

Abstract
Many structurally diverse general anaesthetics enhance inhibitory neurotransmission in the central nervous system by interacting with the GABAA receptor. By contrast, GABA receptors composed of the ρ1 subunit are anaesthetic-insensitive. Here, we demonstrate that both δ-hexachlorocyclohexane (δ-HCH; 1–100 μM), a positive allosteric modulator of the GABAA receptor, and the anaesthetic pentobarbitone (10–600 μM) have no effect on GABA-evoked currents mediated by wild-type ρ1 recombinant receptors (expressed in Xenopus laevis oocytes). By contrast, these agents produce up to a 10 fold enhancement of GABA responses transduced by a ρ1 receptor in which a transmembrane located isoleucine residue is replaced by serine. However, not all general anaesthetics were similarly influenced by this mutation, because propofol and 5β-pregnan-3α-ol-20-one (5β3α) remained ineffective. These data are discussed in relation to the specificity of general anaesthetic action.
Keywords: GABAA receptor, ρ-subunit, general anaesthetic, pentobarbitone, δ-hexachlorocyclohexane
Introduction
Although general anaesthetics have been utilized in clinical practice for over 150 years, the molecular mechanisms by which they cause a rapid depression of central nervous system function remain unknown. The traditional view, held for much of this century, hypothesized that anaesthetics act primarily to perturb neuronal membrane structure. However, a number of recent findings have challenged this concept and have encouraged the assessment of membrane proteins, and transmitter-gated ion channels in particular, as anaesthetic targets (Franks & Lieb 1994). Structurally, general anaesthetics range from chemically inert gases to complex steroidal agents. In view of this chemical diversity, it is surprising that many general anaesthetics used clinically or as experimental agents share, at relevant concentrations, the effect of potentiating the actions of GABA acting at the GABAA receptor (Franks & Lieb, 1994; Belelli et al., 1996a). Although not precluding alternative ion channels as mediators of anaesthetic action, positive allosteric modulation of the GABAA receptor appeals logically as a mechanism by which the various components of the anaesthetic state may occur.
The mammalian GABAA receptor is composed of five subunits drawn from the products of a multigene family (α1–6, β1–3, γ1–3, δ, ε and π) that exhibit a distinct distribution within the CNS (Barnard et al., 1998). The identification of recombinant GABA receptors that respond differentially to general anaesthetics may aid the identification of protein domains which are essential for anaesthetic activity. Utilizing this strategy, our own studies and those of others have recently identified a single amino acid residue located in the second transmembrane region (TM2) of GABAA, glycine and invertebrate GABA (RDL) receptors that affects the anaesthetic pharmacology of these inhibitory amino acid receptors (Belelli et al., 1997; Mihic et al., 1997; Krasowski et al., 1998; McGurk et al., 1998; Pistis et al., 1999). Of particular interest are the pharmacological properties of homo-oligomeric GABA receptors formed from the ρ subunit (ρ1–3). In contrast to GABAA receptors, homo-oligomeric ρ1 GABA receptors are not positively modulated by benzodiazepines, barbiturates, steroids or volatile anaesthetic agents (Shimada et al., 1992; Mihic & Harris, 1996). Here, we report that the barbiturate pharmacology of the ρ1 GABA receptor is governed by the nature of a single amino acid which occupies a position homologous to that identified in related inhibitory amino acid receptors. Pentobarbitone, acting at ρ1 GABA receptors in which a transmembrane located isoleucine residue (wild type) is mutated to serine (the homologous amino acid in GABAA receptor α1–6, β1 and γ1–3 subunits) now produced a large concentration-dependent enhancement of GABA-evoked responses. Whether this amino acid contributes directly to a binding pocket for the anaesthetic on the receptor protein, or alternatively influences transduction of anaesthetic binding, is discussed.
