

Abstract
In the process of cloning the human P2Y2 receptor in order to establish 1321N1 cell lines expressing this receptor, we detected a gene polymorphism characterized by an arginine 334 to cysteine 334 transition.
The frequency distribution of the polymorphism was studied in a European population. We observed that 66% of the tested persons are homozygotes R/R, 29% are heterozygotes R/C and 5% are homozygotes C/C. The frequency of the R allele was 0.8 versus 0.2 for the C allele.
We stably expressed each form of the human P2Y2 receptor into 1321N1 cells and isolated clones by limiting dilution. The effects of nucleotides and antagonists on inositol trisphosphate accumulation and cyclic AMP formation were compared between the two cell lines.
The time-courses of inositol trisphosphate accumulation as well as concentration-response curves characterizing the effects of UTP, ATP, AP4A and ATPγS were mostly similar, except for slight kinetic differences (slower time-course with the 334C form).
The sensitivity to pertussis toxin of inositol trisphosphates accumulation was critically dependent on the agonist concentration and stimulation duration, suggesting the involvement of a Gi,0 protein during the early stimulation by low nucleotide concentrations. No inhibition of cyclic AMP accumulation could be detected. These properties were observed with both polymorphic receptors.
Keywords: Human P2Y2 receptor; polymorphism; palmitoylation; pharmacological characterization; Gi,0 and Gq,11 coupling; antagonists; adenylyl cyclase modulation
Introduction
The nucleotidic receptors or P2 receptors are subdivided in two classes: P2X receptors, which are ATP-gated cation channels, and P2Y receptors, which are heptahelical G-coupled receptors (Abbrachio & Burnstock, 1994). The P2Y family encompasses selective purinoceptors, the P2Y1 (Webb et al., 1993) and P2Y11 receptors (Communi et al., 1997), selective pyrimidinoceptors, the P2Y3 (Webb et al., 1996a), P2Y4 (Communi et al., 1995a), and P2Y6 (Chang et al., 1995) receptors, and nucleotidic receptors responsive to both adenine and uracil nucleotides, such as the P2Y2 (Lustig et al., 1993) and the P2Y8 (Bogdanov et al., 1997) receptors. All these P2Y receptors are coupled to the phospholipase C pathway; moreover the recently cloned P2Y111 receptor is also coupled to adenylyl cyclase stimulation. The inclusion of other receptors, the P2Y5 (Webb et al., 1996b; Li et al., 1997), P2Y9 also called P2Y5-like (Janssens et al., 1997) and P2Y10 receptors (EMBL database: accession number AA747912 ) within the P2Y family remains controversial. As for the P2Y7 receptor (Akbar et al., 1996), it has been recharacterized as the leukotriene B4 receptor (Yokomizo et al., 1997).
The P2Y2 receptor, activated equipotently by UTP and ATP, is widely expressed among tissues and cell lines. In endothelial cells, the activation of the P2Y2 receptor causes the release of prostacyclin (PGI2) and nitric oxide (NO) which act as vasodilators and inhibitors of platelet aggregation (Boeynaems & Pearson, 1990). The P2Y2 receptor is also expressed in airway epithelial cells where its activation increases chloride permeability, via the activation of Ca2+-dependent ORCC channels (Outward Rectifying Chloride Channel) distinct from the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). Therefore UTP and ATP sprays have been advocated as a therapy of cystic fibrosis (for review: Donaldson & Boucher, 1998). These roles of the P2Y2 receptor make it a target for the discovery of new therapeutic agents. In the process of cloning the human P2Y2 gene in view to establish stable recombinant 1321N1 cell lines, we identified two forms of the human P2Y2 receptor differing by the substitution of C base for T at nucleotide 1000, leading to an arginine to cysteine substitution at position 334. In this paper, we have determined the frequency of the polymorphism and compared the pharmacological properties of the two forms of the P2Y2 receptor.
