

Abstract
Experiments were designed to explore the effects of nitric oxide (NO) donors on generation of superoxide (O2.−) and peroxynitrite (ONOO−) in rabbit aortic rings.
Following inhibition of endogenous superoxide dismutase (SOD), significant basal release of O2.− was revealed (0.9±0.01×10−12 mol min−1 mg−1 tissue). Generation of O2.− increased in a concentration-dependent manner in response to NADH or NADPH (EC50=2.34±1.18×10−4 and 6.21±1.79×10−3 M respectively, n=4). NADH-stimulated O2.− chemiluminescence was reduced by approximately 85% in the presence of exogenous SOD (15×103 U ml−1).
Incubation of aortic rings with S-nitrosoglutathione (GSNO; 1×10−5–3×10−3 M) or sodium nitroprusside (SNP; 1×10−8–1×10−3 M), resulted in a concentration-dependent quenching of O2.− chemiluminescence which was proportional to NO release.
ONOO− formation was assessed indirectly by determining protein tyrosine nitration in rabbit aorta using a specific antibody against nitrotyrosine. Basally and in the presence of NADH, a single band was detected. Incubation of aortic rings with either GSNO (1×10−3 M) alone or GSNO with NADH resulted in the appearance of additional nitrotyrosine bands. Incubation of serum albumin with GSNO alone did not cause nitrotyrosine formation. In contrast, incubation with 3-morpholinosydonomine (SIN-1; 1×10−3 M, 10 min), resulted in marked nitration of albumin which was reduced by oxyhaemoglobin or SOD. Incubation of albumin with GSNO and pyrogallol, a O2.− generator, also resulted in protein nitration.
Addition of exogenous NO results in nitrotyrosine formation in rabbit aortic rings. Nitrotyrosine formation is likely to result from the reaction of exogenous NO and basal endogenous O2.− resulting in the formation of ONOO−. Formation of ONOO− and nitration of tyrosine residues potentially could lead to vascular damage and might represent unexpected adverse effects of long-term nitrate therapy.
Keywords: Superoxide anions, nitric oxide, blood vessels, nitrotyrosine, NO donor, peroxynitrite
Introduction
Blood vessels generate free radical species including nitric oxide (NO) and superoxide anion (O2.−). In the vasculature, the enzymatic sources and biological effects of NO have been well characterized. NO is released in small amounts to regulate local blood flow and inhibit interactions between circulating platelets, white cells and the vessel wall (Moncada et al., 1991). In healthy blood vessels NO is synthesized by a calcium/calmodulin-dependent NO synthase present in endothelium (eNOS; Palmer & Moncada, 1989; Mayer et al., 1989). However, during inflammatory episodes, a cytokine-inducible NO synthase is expressed throughout the vessel wall which results in production of larger quantities of NO (Bogle & Vallance, 1996).
In contrast to the extensively characterized biology of NO, the biosynthetic pathways and roles of O2.− in the vessel wall remain unclear. The source of O2.− may vary and potential generating systems include NADH/NADPH oxidases (Pagano et al., 1993; Jones et al., 1996), NO synthases in the absence of L-arginine (Xia et al., 1996), xanthine oxidase (Miyamoto et al., 1996) or arachidonic acid-metabolising enzymes (Cross & Jones, 1991).
NO and O2.− react rapidly at almost diffusion-limited rate (Huie & Padmaja, 1993) to form peroxynitrite (ONOO−). This product may isomerise to form nitrate, which has little biological activity, and this may provide a mechanism to remove and inactivate both NO and O2.−. However, ONOO− is also a powerful oxidant, which can lead to the generation of other reactive radical species and result in cell damage (see Beckman & Koppenol, 1996 for a review). In addition, ONOO− reacts with proteins resulting in nitration of tyrosine residues and formation of 3-nitrotyrosine (Beckman et al., 1994; Oury et al., 1995). Nitration of tyrosine may alter protein function and initiate cellular damage, and the presence of 3-nitrotyrosine has been used as a marker for ONOO−-mediated tissue damage (Martin et al., 1990; Ohshima et al., 1990; Liu et al., 1994; Van-der-Vliet et al., 1996).
Nitrotyrosine has been detected in human atherosclerotic plaques (Beckman et al., 1994; White et al., 1994), in acute lung (Haddad et al., 1994; Kooy et al., 1995) and myocardial injury (Kooy et al., 1997). Its presence is regarded as an indication of the induction of both NO and O2.− generation. However, in some situations only one of the radical species may be up-regulated e.g. O2.− in diabetes (Giuglianio et al., 1996), or concentrations of NO or O2.− may be altered independently by drugs. For example, levels of NO may increase due to exogenous administration of NO donors.
The aim of this study was (i) to estimate O2.− production from isolated rabbit aortic rings in vitro and identify its possible enzymatic sources and (ii) to test the hypothesis that an increase in NO generated from an NO donor would be sufficient to combine with endogenous O2.− to form ONOO−.
Methods
Preparation of rabbit aortic rings
New Zealand White rabbits (3–4 kg) were sacrificed by injection of sodium pentobarbital (125 mg kg−1) via the lateral ear vein. The thoracic aorta was removed and placed in ice-cold bicarbonate buffer. Vessels were cleaned of surrounding fat and adventitia, cut into 5 mm diameter rings, rinsed with ice-cold bicarbonate buffer solution and stored at 4°C until use.
