on the development of morphine tolerance
Intrathecal morphine
In control animals which received an intrathecal saline injection, the baseline latency in the tailflick test was 1.6±0.1 s, and the threshold pressure to induce a paw withdrawal response was 106±7.5 mmHg. Repeated administration of saline over the 7 day test period did not significantly influence these values (Figure 1a and b). Administration of intrathecal morphine (15 μg) to rats on day 1 produced a maximal analgesic response in both the tailflick and paw pressure tests, respectively (Figure 1a and b). However, daily administration of the drug resulted in a progressive decline of the antinociceptive effect which reached baseline value in both tests at the end of the test period. The administration of 30 and 45 μg ketorolac with morphine significantly inhibited this decline in both nociception tests. In groups receiving these doses of ketorolac with intrathecal morphine, the antinociceptive effects elicited on days 3–7 in the tailflick and paw pressure test were significantly greater than those in the morphine group (tolerant group), an effect that was dose-related (Figure 1a and b). When administered without morphine, ketorolac failed to produce an analgesic effect in either test.
Figure 1.
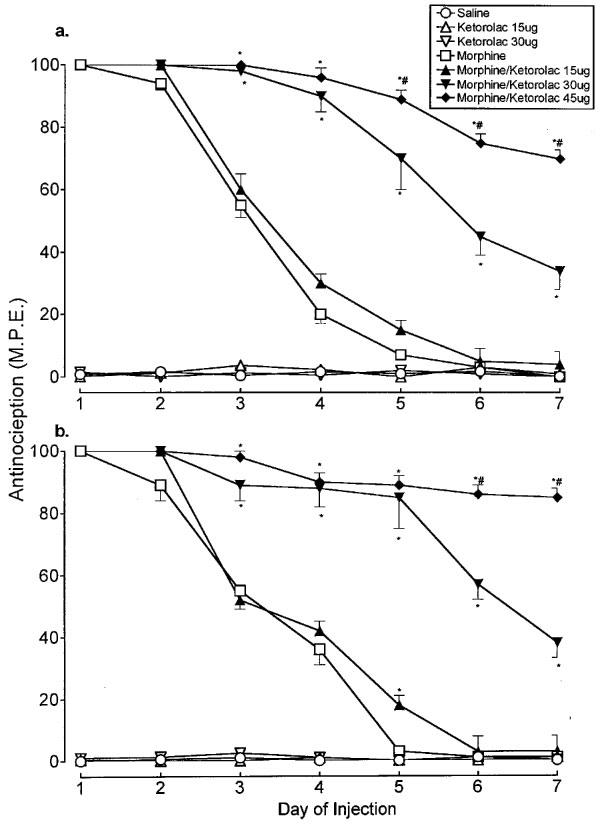
Time course of the antinociceptive effect of daily administration of intrathecal morphine (15 μg) alone and in combination with ketorolac (15, 30 and 45 μg) in the (a) tailflick and (b) paw pressure tests. Morphine and the test agents were administered as a single dose. Nociceptive testing was performed 30 min following each injection. The data are presented as mean±s.e.mean for 5–7 animals. *Significant differences from the action of morphine (P<0.05); #Significant differences from the action of morphine/ketorolac (30 μg) (P<0.05).
Administration of chronic morphine also resulted in the loss of morphine potency, as reflected by an increase in the agonist ED50 value (Table 1). This dose value increased ∼5 fold as a result of 7 day opioid treatment. Co-administration of ketorolac with morphine, depending on the dose used, significantly reduced or blocked the increase in morphine ED50. Indeed, in the group treated with the 45 μg dose of ketorolac and morphine, the ED50 values of acute morphine were comparable to those obtained in the chronic saline treated group. Administration of ketorolac alone for 7 days increased the potency of morphine in the tailflick and paw pressure tests, as reflected by a decrease in the ED50 values of morphine (Table 1). The ED50 values were significantly lower than those in the saline treated group, an effect that was dose related.
Table 1.
Effect of ketorolac and L-NAME on the development of morphine tolerance

Figure 2 shows the effects of intrathecal injections of the ibuprofen isomers on tolerance induced by chronic spinal morphine. In animals treated with the vehicle (5% cyclodextrin) alone for 7 days, the baseline response value, 1.7±0.1 s latency in the tailflick test and 104±7.5 mmHg threshold pressure, did not change significantly over the 7 day administration period (Figure 2a and b). Intrathecal injection of morphine alone (15 μg) on day 1 of the test period produced a maximal analgesic response in both tests which progressively declined to the baseline level by day 7. Co-administration of S(+) ibuprofen (10 μg) with morphine significantly attenuated the decline of the antinociceptive response in the tailflick and paw pressure tests. Similar administration of R(−) ibuprofen (10 μg) also reduced the decline, but to a lesser degree: the responses elicited in the S(+) ibuprofen/morphine group on days 2–7 were significantly greater than corresponding responses in the R(−) ibuprofen/morphine or the morphine (tolerant) group (Figure 2a and b). When administered alone for 7 days, neither ibuprofen isomer produced significant antinociceptive effects.
Figure 2.