Methods
Cloning of the human P2Y2 receptor
The complete open reading frame of the receptor was amplified by polymerase chain reaction (PCR) using specific primers (Forward: atgggaattctggtcagggcgatggcagc; Reverse: atcatctagaagtgttctgctcctacagccgaatg). These primers were synthesized on basis of the 5′ and 3′ extremities of the coding sequence of the human P2Y2 receptor (Parr et al., 1994). The amplified product was subcloned into the expression vector peFIN (Euroscreen) and sequenced using eight specific primers as templates for the sequencing reaction. These primers were chosen at different positions of the receptor allowing us to sequence the receptor on both strands. The technology used for sequencing was the ABI PRISM Dye terminator cycle sequencing Ready reaction kit (Perkin Elmer) on a Applied Biosystems 373 DNA sequencer.
Genotyping
One hundred genomic DNA samples randomly taken among a European population were used in PCR to amplify the complete P2Y2 coding sequence. The 3′ carboxyterminal tail of the PCR product was sequenced using a specific primer located just after the seventh transmembrane region as template. We determined a sequence of approximately 200 bases of the P2Y2 gene, which includes the codon of the amino acid 334.
Cell culture and transfection
The coding sequence of each form was inserted into the expression vector peFIN. Astrocytoma 1321N1 cells were stably transfected with the recombinant peFIN plasmid using the calcium phosphate precipitation method as previously described (Velu et al., 1989). The wild-type cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% FCS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 2.5 μg ml−1 amphotericin B and 1% of sodium pyruvate. The transfected cells were harvested in the same medium supplemented by 400 μg ml−1 G418. From the pool of transfected 1321N1 cells, individual clones were isolated by limiting dilution with the purpose of selecting clones with high expression of the receptor.
Inositol phosphates measurements
150,000 transfected 1321N1 cells were labelled for 24 h with 5 μCi ml−1 myo-D-2[3H]-inositol in inositol free DMEM containing 5% FCS, antibiotics, amphotericin B and sodium pyruvate. The cells were stimulated with the agonist without changing the labelling medium and washing the cells. This procedure avoids the release of intracellular nucleotides by 1321N1 cells, that occurs easily when these cells are mechanically disturbed (Lazarowski et al., 1995). Incubations were terminated by the addition of ice cold 3% perchloric acid solution. Extraction and isolation of inositol trisphosphates (InsP3) on Dowex AG1-X8 columns were performed as described (Communi et al., 1995b). All assays were performed at least twice with triplicate samples. The data are expressed either as percentage of the maximal InsP3 formation, which did not differ by more than 25% between the tested cell clones, or in c.p.m. EC50 values were determined using SigmaPlot v2.0 Curve Fit. Student's t-tests were performed with SigmaPlot v2.0 software.
Cyclic AMP measurements
Stably transfected 1321N1 cells were seeded on Petri dishes (150,000 cells) in complete DMEM medium and cultured overnight. The cells were first preincubated 30 min with 25 μM rolipram, a phosphodiesterase inhibitor, and then incubated for 10 min with forskolin (FK) at 1 μM, in the presence of rolipram 25 μM. Finally, they were stimulated for various times (2–15 min) by nucleotides, in a medium supplemented with rolipram and FK. The incubation was stopped by the addition of HCl 0.1 M. After evaporation to dryness, the samples were diluted in water as required. Cyclic AMP was quantified by radioimmunoassay after acetylation (Brooker et al., 1979). Assays were performed at least three times with triplicate samples.