Measurement and calibration of O2.− production
O2.− production was measured using lucigenin as a chemilumigenic probe (Pagano et al,. 1995; Li et al., 1998b). Aortic rings were equilibrated in oxygenated (95% O2/5% CO2, 37°C) bicarbonate buffer solution for 30 min and placed in a quartz cuvette containing lucigenin (2.5×10−4 M) in a luminometer (Wallac, Model 1250) at 37°C. Chemiluminescence was measured continuously and results expressed in millivolts deflection on a pen recorder. In some experiments endogenous superoxide dismutase (SOD) was inhibited by pre-treatment of rings with diethyldithiocarbamate (DDC; 1×10−2 M; an irreversible inhibitor of copper zinc SOD) for 30 min (Cocco et al., 1981; Kelner et al., 1989). Specificity of lucigenin chemiluminescence was assessed by conducting experiments in the presence of exogenous SOD or 4,5-dihydroxy-1,3-benzene disulfonic acid (tiron), a non-enzymatic O2.− scavenger. In some experiments, substrates for potential O2.− generating enzymes were added to the rings–NADH (1×10−4 M, NADPH (1×10−4 M), xanthine (1×10−3 M), arachidonic acid (1×10−4 M) or succinate (5×10−3 M).
For calibration, and to study the interaction of NO and O2.− in cell-free systems, O2.− was generated using xanthine and xanthine oxidase as described previously (Ohara et al., 1993). Xanthine (1×10−4 M) and lucigenin (2.5×10−4 M) were incubated in HEPES buffer solution (final volume 1 ml) in a quartz cuvette. The production of O2.− was initiated by addition of xanthine oxidase (0.1–1 U ml−1) and chemiluminescence measured as described above. Chemiluminescence signals were calibrated by measuring O2.− generation from xanthine/xanthine oxidase under identical conditions and monitoring the rapid reduction of ferricytochrome c to ferrous cytochrome c by O2.− at 550 nm (Fridovich, 1985). Chemiluminescence signals were converted to O2.− production expressed as nmoles O2.− min−1 mg−1 tissue.
Assay of NO release from NO donors
Release of NO by S-nitrosoglutathione (GSNO) and sodium nitroprusside (SNP) was determined by measurement of the reduction of oxyhaemoglobin to methaemoglobin (Feelisch & Noack, 1987). Oxyhaemoglobin (5×10−6 M) was dissolved in HEPES buffer solution (pH 7.4, 37°C), placed in a quartz cuvette and GSNO was added. The absorbance difference between 401 and 411 nm was measured continuously using a dual-wavelength spectrophotometer (Shimidzu UV-3000). NO production was calculated using an extinction coefficient for oxyhaemoglobin of 16.2 mM−1 cm−1 and a path length of 1 cm according to the following equation (A401–411=[NO]×molar extinction coefficient×path length).
Detection of nitrotyrosine by Western blotting
Rings were homogenized in ice-cold phosphate-buffered saline (pH 7.4) containing Triton X-100 (1% v v−1), phenylmethylsulphonyl fluoride (1×10−3 M), pepstatin A (5×10−5 M) and leupeptin (2×10−4 M) and centrifuged at 10,000×g for 10 min. The supernatant was removed, and mixed with gel loading buffer (1 : 1 v v−1) and boiled for 3 min. The protein content of each sample was determined using Bradford reagent (Bradford, 1976). Equivalent amounts of each sample (50 μg protein) were loaded onto a SDS–PAGE gel (10% SDS) and separated (150 V, 30 mA, 60 min). Proteins were transferred onto a nitrocellulose membrane (0.5 m; Immobilon PVDF transfer membrane, Millipore) for 55 min at 80 V. After transfer, blots were incubated with blocking buffer (5% non-fat dried milk) for 60 min and then with polyclonal rabbit anti-nitrotyrosine antibody (2 μg ml−1) overnight at 4°C. Subsequently, membranes were washed and then incubated with a goat anti-rabbit antibody (peroxidase-linked; 1 : 3000) for 120 min. Secondary antibody binding was detected using diaminobenzidine (0.05% w v−1). To determine the specificity of the anti-nitrotyrosine antibody, the antibody (2 μg ml−1) was incubated with 3-nitro-L-tyrosine (1×10−2 M) overnight.
Nitration of bovine serum albumin
Solutions of bovine serum albumin were prepared (0.7 mg ml−1) in HEPES buffer (pH 7.4). Aliquots (1 ml) were incubated with 3-morpholinosydnonimine (SIN-1; 1×10−3 M), GSNO (1×10−3 M), or combinations of SIN-1 and oxyhaemoglobin (5×10−5 M) or SOD (10–1000 U ml−1) for 10 min at 37°C. In additional experiments bovine serum albumin (dissolved in HEPES buffer, pH 10.8) was incubated with GSNO (1×10−3 M), pyrogallol (2×10−4 M), or GSNO and pyrogallol. Samples (10 μg protein) were loaded onto SDS–PAGE gels, transferred to nylon membranes and blotted for nitrotyrosine as described above.