Methods
The cDNA encoding the human ρ1 GABA subunit contained within the eukaryotic expression vector pcDNA3 (InVitrogen; CA, U.S.A.), under the control of the cytomegalovirus (CMV) promoter was kindly provided by Dr D. Weiss. For site-directed mutagenesis, single stranded template cDNAs were synthesized from the M13 origin of replication and mutation of the isoleucine residue at position 307 to serine (ρ1I307S) was generated using standard procedures (Kunkel et al., 1987). Briefly, the following oligonucleotide encoding the mutated sequence was used to prime the mutagenesis reaction: 5′ - CACGCCCGTGCTGATGGTGGACATGGTTAGCAC-CGTTG-3′. The mutated codon is highlighted in bold whereas the underlined sequence represents a silent mutation removing a Th111I restriction site to facilitate the rapid screening of mutants by restriction analysis. The fidelity of the mutagenesis reaction was confirmed by standard dideoxynucleotide sequencing (fmol DNA Sequencing System Promega, Southampton, U.K.) of both wild type and mutated ρ1 cDNAs.
cDNA (for either the ρ1 or mutated ρ1 subunit ρ1I307S) was injected (20 nl of 0.2 mg ml−1) into Xenopus laevis oocytes (Stage V–VI) defolliculated by pre-treatment with collagenase (see Belelli et al., 1996a, for details). The cDNA was injected intranuclearly using the ‘blind method' (Colman, 1984). Injected oocytes were individually maintained for up to 12 days as previously described and used for experimentation 2–12 days after cDNA injection. Oocytes were held in a recording chamber (0.5 ml) and continuously superfused (7–10 ml min−1) with frog Ringer solution (composition in mM: NaCl 120, KCl 2, CaCl2 1.8, N-2-Hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) 5; adjusted to pH 7.4 with NaOH). Electrical recordings were made from oocytes voltage-clamped at a holding potential of −60 mV using a Gene Clamp 500 amplifier (Axon Instruments, U.S.A.) in the two-electrode voltage-clamp mode. Voltage-sensing and current-passing electrodes were filled with 3 M KCl and had resistances of 1–2 MΩ when measured in frog Ringer solution. Current signals were analysed using the Win WCP program, a Dell pentium computer and an Axon 1200 Digidata A to D converter (Dempster, 1997; Mair et al., 1998). Timed pulses of drugs dissolved in Ringer solution were applied to oocytes via a BPS-4 bath perfusion system (Adams & List Associates, New York) with a four-way manifold. Drug application was computer controlled (see Mair et al., 1998). In all experiments investigating potentiation by modulators, a concentration of GABA which produced a response 10% of the GABA maximum (EC10) was utilized, whereas the inhibitory actions of propofol and 5β3α were assessed against the GABA EC50. The GABA EC10 or EC50 was determined for each oocyte. Concentration-response data for GABA and the enhancement of GABA responses by pentobarbitone and δ-HCH were fitted iteratively with the Hill equation as previously described (McGurk et al., 1998). From such fits, for both pentobarbitone and δ-HCH, the Emax (the amplitude of the response to a GABA EC10 concentration in the presence of a maximally effective concentration of the modulator which is expressed as a percentage of the maximum response to GABA) and the EC50 (the concentration of modulator required to produce a response 50% of the Emax) were derived. Quantitative results are expressed as the arithmetic mean±s.e.mean. Statistical significance of the difference between agonist-evoked response in the absence and presence of antagonists in wild type and mutant receptors was determined by repeated measures ANOVA followed by Newman-Keuls test when warranted. Drugs were obtained from the following sources: pentobarbitone, 5β3α, δ-HCH (Sigma), propofol (Aldrich).