Results
Cloning of the human P2Y2 receptor
In order to amplify the human P2Y2 receptor, PCR were performed on human genomic DNA at high stringency using specific primers, synthesized on basis of the 5′ and 3′ extremities of the published coding sequence of the human P2Y2 receptor (Parr et al., 1994). The amplified open reading frame was subcloned into the peFIN vector and sequenced to detect mutated products generated by the PCR. Twelve amplified fragments derived from four separate PCR (using each time a new DNA sample) were totally sequenced on both strands. A comparison of the obtained sequences to the previously published sequence revealed two differences. In all the amplified fragments, an adenine was present instead of a guanine at position 1049 of the nucleotidic sequence, resulting in the replacement of a glycine by a glutamic acid at position 350 of the amino acid sequence (350G→350E). This difference was later confirmed in the 100 samples of genomic DNA which have been analysed (see below) and indicates a sequencing error in the original sequence. The second difference involves the replacement of a cytosine by a thymine at position 1000, leading to the substitution of arginine 334 by a cysteine (334R→334C). As this difference was observed only in some clones, it was likely to correspond to a polymorphism of the P2Y2 receptor gene (Figure 1).
Figure 1.
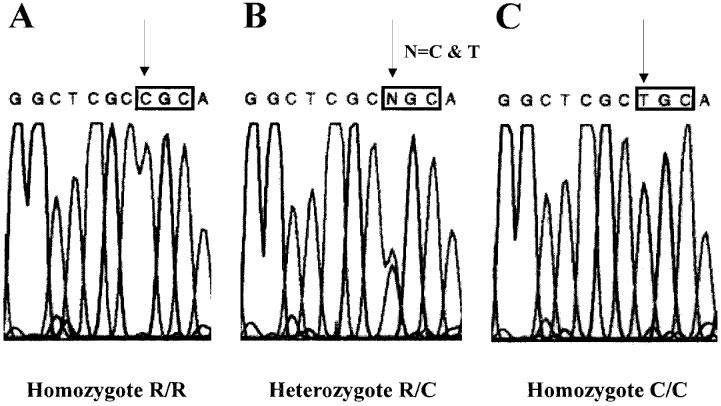
Comparison of the nucleotidic sequences of the two human P2Y2 alleles. The sequencing was performed on three different genomic DNA samples representative of a homozygote R/R (A), a heterozygote R/C (B) and a homozygote C/C (C). The nucleotide 1000 of the coding sequence responsive for the polymorphism is indicated by an arrow. The codon of the amino acid 334 modified by the mutation is represented in a frame.
Frequency of the polymorphism
In order to define the frequency of the polymorphism, we amplified the complete human P2Y2 coding sequence from 100 human genomic DNA samples randomly taken among a European population. The PCR products were sequenced using a primer recognising a region of the gene located just before the region of interest (after the seventh transmembrane region). We determined for each DNA a partial sequence of approximately 200 bases, which includes the amino acid present at position 334. This method allowed us to confirm unambiguously the presence of a polymorphism and also the presence of 350E instead of 350G. Moreover, we determined the frequencies of the different genotypes: R/R=66%, R/C=29%, and C/C=5%; R=0.8 and C=0.2.
Pharmacological characterization of the two human P2Y2 variants: time-course and concentration-response curves
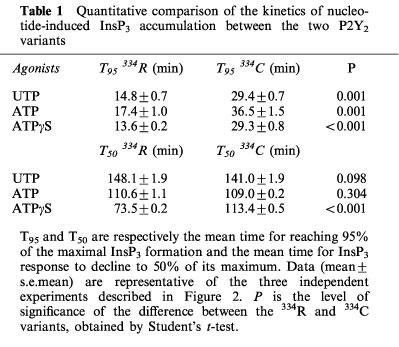
We first studied the time-course of InsP3 accumulation induced by UTP, ATP or ATPγS at 100 μM in the human P2Y2 334R or 334C clones, in absence of LiCl. As can be seen in Figure 2a and b, there was a rapid rise during the first 15 s, a phase of slower accumulation and a plateau followed by a decrease of the intracellular InsP3 level. A comparison between the kinetic curves of the two receptors revealed slight but reproducible differences. With the 334R variant, the time-course was characterized by a faster accumulation of intracellular InsP3: the time to reach 95% of the maximal response (T95), determined by graphic interpolation, was about 15 min for the 334R variant as compared to 30 min for the 334C variant (Table 1). When ATPγS (100 μM) was the agonist, both the onset of the response and its decay were more rapid with the 334R form as compared to the 334C variant (Figure 2a and b; Table 1).