Materials
Bicarbonate buffer solution was of the following composition (mM): NaCl 118.3, KCl 4.7, MgSO4 0.6, KH2 PO4 1.2, CaCl2 2.5, NaHCO3 25, Na2-EDTA 0.026, and D-glucose 5.5. HEPES buffer solution contained (mM): NaCl 119, HEPES 20, KCl 4.6, MgSO4 1, Na2HPO4 0.15, KH2PO4 0.4, NaHCO3 5, CaCl2 1.2, and glucose 5.5; pH 7.4. Gel loading buffer composed of Tris (5×10−2 M), sodium dodecyl sulphate (10% w v−1), glycerol (10% v v−1), 2-mercaptoethanol (10% v v−1) and bromophenol blue (2 mg ml−1). Antimycin A, Bis-N-methyl acridinium nitrate (lucigenin), leupeptin, diaminobenzadine, NADPH, pyrogallol, SOD (from bovine brain), succinate, 4,5-dihydroxy-1,3-benzene disulphonic acid (tiron), xanthine oxidase (from buttermilk), xanthine, wide range colour molecular weight markers, 3-nitro-L-tyrosine and diethyldithiocarbamate (DDC) were obtained from Sigma (Poole, Dorset, U.K.). SIN-1 was obtained from Alexis Corporation (Nottingham, U.K.). NADH was obtained from Boehringer Mannheim (East Sussex, U.K.). Rabbit polyclonal anti-nitrotyrosine IgG was supplied by TCS Biologicals (Buckingham, U.K.). GSNO was synthesized and recrystallized by Dr D. Madge (Medicinal Chemistry, Wolfson Institute for Biomedical Research). Oxyhaemoglobin was a kind gift of Glaxo Wellcome, Stevenage, Herts, U.K. Nitrocellulose membrane (0.45 μm) was purchased from Millipore U.K. Ltd, Hertfordshire, U.K. Stock solutions of GSNO and SIN-1 were prepared in deionized water immediately before use. Solutions of xanthine oxidase, SOD, tiron, NADH, NADPH, arachidonic acid, antimycin A and lucigenin were prepared in HEPES buffer solution. DDC was prepared in bicarbonate buffer solution. Xanthine was dissolved in NaOH (0.2 M) solution containing EDTA (1×10−3 M) at a final pH of 7.4. Pyrogallol was dissolved in HCl (1×10−2 M).
Data analysis and statistics
Results are shown as the mean±s.e.mean of n experiments. Statistical analysis was performed using unpaired two tailed t-test. P<0.05 was considered statistically significant.
Results
Characterization of O2.− production in rabbit aorta
Under basal conditions O2.− production was not detected from rabbit isolated aortic rings. Addition of DDC (1×10−2 M), an inhibitor of copper-zinc SOD unmasked basal O2.− production which reached 0.9±0.01×10−12 mol min−1 mg−1 tissue (n=4), and was completely abolished in the presence of SOD (150 U ml−1; data not shown). All subsequent O2.− measurements were performed in the presence of DDC (1×10−2 M). Addition of NADH (1×10−5–1×10−2 M) or NADPH (1×10−5–3×10−2 M) resulted in a concentration-dependent increase in O2.− production (Figure 1, Table 1). Calculated EC50 values for NADH and NADPH were 2.34±1.18×10−4 and 6.21±1.79×10−3 M respectively (n=4). NADH (3×10−4 M)-stimulated O2.− chemiluminescence was reduced in the presence of SOD (EC50=8.8 U ml−1 Figure 1, inset, n=4). In the absence of tissue, neither NADH nor NADPH increased lucigenin chemiluminescence. Addition of substrates for xanthine oxidase (xanthine, 1×10−4 M), cyclooxygenase/lipoxygenase (arachidonic acid, 1×10−4 M) or mitochondrial complex II (succinate, 5×10−3 M in the presence of the complex III inhibitor antimycin A (3×10−5 M)) did not alter vascular O2.− production from basal levels (Table 1, n=3).
Figure 1.
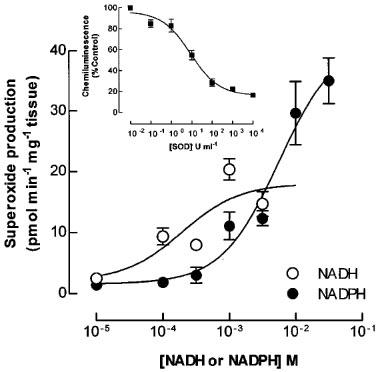
Generation of superoxide by rabbit aortic rings in vitro. Rings of rabbit aorta were placed in a cuvette containing bicarbonate buffer solution and lucigenin. Chemiluminescence was measured in response to increasing concentrations of NADH or NADPH (Inset): Inhibition of NADH stimulated superoxide (O2.−) chemiluminescence by superoxide dismutase (SOD). Results are mean±s.e.mean of data obtained in four experiments.
Table 1.
Substrate-dependence of superoxide production in aortic rings.

Scavenging of O2.− by NO donors
The ability of NO donors to scavenge O2.− was investigated in rabbit aortic rings. Incubation of rings with NADH (3×10−4 M) and increasing concentrations of GSNO (1×10−5–3×10−3 M) or SNP (1×10−8–1×10−3 M) resulted in a significant (P<0.01) reduction of detected O2.− chemiluminescence (Figure 2, n=4). Calculated IC50 values for GSNO and SNP were 3.6±0.1×10−4 and 2.3±0.1×10−6 M respectively.
Figure 2.
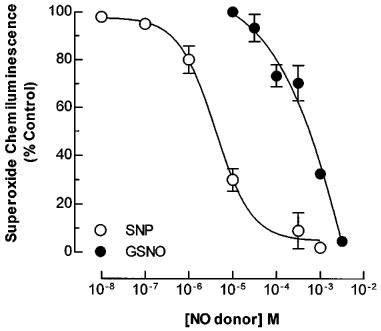
Interaction between nitric oxide and superoxide in rabbit aortic rings. Rings of rabbit aorta were incubated with NADH (3×10−4 M) in the presence of lucigenin and increasing concentrations of S-nitrosoglutathione (GSNO) or sodium nitroprusside (SNP). Superoxide (O2.−) production was measured by chemiluminescence. Results are the mean±s.e.mean of four experiments.