Results
Concentrations of pentobarbitone (10–300 μM) known to produce a large enhancement of GABAA receptor mediated responses (e.g. Belelli et al., 1996a) had no effect on the GABA (EC10)-evoked current recorded from oocytes expressing the wild-type ρ receptor (Figure 1A and B). However, the washout of high concentrations (600 μM–3 mM) of the anaesthetic was associated with the development of a rebound current (Figure 1B). Such currents have also been reported for GABAA receptors (e.g. Wooltorton et al., 1997). The replacement by serine of isoleucine residue 307, located within the second transmembrane (TM2) domain of the ρ1 GABA receptor, (ρ1I307S) produced a 4 fold reduction of the EC50 for GABA from 0.8±0.02 μM (nH=2.4±0.2; n=5) to 0.2±0.01 μM (nH=3.0±0.2; n=5). However, this mutation produced a more fundamental change upon the effect of pentobarbitone. Acting at the ρ1I307S GABA receptor, pentobarbitone produced a concentration-dependent (10–600 μM) enhancement of the GABA (EC10)-evoked current with a calculated EC50 of 226±38 μM (n=4) and an Emax at 600 μM of 99±4% (Figure 1A and C). Hence, concentrations of the anaesthetic that were inert at the wild type receptor produced up to a 10 fold enhancement of the GABA-evoked response of the mutant receptor. At relatively high concentrations, pentobarbitone can directly activate the GABAA receptor (e.g. Belelli et al., 1996a). However the barbiturate (10 μM–3 mM) did not activate either wild type or mutant ρ1 receptors. The ρ1I307S mutation did not influence the actions of other, structurally distinct, anaesthetics.
Figure 1.
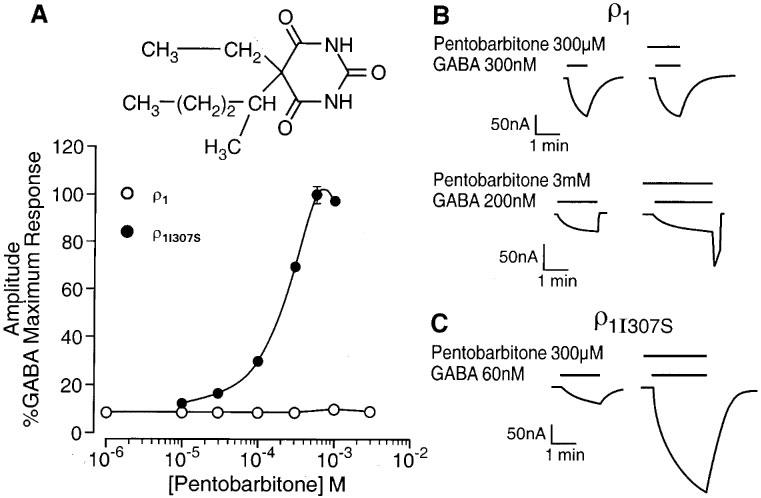
A single transmembrane amino acid governs pentobarbitone sensitivity of the GABA ρ1 receptor. (A) The graph depicts the relationship between the concentration of pentobarbitone (logarithmic scale) and the peak amplitude of the GABA-evoked current (on a linear scale and expressed as percentage of the response to a maximally effective concentration of GABA) at human ρ1 and ρ1I307S GABA receptors. (B) Pentobarbitone (300 μM or 3 mM) has no effect on the GABA (300 or 200 nM respectively)-evoked currents recorded from oocytes expressing human ρ1 receptors. However, upon washout of pentobarbitone 3 mM and GABA 200 nM, a transient inward current (‘rebound') developed. (C) GABA (60 nM)-evoked currents are greatly enhanced by 300 μM pentobarbitone for oocytes expressing human ρ1I307S receptors. The insert shows the chemical structure of pentobarbitone.