Figure 2.
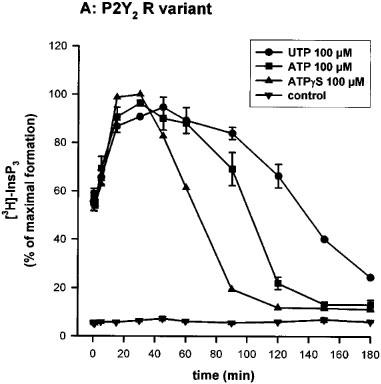
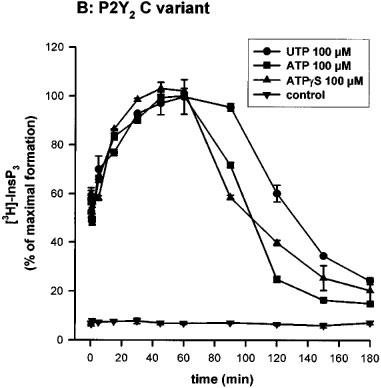
Time-course of InsP3 accumulation in 1321N1 cells expressing the human P2Y2 receptor R variant (A) and C variant (B). 3H-inositol labelled cells were incubated for the indicated time with UTP, ATP or ATPγS (100 μM) in absence of LiCl. The data represent the mean±s.d. of triplicate points and are representative of three independent experiments. 100% of InsP3 formation correspond to 1832 c.p.m. for the 334R variant and 1634 c.p.m. for the 334C variant.
Table 1.
Quantitative comparison of the kinetics of nucleotide-induced InsP3 accumulation between the two P2Y2 variants
Full concentration-response curves were obtained after a 30 s incubation with the most potent nucleotides (UTP, ATP, AP4A and ATPγS) (Lazarowski et al., 1995). For both variants, the order of potency of the agonists was conserved (UTP>ATP>>AP4A>ATPγS) and the EC50 obtained were almost identical (Figure 3a and b). We also tested the effects of other molecules such as UDP, UDP-glucose, ADP, GTP, GDP, AP3A, AP5A and NAD+ at 10 μM. GTP generated an almost maximal effect on the InsP3 accumulation when compared to the UTP response (80% of the UTP response), whereas AP3A increased only slightly the InsP3 level (30% of the UTP response). This last experiment did not reveal differences between the two polymorphic receptors (data not shown).
Figure 3.
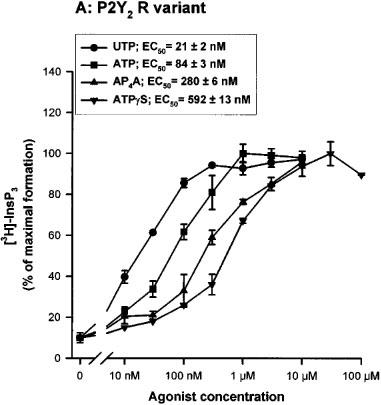
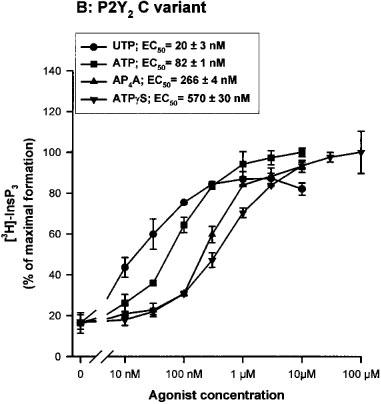
Concentration-response curves of various nucleotides on the InsP3 accumulation in 1321N1 cells expressing the human P2Y2 receptor R variant (A) and C variant (B). The cells, labelled overnight with 3H-inositol, were incubated in presence of various concentrations of UTP, ATP, AP4A and ATPγS for 30 s. The data are the mean±s.d. of triplicate experimental points and are representative of two separate experiments. The EC50 values (means±range) shown in the inset were calculated on basis of the two experiments. 100% of InsP3 formation correspond to 1644 c.p.m. for the 334R variant and 1512 c.p.m. for the 334C variant.