The effects of GSNO on the O2.− chemiluminescence produced by reaction of xanthine and xanthine oxidase were assessed and compared with the release of NO from GSNO. Release of NO by GSNO occurred over a similar concentration range to that which resulted in the quenching of O2.− chemiluminescence (Figure 3). Incubation with reduced glutathione (up to 1×10−3 M), did not affect O2.− detection (data not shown).
Figure 3.
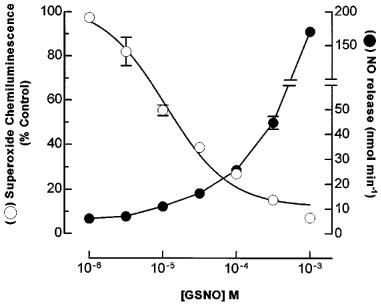
Release of NO and quenching of superoxide by GSNO. Superoxide anions (O2.−) were generated by incubation of xanthine oxidase (0.1 U ml−1) and xanthine (1×10−4 M). The effects of increasing concentrations of S-nitrosoglutathione (GSNO) on O2.− chemiluminescence and release of NO are shown. Results are the mean s.e.mean of four experiments.
Generation of ONOO− assessed by 3-nitrotyrosine formation
Under basal conditions or in the presence of NADH (3×10−4 M) a faint band of nitrotyrosine (corresponding to a molecular weight of approximately 38 kDa) was detected in protein extracts from rabbit aortic rings which may represent a basally nitrated protein. Incubation of rings with GSNO (1×10−3 M, 10 min) alone or GSNO in combination with NADH (3×10−4 M) resulted in the appearance of additional nitrotyrosine bands of which the most prominent had an apparent molecular weight of approximately 30 kDa (Figure 4A). Nitrotyrosine staining was similar whether the tissues were prepared in the presence or absence of DDC. Formation of nitrotyrosine was not inhibited in the presence of SOD (1×104 U ml−1; not shown) or tiron (1×10−2 M, Figure 4A). Specific nitrotyrosine immunoreactivity was not observed when the blot was incubated in the presence of primary antibody and excess 3-nitro-L-tyrosine (1×10−2 M).
Figure 4.
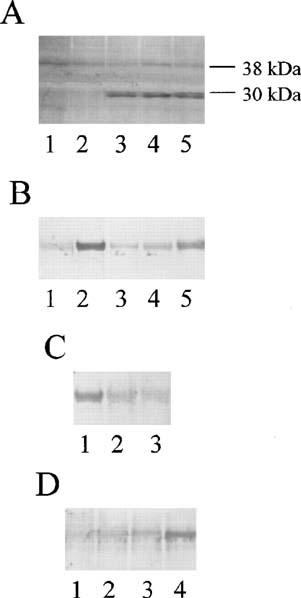
Nitrotyrosine formation by NO donors and superoxide. Peroxynitrite generation was assessed by monitoring nitrotyrosine formation in (A) Rabbit aorta and (B, C and D) serum albumin. (A) 1, control; 2, NADH (3×10−4 M); 3, NADH and GSNO (1×10−3 M); 4, GSNO, (1×10−3 M); 5, GSNO and tiron (1×102 M). (B) Bovine serum albumin was incubated for 10 min with 1, HEPES buffer; 2, SIN-1 (1×10−3 M); 3, SIN-1 and oxyhaemoglobin (5×10−5 M); 4, SIN-1 and SOD (1000 U ml−1); 5, SIN-1, SOD (1000 U ml−1) and GSNO (1×10−3 M). (C) Bovine serum albumin and SIN-1 (1×10−3 M) was incubated for 10 min with 1, SOD (10 U ml−1); 2, SOD (100 U ml−1); 3, SOD (1000 U ml−1). (D) Bovine serum albumin was incubated for 10 min with 1, HEPES buffer; 2, GSNO (1×10−3 M); 3, Pyrogallol (1×10−3 M); 4, GSNO and Pyrogallol. Density of bands are shown above each lane. Results are representative of those obtained in three separate experiments.
The conditions required for tyrosine nitration were examined further using bovine serum albumin as a substrate for nitration. Nitrotyrosine residues were not detected on serum albumin incubated (10 min) with HEPES buffer solution or GSNO alone. Incubation of albumin with SIN-1 (1×10−3 M), a compound which co-generates O2.− and NO (Feelisch et al., 1989), resulted in nitration of albumin (Figure 4B). Treatment of albumin with SIN-1 (1×10−3 M) in the presence of oxyhaemoglobin (5×10−5 M) or a high concentration of SOD (1×103 U ml−1) inhibited nitrotyrosine formation. The effects of SOD to inhibit SIN-1 induced nitrotyrosine formation were concentration-dependent and even at very high concentrations only partial inhibition was possible (Figure 4c). The inhibitory effects of high concentrations of SOD on nitrotyrosine formation were partially reversed in the presence of GSNO (1×10−3 M; Figure 4B). Further experiments were conducted to investigate whether nitrotyrosine formation was dependent on the presence of both NO and O2.−. Incubation of albumin with HEPES buffer, GSNO (1×10−3 M) or pyrogallol (1×10−3 M; a potent donor of O2.−) alone (10 min) did not result in nitrotyrosine formation (Figure 4D) whereas in the presence of GSNO and pyrogallol nitration of albumin occurred (Figure 4D).