We have previously demonstrated that relatively low concentrations (3 nM–1 μM) of the anaesthetic neurosteroid 5β3α potentiate the response to GABA mediated by GABAA receptors (Belelli et al., 1996b). By contrast, 5β3α (0.1–1 μM) caused a modest but significant inhibition (P<0.05 vs control response in the absence of 5β3α) of both wild type and mutant GABA ρ1 receptor, with 1 μM 5β3α reducing the GABA (EC50)-evoked current to 77±6 and 76±2% of control (n=4) respectively. Similarly, concentrations of propofol (3–30 μM) known to greatly enhance GABAA receptor mediated responses (Belelli et al., 1996a), produced a limited but significant depression (P<0.05 vs control response in the absence of propofol) of both wild type and mutant GABA ρ1 receptors, with 30 μM of the anaesthetic reducing the GABA (EC50)-evoked current to 86±5 and 83±1% of control (n=3) respectively. However, neither 5β3α nor propofol inhibition of the GABAA-evoked response was significantly different (P>0.05) between wild type and mutant ρ receptors. The insecticide lindane (γ-hexachlorocyclohexane) is a non-competitive antagonist of GABAA receptors, whereas the δ-isomer (δ-HCH) is a potent positive allosteric modulator (Woodward et al., 1992; Belelli et al., 1996a). δ-HCH (0.1–100 μM) had no effect on GABA (EC10)-evoked currents recorded from oocytes expressing the wild type ρ1 receptor (n=4; Figure 2A and B). However, δ-HCH (1–100 μM) greatly enhanced GABA-induced responses mediated by the mutant ρ1I307S receptor (EC50=38±2 μM; Emax=94±8%; n=4; see Figure 2C). Hence, as found for pentobarbitone, this single amino acid mutation has revealed a potent, positive allosteric action of δ-HCH.
Figure 2.
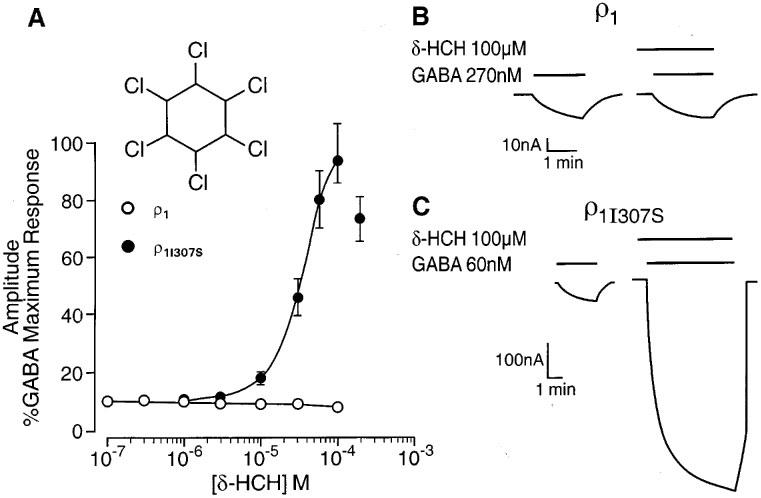
A single transmembrane amino acid governs δ-HCH sensitivity of the GABA ρ1 receptor. (A) The graph depicts the relationship between the concentration of δ-HCH (logarithmic scale) and the peak amplitude of the GABA-evoked current (on a linear scale and expressed as percentage of the response to a maximally effective concentration of GABA) at human ρ1 and ρ1I307S GABA receptors. (B) δ-HCH (100 μM) has no effect on the GABA (270 nM)-evoked current recorded from oocytes expressing ρ1 receptors. (C) GABA (60 nM)-evoked currents are greatly enhanced by 100 μM δ-HCH at oocytes expressing human ρ1I307S receptors. The insert shows the chemical structure of δ-HCH.