Time course of the antinociceptive effect of daily administration of intrathecal morphine (15 μg) alone and in combination with S(+) and R(−) ibuprofen (10 μg) in the (a) tailflick and (b) paw pressure tests. Morphine and the test agents were administered as a single dose. Nociceptive testing was performed 30 min following each injection. The data are presented as mean±s.e.mean for 5–7 animals. *Significant differences from the action of morphine (P<0.05); #Significant differences from the action of morphine/R(−) ibuprofen alone (P<0.05).
The effects of ibuprofen isomers on the potency of acute morphine are represented in Table 2A. In animals treated with the opioid agonist for 7 days the acute ED50 values increased ∼3 and ∼8 fold in the tailflick and paw pressure tests, respectively. Co-treatment with S(+) ibuprofen prevented this increase: the morphine ED50 values in this group, obtained in both tests, were not significantly different from those in the vehicle treated group. In marked contrast, co-treatment with R(−) ibuprofen did not significantly influence the increase in morphine ED50 value; the values in this group of animals were comparable to those in the morphine-treated (tolerant) group and were significantly greater than those in the S(+) ibuprofen/morphine or the vehicle group. Thus, ibuprofen exerted a stereoselective effect on both the decline of the morphine effect and its antinociceptive potency. Additionally, chronic treatment with S(+) ibuprofen, but not R(−) ibuprofen, administered in the absence of morphine for 7 days significantly decreased the ED50 values of acute morphine from those in the vehicle treated group.
Table 2.
Effect of S(+) and R(−) ibuprofen on the development of morphine tolerance

A visual assessment of animals given chronic treatment with ketorolac or ibuprofen isomers, alone or in combination with intrathecal morphine, showed no signs of motor impairment. Two of the seven animals in the group receiving chronic ketorolac (45 μg) with morphine developed signs of irritability and were excluded from experiments.
Systemic morphine
The effects of ibuprofen isomers were also evaluated on the development of tolerance induced by daily administration of systemic morphine (15 mg kg−1 i.p.). The results of these experiments are represented in Figure 3a and b. The injection of the ibuprofen vehicle or S(+) ibuprofen (10 mg kg−1 i.p.) alone for 7 days did not alter the baseline response in the tailflick or paw pressure test. The administration of morphine (15 mg kg−1 i.p.) on day 1 yielded a maximal antinociceptive response in both the tailflick and paw pressure test and this response progressively declined to baseline levels at the end of 7 day test period. Co-administration of S(+) ibuprofen with morphine significantly reduced the decline of morphine-induced antinociception in both the tailflick and paw pressure test, but a similar administration of R(−) ibuprofen failed to attenuate this decline. In the S(+) ibuprofen/morphine group, the antinociceptive responses obtained on days 4–7 in the tailflick test, and on days 6–7 in the paw pressure test, were significantly greater than the corresponding response in the R(−) ibuprofen/morphine group or in the morphine (tolerant) group.
Figure 3.

Time course of the effects of S(+) and R(−) ibuprofen (10 mg kg−1) administered by intraperitoneal injection in 5% cyclodextrin on the antinociceptive response produced by daily administration of morphine (15 mg kg−1) in the (a) tailflick and (b) paw pressure tests. Nociceptive testing was performed 30 min following each injection. The data are presented as mean±s.e.mean for 5–7 animals. *Significant differences from the action of morphine/cyclodextrin (P<0.05); #Significant differences from the action of morphine/R(−) ibuprofen (P<0.05).
The ED50 values of acute systemic morphine obtained in these groups at the end of the 7 day test period are represented in Table 2B. Following chronic morphine treatment, the ED50 values of acute morphine increased ∼8 fold compared to the values obtained with vehicle treatment. In both tests, the co-administration of S(+) ibuprofen with chronic morphine significantly inhibited the increase in morphine ED50 value. Treatment with R(−) ibuprofen also inhibited the increase in morphine ED50 value but to a lesser extent than did the S(+) isomer; the morphine ED50 values in animals receiving the R(−) ibuprofen were significantly greater than that in animals receiving the S(+) isomer. Thus, as was observed in intrathecal experiments, ibuprofen exerted stereoselective effects on systemic morphine tolerance.
Figure 4 shows the effects of L-NAME on the development of tolerance to spinal morphine tolerance. Co-administration of morphine (15 μg) with L-NAME (100 μg) produced a maximal antinociceptive response on day 1, but this response declined to baseline values in both tests (Figure 4a and b). In the paw pressure test, however, the response was significantly greater than that produced by morphine alone on days 3, 4 and 5, but declined to baseline by day 6 (Figure 4b). To determine if the decline in response to morphine could be completely prevented, the opioid agonist was combined with ketorolac (30 μg) and L-NAME (100 μg). The lower dose of ketorolac was chosen as it was thought that few complications would arise than if the higher dose were used (see above). The response produced by morphine/ketorolac/L-NAME combination was maximal on day 1 and significantly greater than that produced by morphine alone on days 2 through 6 (Figure 4a and b). However, the effect of this combination was not significantly different from the morphine/ketorolac combination. Thus, ketorolac/L-NAME combination significantly slowed the decline of morphine effect, but did not completely prevent it.
Figure 4.