Discussion
This study investigated the interaction between O2.− and NO in rabbit aorta. Under normal conditions intracellular O2.− concentrations are kept at low levels because eukaryotic cells contain large amounts of SOD (4–10×10−6 M; Fridovich, 1978), and in the present study endogenous generation of O2.− by aortic rings was not evident unless intrinsic SOD activity was inhibited. Once SOD was inhibited, the amount of O2.− generated in rabbit aorta was in the order of 1 pmol min−1 mg−1 tissue. However, under certain conditions autooxidation of the lucigenin cation radical may result in redox cycling and O2.− generation from lucigenin itself (Li et al., 1998b) and thus the figure of 1 pmol min−1 mg−1 tissue O2.− generation should be considered an estimate rather than precise value. Nonetheless, the results of this and other studies (Pagano et al., 1993; Jones et al., 1996) are compatible with significant vascular generation of O2.− which may greatly exceed the capacity of the endothelium to generate NO (Guo et al., 1996; Kelm et al., 1997).
Potential sources of O2.− in blood vessels include xanthine oxidase, mitochondrial enzymes, cyclooxygenases, NO synthases and enzymes similar to the leukocyte-NADPH oxidase. Addition of NADH or NADPH, but not substrates selective for the other enzyme systems, results in increased O2.− production from aortic rings and this was inhibited in the presence of SOD. In leukocytes, NADPH oxidase is a multi-component enzyme system capable of generating rapidly large amounts of O2.− (nmol min−1) following cellular activation (Cross et al., 1984). In blood vessels and cultured endothelial cells (Jones et al., 1996) NADH/NADPH-oxidase like systems have been identified (Pagano et al., 1993; Mohazzab et al., 1994) and our experiments clearly demonstrate that O2.− generation by aortic rings can be stimulated by NADH or NADPH. We used endothelium-intact aortic rings throughout this study and thus have not assessed the contribution of the endothelium to the measured O2.− release but previous studies suggest that most vascular O2.− may be released from the adventitia (Wang et al., 1998).
The NO donors GSNO and SNP quenched O2.− chemiluminescence originating from either aortic rings stimulated with NADH or from xanthine/xanthine oxidase used as an artificial cell-free O2.− generating system. These effects occurred over the concentration range 1×10−5–3×10−3 M for GSNO and 1×10−8–1×10−3 M for SNP. We determined the relationship between NO release and quenching of O2.− and found that the quantity of NO released from GSNO in this acellular system was directly proportional to the quenching of O2.− chemiluminescence (Figure 3), suggesting that the quenching of the signal occurred as a result of interaction between the two radicals. Quenching of O2.− occurred at relatively low concentrations of SNP and would be expected to occur at therapeutic doses. The apparently greater potency of SNP may be related to its ability to release NO spontaneously without metabolism, whereas GSNO is more stable in solution (Jourd'heuil et al., 1998) unless cells are present. Interestingly, NADH-stimulated O2.− production was not affected by stimulating endogenous NO release from vascular endothelial cells with acetylcholine or inhibition of basal NO release with Nω-nitro-L-arginine methyl ester (C. Amirmansour, unpublished observations). These findings suggest that the amounts of NO generated basally or by agonist stimulation from endothelium may be insufficient to significantly reduce the large amounts of O2.− generated.
In aortic rings no O2.− was detected in the absence of DDC suggesting that metabolism of O2.− by SOD is an important regulator of basal O2.− levels. Addition of GSNO resulted in nitrotyrosine formation and this was not increased by inhibition of SOD. The rapid reaction between NO and O2.− (see later) would result in a relatively small contribution of SOD to the disposal of O2.−. Thus in the presence of a NO donor inhibition of SOD would not be expected to alter peroxynitrite formation and thus tyrosine nitration. Incubation of aortic rings with NADH and GSNO did not increase tyrosine nitration compared to that observed in the presence of GSNO alone. This may reflect the relatively small increase in superoxide production with NADH and the semi-quantitative nature of the Western blotting technique for detection of nitrotyrosine.
The reaction between NO and O2.− results in the formation of ONOO− which is capable of nitrating tyrosine (Beckman et al., 1994) and addition of GSNO to aortic rings resulted in tyrosine nitration, suggesting that ONOO− formation was occurring. However, attempts to scavenge endogenous O2.− with SOD did not prevent the appearance of nitrotyrosine in aortic rings incubated with GSNO. SOD does not cross cell membranes and this may explain why it was unable to reduce nitrotyrosine formation. However, tiron, a relatively weak scavenger of O2.− (1×10−7–5×10−8 M−1 s−1; Greenstock & Miller, 1975; Bors et al., 1979; Mok et al., 1998), but one which is cell permeable, also did not inhibit nitrotyrosine formation. One hypothesis is that GSNO can nitrate tyrosine residues directly, independent of its reaction with O2.−. Alternatively, the reaction between NO and O2.− might be particularly resistant to SOD under certain conditions. To test these hypotheses we used a cell-free system.