Discussion
In contrast to GABAA receptors, homomeric GABA ρ receptors are insensitive to the positive allosteric actions of benzodiazepines and a number of structurally diverse general anaesthetics including barbiturates, steroids, propofol and isoflurane (Shimada et al., 1992; Mihic & Harris, 1996). The cardinal finding of the present study is that the mutation of a single amino acid from isoleucine (wild type) to serine (the homologous amino acid for α1–6, γ1–3 and β1, GABAA receptor subunits) makes the GABA ρ1 receptor barbiturate-sensitive, such that concentrations of pentobarbitone that had little effect on the wild type recombinant receptor, now produce up to a 10 fold enhancement of the GABA response mediated by the mutant receptor. However, not all general anaesthetics were similarly influenced, as propofol and 5β3α caused modest inhibition of both the wild type and mutant receptors. This study does not address whether the identified amino acid participates directly in the binding of pentobarbitone to the receptor protein, or alternatively influences the transduction of the allosteric actions of the anaesthetic. However, the effects of the mutation were not exclusive to the barbiturate, as δ-HCH also enhanced GABA function at mutant, but not wild type ρ1 receptors. As pentobarbitone and δ-HCH exhibit little structural similarity, it is difficult to conceive that these compounds bind to a common site. Alternatively, the wild type ρ1 receptor may have distinct binding sites for pentobarbitone and δ-HCH, but only in the mutant receptor does their binding enhance GABA receptor function. The mutation also increased the apparent affinity of GABA for the receptor. The proposed transmembrane location of this amino acid (Wick et al., 1998) is difficult to reconcile with the residue contributing to the GABA recognition site directly (Amin & Weiss, 1994), but again favours the mutation modifying the transduction properties of the receptor. Irrespective of whether the identified amino acid participates directly in anaesthetic binding, modifies transduction, or both, it is evident that the anaesthetic pharmacology of GABAA, glycine, invertebrate GABA (RDL) receptors and now ρ GABA receptors is influenced by the nature of the amino acid that occupies the homologous position in these related receptors. Finally, the distinct agonist and antagonist pharmacology of GABA ρ receptors, coupled with their insensitivity to benzodiazepines and a variety of general anaesthetics, has led to them being classified as GABAC receptors (Shimada et al., 1992). However, the present demonstration, that at least part of this distinct pharmacology may be dependent upon a single amino acid warrants caution in such a categorization, and supports the proposal that these ionotropic receptors should, as suggested by sequence homology, be considered as a ‘specialized set of GABAA receptors' (Barnard et al., 1998).
Acknowledgments
We thank Dr D. Weiss for supplying the ρ1 cDNA. Supported by an EC Biomedicine & Health Grant BMH4-CT97-2359.
Abbreviations
- δ-HCH
δ-hexachlorocyclohexane
- EC10
the concentration of GABA which produced a response 10% of the GABA maximum
- Emax
the amplitude of the response evoked by GABA at EC10 in the presence of a maximally effective concentration of the modulator
- HEPES
N-2-Hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- 5β3α
5β-pregnan-3α-ol-20-one
References
- AMIN J., WEISS D.S. Homomeric ρ1 GABA channels: activation properties and domains. Receptors Channels. 1994;2:227–236. [PubMed] [Google Scholar]