Time course of the antinociceptive effect of daily administration of intrathecal morphine (15 μg) alone and in combination with ketorolac (30 μg) or L-NAME (100 μg) in the (a) tailflick and (b) paw pressure tests. Morphine and the test agents were administered as a single dose. Nociceptive testing was performed 30 min following each injection. The data are presented as mean±s.e.mean for 5–7 animals. *Significant differences from the action of morphine (P<0.05); #Significant differences from the action of morphine/L-NAME (P<0.05).
The effects of L-NAME on the potency of acute morphine are presented in Table 1. The ED50 values in the morphine/L-NAME group were significantly lower than those in the morphine alone group, but were also significantly greater than those obtained with saline treatment (Table 1). The effects seen with morphine/ketorolac/L-NAME combination were similar to those produced with morphine/ketorolac treatment.
Study 2: The effect of COX inhibition on acute intrathecal and systemic morphine
The possibility that ketorolac or ibuprofen augmented the acute antinociceptive effect of intrathecal morphine was evaluated by combining these agents with a sub-maximal dose of acute morphine (7.5 μg) and testing their effects in the tailflick and paw pressure tests in drug naïve animals. The results of these tests are represented in Figure 5a and b. When administered at doses which inhibited morphine tolerance (see above), neither ketorolac (30, 45 μg) nor S(+) ibuprofen (10 μg) significantly influenced the effect of morphine (7.5 μg) in the tailflick test (Figure 5a). Administration of S(+) ibuprofen, but not ketorolac, augmented the effect of morphine in the paw pressure test (Figure 5b).
Figure 5.

Effect of ketorolac and ibuprofen isomers on the analgesic response to acute intrathecal morphine in drug naïve animals. Ketorolac and ibuprofen were co-injected with a single dose of intrathecal morphine. The response was determined 30 min after drug injection using the (a) tailflick and (b) paw pressure tests. The data are presented as mean±s.e.mean for 4–5 animals. *Significant differences from the action of morphine/cyclodextrin (P<0.05).
Systemic pretreatment with S(+) ibuprofen (10 mg kg−1) did not produce a shift in the dose-response curve for the acute action of systemic morphine in the tailflick and paw pressure tests. The acute morphine ED50 values in the drug treated animals (6.35±2.7 (tailflick), 6.24±2.3 (paw pressure)) were not different from those in the vehicle pretreated animals, as shown in Table 2B.
Study 3: Effects of ketorolac and L-NAME on established morphine tolerance
The effects of ketorolac (30 μg) and L-NAME (100 μg) on established morphine tolerance in the paw pressure test are illustrated in Figure 6a and b. Although intrathecal injections of morphine (15 μg) were administered for 10 days, the morphine response reached baseline levels on day 5 and remained at this level for the next 5 days, reflecting the development and maintenance of tolerance (Figure 6a). Addition of ketorolac (30 μg) to morphine on days 6 through 10 produced an antinociceptive effect which recovered to 75% of the original response observed on day 1, at the end of the test period (Figure 6a). Similar addition of L-NAME (100 μg) to morphine restored the effects of morphine to 45% of the original value (Figure 6a). However, the antinociceptive response elicited in the ketorolac group on days 6, 7, 8 and 10 was significantly greater than that in the L-NAME group. Thus, intrathecal ketorolac and L-NAME partially restored the antinociceptive effects of morphine in tolerant animals, but ketorolac exerted a stronger effect. Intervention with saline, ketorolac (30 μg) or L-NAME (100 μg) without morphine on days 6 through 10 did not produce a recovery in morphine effect (Figure 6b).
Figure 6.
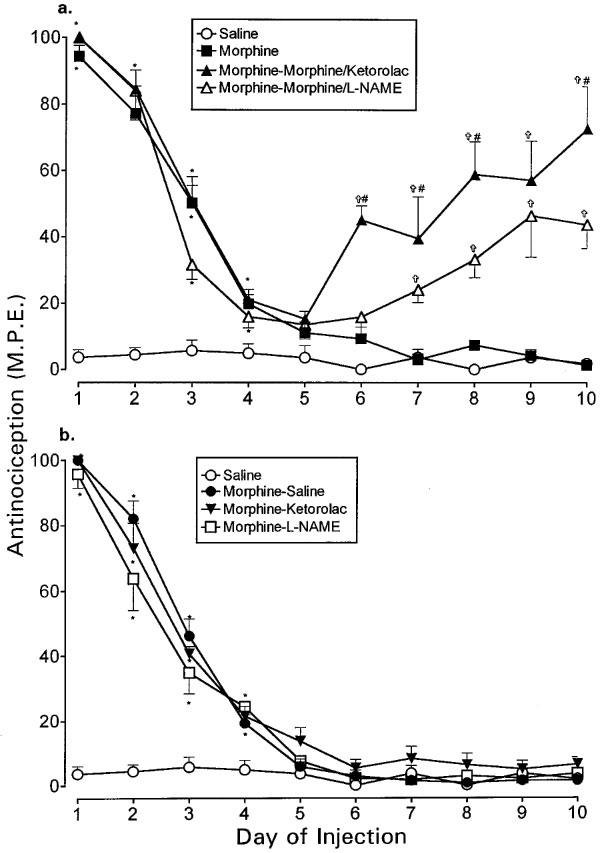
The effects of intrathecal ketorolac (30 μg) and L-NAME (100 μg) on established tolerance to intrathecal morphine in the paw pressure test. Tolerance was induced by administration of single morphine (15 μg) injections from days 1–5. Ketorolac or L-NAME was administered (a) with morphine or (b) without morphine from days 6–10. Morphine and the test agents were given as a single injection followed by nociceptive testing 30 min after each injection. Although not shown here, the tailflick test produced similar results. The data are presented as mean±s.e.mean for 5–7 animals. *Significant differences from the action of saline (P<0.05); †Significant differences from the action of morphine alone (10 days) (P<0.05); #Significantly different from the action of morphine (5 days)-morphine/L-NAME (5 days) (P<0.05).