Incubation of serum albumin with GSNO did not result in nitrotyrosine formation suggesting that a direct reaction does not occur at least on tyrosine residues on albumin. We examined the dependence of nitrotyrosine formation on O2.− and NO using a O2.− donor, pyrogallol. This agent releases O2.− at alkaline pH in a reproducible manner and using serum albumin as a substrate for nitration we found that GSNO and pyrogallol in combination but not alone resulted in nitrotyrosine formation. These results are consistent with the hypothesis that nitration of albumin is dependent on the presence of both NO and O2.−. Experiments were conducted with SIN-1, a co-donor NO and O2.−. GSNO and SIN-1 at a concentration of 1×10−3 M release similar amounts of NO (1.02 and 1.23×10×6 M respectively; Kelm et al., 1997) but SIN-1 will also liberate O2.− (Feelisch et al., 1989). Oxyhaemoglobin (a scavenger of NO) or SOD (a scavenger of O2.−) inhibited nitrotyrosine formation by SIN-1, suggesting that both NO and O2.− are required for tyrosine nitration. However, whereas oxyhaemoglobin readily inhibited nitrotyrosine formation by SIN-1, high concentrations of SOD (>100 U ml−1) were required to suppress SIN-1-induced nitrotyrosine formation on serum albumin and its IC50 in this system lies somewhere between 100–1000 U ml−1. This is in marked contrast to the calculated IC50 (8.8 U ml−1) for SOD inhibition of NADH-stimulated lucigenin chemiluminescence measured in aortic rings (Figure 1; inset). Furthermore, even in the presence of 1000 U ml−1 of SOD, addition of GSNO to the SIN-1 solution increased nitrotyrosine formation. The reaction between O2.− and NO is effectively diffusion limited (6.7×109 M−1 s−1) and is three times faster than the rate constant for reaction of O2.− and SOD (2×109 M−1 s−1; Huie & Padmaja, 1993). Thus, in a system in which NO, O2.− and SOD are all present, the dominant reaction will be critically dependent on the concentration of each reagent (Figure 5). At low NO concentrations the dismutation reaction between SOD and O2.− would be expected to predominate, but when the NO concentration rises the reaction between NO and O2.− will predominate and SOD will become progressively less able to compete (Figure 5). Thus, the failure of SOD or tiron to prevent GSNO-induced nitrotyrosine formation in the rabbit aorta is most likely to be due to the inability of these O2.− scavengers to compete for O2.− in the presence of the large amounts of O2.− generated basally and the large amounts of NO liberated by the NO-donor. However, we cannot exclude the possibility that GSNO might directly nitrate proteins present in aortic rings, even though it was unable to do so to albumin. The results of this study show that GSNO, a NO donor, leads to nitrotyrosine formation in the vessel wall. This effect occurs because as the concentration of NO rises, O2.− reacts preferentially with NO and SOD cannot compete. Alternatively, it is possible that the SOD is inactivated by the NO or ONOO−. Whichever mechanism is correct, our results suggest that as the concentration of NO rises, the endogenous SOD system is unable to keep oxidant stress under control and this might have implications for pathophysiology. However, these studies were performed in vitro in a haemoglobin-free environment and it remains to be determined whether NO donors cause nitrotyrosine formation in vivo, especially under conditions where oxyhaemoglobin may scavenge NO.
Figure 5.
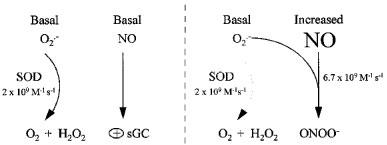
Interactions of NO and superoxide. Formation of peroxynitrite (ONOO−) is dependent on the concentration of superoxide (O2.−) and nitric oxide (NO). When levels of NO rise above a critical level, the reaction between NO and O2.− predominates and superoxide dismutase (SOD) is unable to compete.
The biological significance of protein tyrosine nitration remains unclear. Several reports suggest that nitration may alter the function of important proteins. In murine macrophages, ONOO− nitrates the regulatory subunit of phosphatidylinositol 3-kinase. This protein is involved in the signal transduction cascade initiated by many agonists including growth factors (Helberg et al., 1998). Similarly, in human neuroblastoma cells nitration of cytosolic proteins inhibits phosphoinositide hydrolysis in response to carbachol (Li et al., 1998a). Modification of enzymes such as glutamine synthetase by nitration results in loss of enzyme activity and may alter cellular metabolism (Berlett et al., 1996). Protein nitration may well have marked biological effects on regulatory enzymes and signal transduction pathways. Furthermore, the finding of nitrotyrosine staining in the aorta of patients with atheroma (Beckman et al., 1994) might be due to nitrovasodilator therapy per se rather than inflammatory induction of endogenous NO and O2.− in the vessel wall. Whilst NO donors have been used extensively in the management of ischaemic heart disease evidence suggests that long term treatment with these agents may increase cardiac events, especially in patients with healed myocardial infarction (Ishikawa et al., 1996).
Acknowledgments
We thank Drs Martin Feelisch and Amrita Ahluwalia for helpful advice and discussions. Charles Amirmansour is a recipient of a British Heart Foundation PhD studentship.