- BARNARD E.A., SKOLNICK P., OLSEN R.W., MOHLER H., SIEGHART W., BIGGIO G., BRAESTRUP C., BATESON A.N., LANGER S.Z. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acid A receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- BELELLI D., CALLACHAN H., HILL VENNING C., PETERS J.A., LAMBERT J.J. Interaction of positive allosteric modulators with human and Drosophila recombinant GABA receptors expressed in Xenopus laevis oocytes. Br. J. Pharmacol. 1996a;118:563–576. doi: 10.1111/j.1476-5381.1996.tb15439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELELLI D., LAMBERT J.J., PETERS J.A., GEE K.W., LAN N.C. Modulation of human recombinant GABAA receptors by pregnanediols. Neuropharmacology. 1996b;35:1223–1231. doi: 10.1016/s0028-3908(96)00066-4. [DOI] [PubMed] [Google Scholar]
- BELELLI D., LAMBERT J.J., PETERS J.A., WAFFORD K., WHITING P.J. The interaction of the general anesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLMAN A.Expression of exogenous DNA in Xenopus oocytes Transcription and translation: A Practical approach 1984IRL Press Ltd, Oxford; 49–68.eds. Hames, B.D. & Higgins, S.J. pp [Google Scholar]
- DEMPSTER J. A new version of the Strathclyde Electrophysiology Software package running within the Windows environment. J. Physiol. 1997;504:P57. [Google Scholar]
- FRANKS N.P., LIEB W.R. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- KRASOWSKI M.D., KOLTCHINE V.V., RICK C.E., YE Q., FINN S.E., HARRISON N.L. Propofol and other intravenous anesthetics have sites of action on the γ-aminobutyric acid type A receptor distinct from that for isoflurane. Mol. Pharmacol. 1998;53:530–538. doi: 10.1124/mol.53.3.530. [DOI] [PubMed] [Google Scholar]
- KUNKEL T.A., ROBERTS J.D., ZAKOUR R.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- MAIR I.D., LAMBERT J.J., YANG J., DEMPSTER J., PETERS J.A. Pharmacological characterisation of a rat 5-hydroxytryptamine type3 receptor subunit (r5-HT3A(b)) expressed in Xenopus laevis oocytes. Br. J. Pharmacol. 1998;124:1667–1674. doi: 10.1038/sj.bjp.0702037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGURK K.A., PISTIS M., BELELLI D., HOPE A.G., LAMBERT J.J. The effect of a transmembrane amino acid on etomidate sensitivity of an invertebrate GABA receptor. Br. J. Pharmacol. 1998;124:13–20. doi: 10.1038/sj.bjp.0701787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIHIC S.J., HARRIS R.A. Inhibition of ρ1 receptor GABA-ergic currents by alcohols and volatile anesthetics. J. Pharmacol. Exp. Ther. 1996;277:411–416. [PubMed] [Google Scholar]
- MIHIC S.J., YE Q., WICK M.J., KOLTCHINE V.V., KRASOWSKI M.D., FINN S.E., MASCIA M.P., VALENZUELA C.F., HANSON K.K., GREENBLATT E.P., HARRIS R.A., HARRISON N.L. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- PISTIS M., BELELLI D., MCGURK K., PETERS J.A., LAMBERT J.J. Complementary regulation of anaesthetic activation of human (α6β3γ2L) and Drosophila (RDL) GABA receptors by a single amino acid residue. J. Physiol. 1999;515:3–18. doi: 10.1111/j.1469-7793.1999.003ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMADA S., CUTTING G., UHL G.R. γ-Aminobutyric acid A or C receptor? γ-Aminobutyric acid ρ1 receptor RNA induces bicuculline-, barbiturate-, and benzodiazepine-insensitive gamma-aminobutyric acid responses in Xenopus oocytes. Mol. Pharmacol. 1992;41:683–687. [PubMed] [Google Scholar]
- WICK M.J., MIHIC S.J., UENO S., MASCIA M.P., TRUDELL J.R., BROZOWSKI S.J., YE Q., HARRISON N.L., HARRIS R.A. Mutations of γ-aminobutyric acid and glycine receptors change alcohol cutoff: evidence for an alcohol receptor. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6504–6509. doi: 10.1073/pnas.95.11.6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOLTORTON J.R.A., MOSS S.J., SMART T.G. Pharmacological and physiological characterization of murine homomeric β3 GABAA receptor. Eur. J. Neurosci. 1997;9:2225–2235. doi: 10.1111/j.1460-9568.1997.tb01641.x. [DOI] [PubMed] [Google Scholar]
- WOODWARD R.M., POLENZANI L., MILEDI R. Effects of hexachlorocyclohexanes on γ-aminobutyric acid receptors expressed in Xenopus oocytes by RNA from mammalian brain and retina. Mol. Pharmacol. 1992;41:1107–1115. [PubMed] [Google Scholar]