The ED50 values of acute morphine obtained on day 11 in the paw pressure as well as the tailflick test are represented in Table 3. Chronic administration of intrathecal morphine for 10 days produced a 5.5 fold increase in the ED50 values, reflecting a substantial loss of opioid potency. However, the addition of ketorolac to morphine after 5 days significantly reduced the ED50 values. L-NAME exerted similar but weaker effects on the morphine potency. Animals given saline on days 6–10 still showed an increase in the ED50 values, suggesting the persistence of tolerance despite discontinuation of morphine treatment for 5 days (Table 3). Treatment with ketorolac alone on days 6–10 partially reduced the increase in ED50 value in both tests. L-NAME treatment alone also partially blocked the increase in ED50 values, but only in the tailflick test. Thus, ketorolac and L-NAME partially restored morphine potency with the former exerting a stronger effect.
Table 3.
Effect of ketorolac and L-NAME on the reversal of morphine tolerance

Discussion
The COX inhibitors, ketorolac and ibuprofen, and the NOS inhibitor L-NAME all have the ability to functionally antagonize the activity of the NMDA receptor, which has been implicated in the development of opioid analgesic tolerance. In this study, we sought to determine the effects of ketorolac and ibuprofen on the development and reversal of tolerance to spinal and systemic morphine, and to compare these effects with the action of L-NAME. The results of the present study demonstrated that intrathecal administration of ketorolac or ibuprofen effectively inhibits the decrease in spinal morphine potency. Moreover, systemic administration of ibuprofen with morphine also produced this effect. Additionally, ketorolac partially restored the potency of morphine in animals with established tolerance to spinal morphine. Thus, to the extent that a decrease in opioid potency effects tolerance, treatment with COX inhibitors prevents the development of this phenomenon. However, the actions of L-NAME on the development and reversal of spinal opioid tolerance were significantly weaker than those of the COX inhibitors. These results suggest that spinal prostaglandins and NO influence the development of tolerance, but prostaglandins appear to play a more significant role.
Ketorolac was selected as the test agent as it is a potent, water soluble and relatively non-selective COX inhibitor (Cryer & Feldman, 1998). Additionally, at the dose used in this study, ketorolac was previously shown to inhibit NMDA-induced hyperalgesia (Malmberg & Yaksh, 1992a; 1992b). In order to determine if the effects of ketorolac were related to COX inhibition we used isomers of ibuprofen which exert a differential action on the COX enzyme (Adams et al., 1976; Boneberg et al., 1996). The NOS inhibitor L-NAME was chosen as several different studies have previously demonstrated its ability to inhibit systemic morphine tolerance (Dambisya & Lee, 1996; Majeed et al., 1994; Xu et al., 1998), and the intrathecal dose used in the present study was reported to inhibit NMDA-induced pain behaviours (Malmberg & Yaksh, 1993b) without producing antinociception on its own.
The effect of both COX inhibitors on tolerance was observed at doses that did not elicit antinociception on their own or, when given acutely, produce a generalized potentiation of the morphine action. Thus, the inhibition of tolerance is not likely a result of simple additivity between the acute antinociceptive actions of morphine and ketorolac or ibuprofen. Their action, however, can be attributed to a mechanism which involves COX inhibition as both ketorolac and S(+) ibuprofen, which possess this property (Cryer & Feldman, 1998), impaired tolerance whereas R(−) ibuprofen, a weaker enzyme inhibitor, exerted a lesser effect. Since both agents inhibited the loss of morphine potency when administered intrathecally, we conclude that a decrease in prostaglandin synthesis at spinal sites underlies this effect. Indeed, both ketorolac and S(+) ibuprofen, but not R(−) ibuprofen, have been found to inhibit prostaglandin E2 release from the rat spinal cord (Malmberg & Yaksh, 1994; Sorkin, 1993). Such a mechanism may also explain the inhibition of tolerance to systemic morphine following intraperitoneal injections of ibuprofen although in this case additional involvement of a supraspinal site of action cannot be excluded.
Since previous evidence suggests that prostaglandins act as intermediaries in the expression by hyperalgesia produced by the activity of spinal NMDA receptors (Malmberg & Yaksh, 1992a; 1992b), and this activity also mediates spinal opioid tolerance (Dunbar & Yaksh, 1996a; Mao et al., 1994), we predicted that agents which inhibit prostanoid synthesis would impair the NMDA receptor function and thus inhibit morphine tolerance. This prediction was indeed borne out by the results of the present study: the two COX inhibitors used here mimicked the previously described inhibitory effects of MK 801 on both the spinal (Dunbar & Yaksh, 1996a; Mao et al., 1994) and systemic morphine tolerance (Trujillo & Akil, 1991). The effects of ketorolac and ibuprofen can be interpreted in terms of an inhibition of intracellular NMDA signalling pathways preventing the production of pro-nociceptive prostaglandins which antagonize morphine action. Thus, the NMDA receptor-mediated neural adaptation producing opioid analgesic tolerance may involve an increase in the release or activity of prostaglandins at the spinal sites mediating nociceptive signalling. This proposal is favoured by previous studies on other models (see below) which show that opioids can promote prostanoid activity.