Abbreviations
- DDC
Diethyldithiocarbamate
- GSNO
S-nitrosoglutathione
- NO
nitric oxide
- O2.−
superoxide
- ONOO−
peroxynitrite
- SIN-1
3-morpholinosydnonimine
- SNP
sodium nitroprusside
- SOD
superoxide dismutase
References
- BECKMAN J.S., KOPPENOL W.H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., YE Y.Z., ANDERSON P.G., CHEN J., ACCAVITTI M.A., TARPEY M.M., WHITE C.R. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol. Chem. Hoppe Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- BERLETT B.S., FRIGUET B., YIM M.B., CHOCK P.B., STADTMAN E.R. Peroxynitrite-medicated nitration of tyrosine residues in Escherichia coli glutamine synthetase mimics adenylation: relevance to signal transduction. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1776–1780. doi: 10.1073/pnas.93.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOGLE R.G., VALLANCE P.Regulation of vascular smooth muscle tone in sepsis Pharmacology of vascular smooth muscle 1996Oxford Press; 369–386.ed. Garland, C.J. & Argues, J.A. pp [Google Scholar]
- BORS W., SAREN M., MICHEL C. Pulse radiolytic investigation of catechols and catecholamines. II Reactions of tiron with oxygen radical species. Biochim. Biophys. Acta. 1979;582:537–544. doi: 10.1016/0304-4165(79)90145-4. [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- COCCO D., CALABRESE L., RIGO A., ARGESE W., ROTILO G. Re-examination of the reaction of diethyldithiocarbamate with the copper of superoxide dismutase. J. Biol. Chem. 1981;256:8983–8986. [PubMed] [Google Scholar]
- CROSS A.R., JONES O.T.G. Enzymatic mechanisms of superoxide production. Biochim. Biophys. Acta. 1991;1057:291–298. doi: 10.1016/s0005-2728(05)80140-9. [DOI] [PubMed] [Google Scholar]
- CROSS A.R., PARKINSON J.F., JONES O.T.G. The superoxide-generating oxidase of leukocytes-NADPH-dependent reduction of flavin and cytochrome-b in solubilized preparations. Biochem. J. 1984;223:337–344. doi: 10.1042/bj2230337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEELISCH M., NOACK E.A. Nitric oxide formation from nitrovasodilators occurs independently of haemoglobin or non-heme iron. Eur. J. Pharmacol. 1987;129:465–469. doi: 10.1016/0014-2999(87)90090-2. [DOI] [PubMed] [Google Scholar]
- FEELISCH M., OSTROWSKI J., NOACK E. On the mechanism of NO release from sydnonimines. J. Cardiovasc. Pharmacol. 1989;14:S13–S22. [PubMed] [Google Scholar]
- FRIDOVICH I. Superoxide dismutases: defence against endogenous superoxide radical. Ciba Found Symp. 1978;65:77–93. doi: 10.1002/9780470715413.ch6. [DOI] [PubMed] [Google Scholar]
- FRIDOVICH I.CRC Handbook of Methods or Oxygen Radical Research 1985CRC Press Inc, Boca Raton, Florida; 121–122.ed. Greenwald, R. pp [Google Scholar]
- GIUGLIANO D., CERIELLO A., PAOLISSO G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- GUO J.P., MUROHARA T., BUERKE M., SCALIA R., LEFER A.M. Direct measurement of nitric oxide release from vascular endothelial cells. J. Appl. Physiol. 1996;81:774–779. doi: 10.1152/jappl.1996.81.2.774. [DOI] [PubMed] [Google Scholar]
- GREENSTOCK C.L., MILLER R.W. The oxidation of tiron by superoxide anions. Kinetics of the reaction in aqueous solution of chloroplasts. Biochim. Biophys. Acta. 1975;396:11–16. doi: 10.1016/0005-2728(75)90184-x. [DOI] [PubMed] [Google Scholar]
- HADDAD I., PATAKI G., HU P., GALLIANI C., BECKMAN J.S., MATALON S. Quantification of nitrotyrosine levels in lung sections of patients and animals with acute lung injury. J. Clin. Invest. 1994;94:2407–2413. doi: 10.1172/JCI117607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HELLBERG C.B., BOGGS S.E., LAPETINA E.G. Phosphatidylinositol 3-kinase is a target for protein tyrosine nitration. Biochem. Biophys. Res. Commun. 1998;252:313–317. doi: 10.1006/bbrc.1998.9581. [DOI] [PubMed] [Google Scholar]
- HUIE R.E., PADMAJA S. The reaction rate of nitric oxide with superoxide. Free Radical Res. Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- ISHIKAWA K., KANAMASA K., OGAWA I., TAKENAKA T., NAITO T., KAMATA N., YAMAMOTO T., NAKAI S., HAMA J., OYAIZU M., KIMURA A., YAMAMOTO K., ASO N., ARAI M., YANUSHITA H., KATORI Y. Long-term nitrate treatment increases cardiac events in patients with healed myocardial infarction. Secondary Prevention Group. Jpn. Circ. J. 1996;60:779–788. doi: 10.1253/jcj.60.779. [DOI] [PubMed] [Google Scholar]
- JONES S.A., O'DONNELL V.B., WOOD J.D., BROUGHTON J.P., HUGHES E.J., JONES O.T.G. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am. J. Physiol. 1996;271:H1626–H1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- JOURD'HEUIL D., MILLS L., MILES A.M., GRISHAM M.B. Effect of nitric oxide on hemoprotein-catalysed oxidative reactions. Nitric Oxide: Biol. & Chem. 1998;2:37–44. doi: 10.1006/niox.1998.0167. [DOI] [PubMed] [Google Scholar]
- KELNER M.J., BAGNELL R., HALE B., ALEXANDER N.M. Inactivation of intracellular copper-zinc superoxide dismutase by copper chelating agents without gluthathione depletion and methemoglobin formation. Free Radical Biol. Med. 1999;6:355–360. doi: 10.1016/0891-5849(89)90079-8. [DOI] [PubMed] [Google Scholar]