Chronic opioid treatment may augment prostanoid activity by increasing the release of pronociceptive prostaglandins or by increasing sensitivity of the presynaptic prostanoid receptor sites that have been localized on primary afferents signalling nociception in the dorsal horn (Matsumura et al., 1992). Evidence obtained in studies on other models of opioid activity favours both possibilities: (i) opioid receptors in the periaqueductal area, an important site of suprasinal opioid analgesia, are coupled to a potassium conductance via the arachidonic acid cascade and there is evidence that COX activity inhibits opioid function (Vaughan et al., 1997); (ii) opioid agonists stimulate calcium-dependent release of arachidonic acid from the Chinese hamster ovary cells expressing opioid receptors, including the morphine-sensitive mμ receptors (Fukuda et al., 1996); and (iii) chronic exposure to morphine increases the ability of prostaglandin E1 to stimulate adenylate cyclase activity in the human neuroblastoma SH-SY5Y cells bearing opioid receptors (Ammer & Schulz, 1996). This last observation is significant in that acute opioid activity inhibits adenylate cyclase activity (Sharma et al., 1975) but an increase in the prostaglandin receptor sensitivity would antagonize the inhibition of enzyme activity. Indeed, there is a basis for a physiological antagonism between opioids and prostanoids at the level of primary afferent terminals that provide nociceptive input to the spinal cord. These terminals express both opioid and prostanoid receptors which operate in opposite direction: the activity of opioid receptors inhibits whereas that of prostanoid receptors stimulates release of nociceptive transmitters such as substance P and calcitonin gene-related peptide (CGRP) from these terminals (Vasko et al., 1994; Vasko, 1995; Nicol et al., 1992). Since the inhibition of this release partly underlies spinal opioid analgesia (Yaksh et al, 1980), an increase in the release of prostaglandins or receptor sensitivity would augment transmitter release and reduce opioid inhibition. The COX inhibitors, by blocking the prostanoid synthesis, would prevent the loss of this inhibition and preserve the analgesic effect of opioids. The status of spinal prostaglandin release or activity during chronic exposure to opioids is unclear but this merits study in future experiments.
Although acute ketorolac and ibuprofen produced no antinociceptive potentiation of acute morphine, chronic treatment produced a leftward shift in the morphine dose response curve and a decrease in the acute morphine ED50 value relative to vehicle treatment. The magnitude of the decrease in ED50 values COX inhibition produced in morphine naïve animals was comparable to the decrease COX inhibition produced in tolerant animals. Thus, a sensitization of opioid receptors possibly underlies the restoration of morphine potency in tolerant animals. The mechanism underlying this action is not known, but the effect is related to COX inhibition as it showed stereoselectivity in the spinal ibuprofen experiments. Remarkably, in a previous study chronic treatment with MK-801 alone was also found to shift the acute morphine dose-response curve to the left and lower the morphine ED50 value (Dunbar & Yaksh, 1996a). Thus, inhibition of NMDA receptor activity or COX activity appears to sensitize the spinal cord to the antinociceptive action of morphine. The factors that produce this sensitization are not known.
In this study, the development of tolerance to spinal morphine was characterized by both a decline in the magnitude of morphine-induced antinociception and a reduction in morphine potency, as reflected in the ED50 values of acute morphine. The results of this study demonstrated that ketorolac and ibuprofen influenced both indices of tolerance: it significantly attenuated the progressive decline in antinociceptive response and completely inhibited the reduction in morphine potency. However, these agents did not completely eliminate the decline in the morphine antinociceptive response. This suggests that either ketorolac/ibuprofen produced an incomplete inhibition of COX-1 and/or COX-2, or that there are additional mechanisms contributing to the expression of tolerance. We considered that one of these mechanisms might involve NO, as previous studies have proposed a role for NO in the development of systemic morphine tolerance (Kolesnikov et al., 1992; 1993; Majeed et al., 1994). However, Dunbar & Yaksh (1996b) showed that continuous intrathecal infusion of L-NAME with morphine did not substantially prevent the decline in antinociceptive response. The total daily dose of L-NAME they infused intrathecally was 30 times greater than the single dose we administered daily in the present study. In that study, using a single acute dose of morphine, they also demonstrated a modest return of sensitivity to morphine in the morphine/L-NAME group. In this study, involving daily intrathecal administration of the test agent, L-NAME also appeared to partially inhibit morphine tolerance. While L-NAME did not completely inhibit the decline in antinociception, it partially reduced the loss in morphine potency. Thus, our results are consistent with the findings of Dunbar & Yaksh (1996b) that NOS inhibition exerts a modest effect on tolerance at the spinal level.