- KELM M., DAHMANN R., WINK D., FEELISCH M. The nitric oxide/superoxide assay. Insights into the biological chemistry of the nitric oxide/superoxide interaction. J. Biol. Chem. 1997;272:9922–9932. doi: 10.1074/jbc.272.15.9922. [DOI] [PubMed] [Google Scholar]
- KOOY N.W., ROYALL J.A., YE Y.Z., KELLY D.R., BECKMAN J.S. Evidence for in vivo peroxynitrite production in human acute lung injury. Am. J. Respir. Crit. Care Med. 1995;151:1250–1254. doi: 10.1164/ajrccm/151.4.1250. [DOI] [PubMed] [Google Scholar]
- KOOY N.W., LEWIS S.J., ROYALL J.A., YE Y.Z., KELLY D.R., BECKMAN J.S. Extensive tyrosine nitration in human myocardial inflammation: evidence for the presence of peroxynitrite. Crit. Care Med. 1997;25:812–819. doi: 10.1097/00003246-199705000-00017. [DOI] [PubMed] [Google Scholar]
- LI X., DE SARNO P., SONG L., BECKMAN J.S., JOPE R.S. Peroxynitrite modulates tyrosine phosphorylation and phosphoinositide signalling in human neuroblastoma SH-SY5Y cells: Attenuated effects in human 1321N1 astrocytoma cells. Biochem. J. 1998a;331:599–606. doi: 10.1042/bj3310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI Y., ZHU H., KUPPUSAMY P., ROUBAUD V., ZWEIER J.L., TRUSH M.A. Validation of lucigenin (Bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular source. J. Biol. Chem. 1998b;273:2015–2023. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- LIU S.Y., BECKMAN J.S., KU D.D. Peroxynitrite, a product of superoxide and nitric oxide, produces coronary vasodilation in dogs. J. Pharmacol. Exp. Therap. 1994;268:1114–1120. [PubMed] [Google Scholar]
- MARTIN B., WU D., JAKES S., GRAVES D.J. Chemical influences on the specificity of tyrosine phosphorylation. J. Biol. Chem. 1990;265:7108–7111. [PubMed] [Google Scholar]
- MAYER B., SCHMIDT K., HUMBERT P., BOHME E. Biosynthesis of endothelium-derived relaxing factor: A cytosolic enzyme in porcine aortic endothelial cells Ca2+-dependently converts L-arginine into an activator of soluble guanylate cyclase. Biochem. Biophys. Res. Commun. 1989;164:678–685. doi: 10.1016/0006-291x(89)91513-1. [DOI] [PubMed] [Google Scholar]
- MIYAMOTO Y., AKAIKE T., YOSHIDA M., GOTO S., HORIE H., MAEDA H. Potentiation of nitric oxide-mediated vasorelaxation by xanthine oxidase inhibitors. Proc. Soc. Exp. Biol. Med. 1996;211:366–373. doi: 10.3181/00379727-211-43982. [DOI] [PubMed] [Google Scholar]
- MOHAZZAB K.M., KAMINSKI P.M., WOLIN M.S. NADH oxidoreductase is a major source of superoxide in bovine coronary artery endothelium. Am. J. Physiol. 1994;266:H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- MOK J.S.L., PAISLEY K., MARTIN W. Inhibition of nitrergic neurotransmission in the bovine retractor penis by an oxidant stress: effects of superoxide dismutase mimetics. Br. J. Pharmacol. 1998;115:993–1000. doi: 10.1038/sj.bjp.0701809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONCADA S., RADOMSKI M.W., HIGGS E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;443:109–142. [PubMed] [Google Scholar]
- OHARA Y., PETERSON T.E., HARRISON D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHSHIMA H., FRIESEN M., BROUET I., BARTSCH H. Nitrotyrosine as a new marker for endogenous nitrosation and nitration of proteins. Food Chem. Toxicol. 1990;28:647–652. doi: 10.1016/0278-6915(90)90173-k. [DOI] [PubMed] [Google Scholar]
- OURY T.D., TATRO L., GHIO A.J., PIANTADOSI C.A. Nitration of tyrosine by hydrogen peroxide and nitrite. Free Radical Res. Commun. 1995;23:537–547. doi: 10.3109/10715769509065275. [DOI] [PubMed] [Google Scholar]
- PAGANO P.J., ITO Y., TORNHEIM K., GALLOP P.M., TAUBER A.I., COHEN R.A. An NADPH oxidase superoxide-generating system in the rabbit aorta. Am. J. Physiol. 1995;268:H2274–H2280. doi: 10.1152/ajpheart.1995.268.6.H2274. [DOI] [PubMed] [Google Scholar]
- PAGANO P.J., TORNHEIM K., COHEN R.A. Superoxide anion production by rabbit thoracic aorta: effect of endothelium-derived nitric oxide. Am. J. Physiol. 1993;265:H707–H712. doi: 10.1152/ajpheart.1993.265.2.H707. [DOI] [PubMed] [Google Scholar]
- PALMER R.M.J., MONCADA S. A novel citrulline-forming enzyme implicated in the formation of nitric oxide by vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989;158:348–352. doi: 10.1016/s0006-291x(89)80219-0. [DOI] [PubMed] [Google Scholar]
- VAN-DER-VLIET A., EISERICH J.P., KAUR H., CROSS C.E., HALLIWELL B. Nitrotyrosine as biomarker for reactive nitrogen species. Methods Enzymol. 1996;269:175–184. doi: 10.1016/s0076-6879(96)69019-3. [DOI] [PubMed] [Google Scholar]
- WANG H.D., PAGANO P.J., DU Y., CAYATTE A.J., QUINN M.T., BRECHER P., COHEN R.A. Superoxide anion from the adventitia of the rat thoracic aorta inactivates nitric oxide. Circ. Res. 1998;82:810–818. doi: 10.1161/01.res.82.7.810. [DOI] [PubMed] [Google Scholar]
- WHITE C.R., BROCK T.A., CHANG L.Y., CRAPO J., BRISCOE P., KU D., BRADLEY W.A., GIANTURCO S.H., GORE J., FREEMAN B.A. Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1044–1048. doi: 10.1073/pnas.91.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIA Y., DAWSON V.L., DAWSON T.M., SNYDER S.H., ZWEIER J.L. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]