We reasoned that the addition of L-NAME to ketorolac might completely eliminate the decline in morphine effect. However, when these two agents were combined with morphine the decline in antinociception still persisted. Therefore, it would appear that prostaglandins and NO are not the only factors contributing to the time dependent loss of opioid activity in tolerance. Indeed, recent studies have reported the involvement of CGRP in the development of spinal opioid tolerance (Menard et al., 1995a; 1996). Continuous intrathecal co-infusion of the CGRP receptor antagonist CGRP8–37 with morphine has been shown to attenuate the decline in morphine-induced antinociception and inhibit the loss of opioid potency (Menard et al., 1996). Since CGRP can enhance spinal glutamate and substance P release (Kangrga et al., 1990; Oku et al., 1987), it has potential to indirectly increase the production of prostaglandins and NO. Therefore, CGRP may modulate the development of tolerance partly through the production of prostaglandins or NO, however, the latter is a less likely candidate in this respect (Menard et al., 1995b).
Previous work shows that tolerance to systemic morphine can be reversed by anti-NMDA agents (Elliott et al., 1994; Shimoyama et al., 1996; Tiseo & Inturrisi, 1993). If the accumulation of prostaglandins due to NMDA activity contributes to tolerance, we reasoned that intervention with ketorolac would reverse established tolerance. Indeed, intervention with ketorolac in animals showing loss of opioid potency restored the size of antinociceptive response and morphine potency to 75 and 50% of the original value, respectively, indicating the potential to reverse established tolerance. This recovery of morphine sensitivity is comparable to that previously observed with the NMDA receptor antagonists ketamine, dextromethorphan and LY274614 in animals tolerant to systemic morphine (Elliott et al., 1994; Tiseo & Inturrisi, 1993; Shimoyama et al., 1996). Thus, ketorolac behaves as a functional NMDA receptor antagonist blocking the development of spinal morphine tolerance and reversing established tolerance.
We also examined the effect of L-NAME on established tolerance. This agent partially restored the effect of morphine and its potency after the induction of tolerance, but it was less effective than treatment with ketorolac. This suggests that spinal prostanoids may have a greater role in the expression of tolerance than spinal NO. The differences observed between the effects of NOS inhibition on the development of systemic versus spinal tolerance may be explained by a recent study which identified two NOS splice variants (NOS-1 and NOS-2) that appear to differentially modulate morphine analgesia (Kolesnikov et al., 1997). It has been suggested that NOS-1 is abundant in the brain and its activity antagonizes the actions of morphine, whereas NOS-2 is predominantly expressed in the spinal cord and its activity enhances the analgesic effects of morphine. Thus, intrathecal administration of L-NAME may partly inhibit a NOS form that facilitates the actions of morphine.
In conclusion, the current study suggests that morphine tolerance is mediated by spinal COX activity. Two different isoforms of this enzyme, COX-1 and COX-2, have been identified and are represented in the spinal cord (Willingale et al., 1997). Ketorolac and ibuprofen inhibit both of these forms (Cryer & Feldman, 1998). Thus, it would be of interest to determine the importance of COX-1 and COX-2 in spinal morphine tolerance. Although NOS activity appears to be involved in spinal tolerance, its contribution to the induction and maintenance of tolerance appears to be less than that of spinal COX activity. Thus, pharmacological intervention with COX inhibitors may provide a more attractive approach for the prevention and reversal of opioid tolerance. Given the clinical safety and utility of NSAIDs, which inhibit COX activity, their use with opioids may present a better option for inhibition of clinical tolerance than the use of NMDA receptor antagonist.
Acknowledgments
The authors acknowledge financial support from the Medical Research Council of Canada and the Canadian Anesthetists Society. K.J. Powell was supported by a grant from Ferring. We would like to thank Ms Maaja Sutak for her technical assistance.
Abbreviations
- CGRP
calcitonin gene-related peptide
- COX
cyclo-oxygenase
- L-NAME
NG-nitro-L-arginine methyl ester
- L-NOARG
NG-nitro-L-arginine
- NMDA
N-methyl-D-Aspartate
- NO
nitric oxide
- NOS
nitric oxide synthase
References
- ADAMS S.S., BRESLOFF P., MASON C.G. Pharmacological differences between the optical isomers of ibuprofen: evidence for metabolic inversion of the (−)-isomer. J. Pharm. Pharmacol. 1976;28:256–257. doi: 10.1111/j.2042-7158.1976.tb04144.x. [DOI] [PubMed] [Google Scholar]
- AMMER H., SCHULZ R. Morphine dependence in human neuroblastoma SH-SY5Y cells is associated with adaptive changes in both the quantity and functional interaction of PGE1 receptors and stimulatory G proteins. Brain Res. 1996;707:235–244. doi: 10.1016/0006-8993(95)01265-6. [DOI] [PubMed] [Google Scholar]
- BONEBERG E.M., ZOU M.H., ULLRICH V. Inhibition of cyclooxygenase-1 and -2 by R(−)- and S(+)-ibuprofen. J. Clin. Pharmacol. 1996;36 Suppl:19S. [PubMed] [Google Scholar]
- CRYER B., FELDMAN M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998;104:413–421. doi: 10.1016/s0002-9343(98)00091-6. [DOI] [PubMed] [Google Scholar]
- D'AMOUR F.E., SMITH D.L. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72:74–79. [Google Scholar]
- DAMBISYA Y.M., LEE T.L. Role of nitric oxide in the induction and expression of morphine tolerance and dependence in mice. Br. J. Pharmacol. 1996;117:914–918. doi: 10.1111/j.1476-5381.1996.tb15280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNBAR S., YAKSH T.L. Concurrent spinal infusion of MK801 blocks spinal tolerance and dependence induced by chronic intrathecal morphine in the rat [published erratum appears in Anesthesiology 1996 Sep;85(3):695] Anesthesiology. 1996a;84:1177–1188. doi: 10.1097/00000542-199605000-00020. [DOI] [PubMed] [Google Scholar]
- DUNBAR S., YAKSH T.L. Effect of spinal infusion of L-NAME, a nitric oxide synthase inhibitor, on spinal tolerance and dependence induced by chronic intrathecal morphine in the rat. Neurosci. Lett. 1996b;207:33–36. doi: 10.1016/0304-3940(96)12481-2. [DOI] [PubMed] [Google Scholar]
- ELLIOTT K., HYNANSKY A., INTURRISI C.E. Dextromethorphan attenuates and reverses analgesic tolerance to morphine. Pain. 1994;59:361–368. doi: 10.1016/0304-3959(94)90022-1. [DOI] [PubMed] [Google Scholar]
- FOLEY K.M. The treatment of cancer pain. New England J. Med. 1985;313:84–95. doi: 10.1056/NEJM198507113130205. [DOI] [PubMed] [Google Scholar]
- FUKUDA K., KATO S., MORIKAWA H., SHODA T., MORI K. Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J. Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- GARTHWAITE J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends in Neurosciences. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- KANGRGA I., LAREW J.S., RANDIC M. The effects of substance P and calcitonin gene-related peptide on the efflux of endogenous glutamate and aspartate from the rat spinal dorsal horn in vitro. Neurosci. Lett. 1990;108:155–160. doi: 10.1016/0304-3940(90)90723-m. [DOI] [PubMed] [Google Scholar]
- KOLESNIKOV Y.A., PAN Y.X., BABEY A.M., JAIN S., WILSON R., PASTERNAK G.W. Functionally differentiating two neuronal nitric oxide synthase isoforms through antisense mapping: evidence for opposing NO actions on morphine analgesia and tolerance. Proc. Natl. Acad. Sci. U.S.A. 1997;94:8220–8225. doi: 10.1073/pnas.94.15.8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLESNIKOV Y.A., PICK C.G., CISZEWSKA G., PASTERNAK G.W. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5162–5166. doi: 10.1073/pnas.90.11.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLESNIKOV Y.A., PICK C.G., PASTERNAK G.W. NG-nitro-L-arginine prevents morphine tolerance. Eur. J. Pharmacol. 1992;221:399–400. doi: 10.1016/0014-2999(92)90732-j. [DOI] [PubMed] [Google Scholar]
- LOOMIS C.W., CERVENKO F.W., JHAMANDAS K., SUTAK M., MILNE B. Analgesia and autonomic function following intrathecal administration of morphine and norepinephrine to the rat. Can. J. Physiol. Pharmacol. 1985;63:656–662. doi: 10.1139/y85-109. [DOI] [PubMed] [Google Scholar]
- LOOMIS C.W., MILNE B., CERVENKO F.W. Determination of cross tolerance in rat spinal cord using intrathecal infusion via sequential mini-osmotic pumps. Pharmacol. Biochem. Behav. 1987;26:131–139. doi: 10.1016/0091-3057(87)90545-4. [DOI] [PubMed] [Google Scholar]
- MAJEED N.H., PRZEWLOCKA B., MACHELSKA H., PRZEWLOCKI R. Inhibition of nitric oxide synthase attenuates the development of morphine tolerance and dependence in mice. Neuropharmacology. 1994;33:189–192. doi: 10.1016/0028-3908(94)90006-x. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Antinociceptive actions of spinal nonsteroidal anti-inflammatory agents on the formalin test in the rat. J. Pharmacol. Exp. Ther. 1992a;263:136–146. [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992b;257:1276–1279. doi: 10.1126/science.1381521. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Spinal actions of non-steroidal anti-inflammatory drugs: evidence for a central role of prostanoids in nociceptive processing. Progress in Pharmacology and Clinical Pharmacology. 1993a;10:91–110. [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Spinal nitric oxide synthesis inhibition blocks NMDA-induced thermal hyperalgesia and produces antinociception in the formalin test in rats. Pain. 1993b;54:291–300. doi: 10.1016/0304-3959(93)90028-N. [DOI] [PubMed] [Google Scholar]
- MALMBERG A.B., YAKSH T.L. Capsaicin-evoked prostaglandin E2 release in spinal cord slices: relative effect of cyclooxygenase inhibitors. Eur. J. Pharmacol. 1994;271:293–299. doi: 10.1016/0014-2999(94)90786-2. [DOI] [PubMed] [Google Scholar]
- MAO J., PRICE D.D., MAYER D.J. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J. Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATSUMURA K., WATANABE Y., IMAI-MATSUMURA K., CONNOLLY M., KOYAMA Y., ONOE H. Mapping of prostaglandin E2 binding sites in rat brain using quantitative autoradiography. Brain Res. 1992;582:292–298. doi: 10.1016/0006-8993(92)90720-t. [DOI] [PubMed] [Google Scholar]
- MELLER S.T., GEBHART G.F. Nitric oxide (NO) and nociceptive procesing in the spinal cord. Pain. 1993;52:127–136. doi: 10.1016/0304-3959(93)90124-8. [DOI] [PubMed] [Google Scholar]
- MENARD D.P., VAN ROSSUM D., KAR S., QUIRION R. Alteration of calcitonin gene related peptide and its receptor binding sites during the development of tolerance to mu and delta opioids. Can. J. Physiol. Pharmacol. 1995a;73:1089–1095. doi: 10.1139/y95-156. [DOI] [PubMed] [Google Scholar]
- MENARD D.P., VAN ROSSUM D., KAR S., QUIRION R. Chronic intrathecal treatment with nitric oxide (NO) modulators alters calcitonin-gene-related peptide (CGRP) markers in the rat spinal cord. Soc. Neuroscience. Abst. 1995b;3:155. [Google Scholar]
- MENARD D.P., VAN ROSSUM D., KAR S., ST PIERRE S., SUTAK M., JHAMANDAS K., QUIRION R. A calcitonin gene-related peptide receptor antagonist prevents the development of tolerance to spinal morphine analgesia. J. Neurosci. 1996;16:2342–2351. doi: 10.1523/JNEUROSCI.16-07-02342.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICOL G.D., KLINGBERG D.K., VASKO M.R. Prostaglandin E2 increases calcium conductance and stimulates release of substance P in avian sensory neurons. J. Neurosci. 1992;12:1917–1927. doi: 10.1523/JNEUROSCI.12-05-01917.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKU R., SATOH M., FUJII N., OTAKA A., YAJIMA H., TAKAGI H. Calcitonin gene-related peptide promotes mechanical nociception by potentiating release of substance P from the spinal dorsal horn in rats. Brain Res. 1987;403:350–354. doi: 10.1016/0006-8993(87)90074-6. [DOI] [PubMed] [Google Scholar]
- OWEN J.A., MILNE B., JHAMANDAS K., NAKATSU K. Assembly of an inexpensive tail flick analgesia meter. J. Pharmacol. Methods. 1981;6:33–37. doi: 10.1016/0160-5402(81)90081-4. [DOI] [PubMed] [Google Scholar]
- SHARMA S.K., KLEE W.A., NIRENBERG M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMOYAMA N., SHIMOYAMA M., INTURRISI C.E., ELLIOTT K.J. Ketamine attenuates and reverses morphine tolerance in rodents. Anesthesiology. 1996;85:1357–1366. doi: 10.1097/00000542-199612000-00017. [DOI] [PubMed] [Google Scholar]
- SORKIN L.S.IT Ketorolac blocks NMDA-evoked spinal release of Prostaglandin E2 (PGE2) and Thromboxane B2 (TXB2) Anesthesiology 199379A909(Abstract) [Google Scholar]
- TISEO P.J., INTURRISI C.E. Attenuation and reversal of morphine tolerance by the competitive N-methyl-D-aspartate receptor antagonist, LY274614. J. Pharmacol. Exp. Ther. 1993;264:1090–1096. [PubMed] [Google Scholar]
- TRUJILLO K.A., AKIL H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- TRUJILLO K.A., AKIL H. Inhibition of opiate tolerance by non-competitive N-methyl-D-aspartate receptor antagonists. Brain Res. 1994;633:178–188. doi: 10.1016/0006-8993(94)91538-5. [DOI] [PubMed] [Google Scholar]
- TWYCROSS R.G. Opioid analgesics in cancer pain: current practice and controversies. Cancer Surveys. 1988;7:29–53. [PubMed] [Google Scholar]
- VASKO M.R. Prostaglandin-induced neuropeptide release from spinal cord. Progr. Brain Res. 1995;104:367–380. doi: 10.1016/s0079-6123(08)61801-4. [DOI] [PubMed] [Google Scholar]
- VASKO M.R., CAMPBELL W.B., WAITE K.J. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J. Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAUGHAN C.W., INGRAM S.L., CONNOR M.A., CHRISTIE M.J. How opioids inhibit GABA-mediated neurotransmission [see comments] Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- WILLINGALE H.L., GARDINER N.J., MCLYMONT N., GIBLETT S., GRUBB B.D. Prostanoids synthesized by cyclo-oxygenase isoforms in rat spinal cord and their contribution to the development of neuronal hyperexcitability. Br. J. Pharmacol. 1997;122:1593–1604. doi: 10.1038/sj.bjp.0701548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU J.Y., HILL K.P., BIDLACK J.M. The nitric oxide/cyclic GMP system at the supraspinal site is involved in the development of acute morphine antinociceptive tolerance. J. Pharmacol. Exp. Ther. 1998;284:196–201. [PubMed] [Google Scholar]
- YAKSH T.L., JESSELL T.M., GAMSE R., MUDGE A.W., LEEMAN S.E. Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature. 1980;286:155–157. doi: 10.1038/286155a0. [DOI] [PubMed] [Google Scholar]
- YAKSH T.L., RUDY T.A. Chronic catheterization of the subarachnoid space. Physiol. Behav. 1976;7:1032–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]