Study of the effects of the antagonists
The inhibitory action of suramin, pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) and reactive blue 2 (RB2) on the stimulation of phospholipase C induced by UTP was investigated in the P2Y2 334R or 334C 1321N1 transfected cells. These cells were preincubated 30 s with the antagonists at a concentration of 100 μM before the addition of UTP at various concentrations (30, 100 and 300 nM) for 30 s. As represented in Figure 4, the rank order of inhibition was RB2>>suramin>PPADS. Exactly the same order and degree of inhibition were observed for both variants (data not shown).
Figure 4.
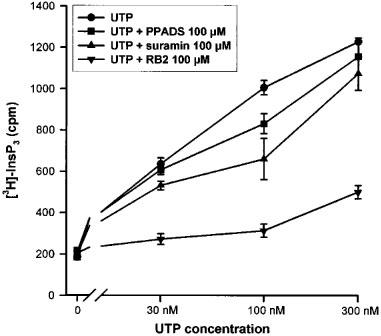
Effect of suramin, PPADS and RB2 (100 μM) in the InsP3 formation generated by the stimulation of the human P2Y2 R variant (closely similar results were observed for the P2Y2 C variant) with UTP. The 3H-inositol labelled cells were preincubated 30 s with the antagonist, before the addition of UTP (30, 100 or 300 nM) for another 30 s. The data represent the mean±s.d. of triplicate points and are representative of two independent experiments.
Study of the P2Y2 coupling to the Gi,0 protein
The effect of pertussis toxin (PTX) (24 h pretreatment at 100 ng ml−1) was tested on the action of UTP and ATPγS at different concentrations (10 nM–100 μM) and for different periods of stimulation (30 s and 15 min). After 30 s of stimulation, PTX produced a parallel shift in concentration-response curves with no change in maximum. After a 15 min stimulation, the concentration-response curves were shifted to higher concentrations, both for UTP and ATPγS, and the PTX effect was no longer detectable (Figure 5a and b). The two variants had the same sensitivity to PTX (data not shown).
Figure 5.
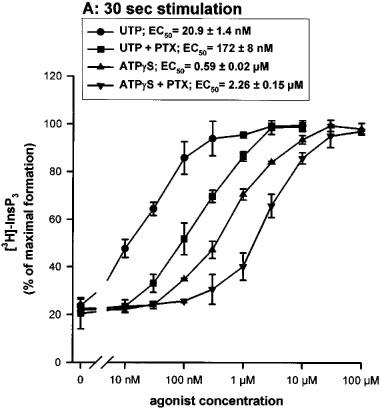
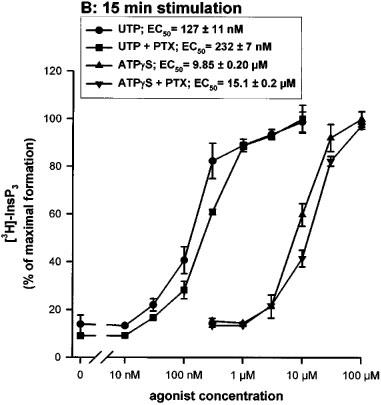
Effect of pertussis toxin on the UTP or ATPγS-induced accumulation of InsP3 in 1321N1 cells expressing the human P2Y2 C variant (similar results were obtained with the R variant). The cells were preincubated or not overnight in the presence of 100 ng ml−1 of pertussis toxin (PTX) and were then incubated in the presence of various concentrations of UTP or ATPγS for 30 s (A) or 15 min (B). The data represent the mean±s.d. of triplicate points and are representative of three independent experiments. The EC50 values (means±s.e.mean) shown in the inset were calculated on basis of the three experiments.
Study of the modulation of the adenylyl cyclase pathway
The transfected cells, preincubated with forskolin (FK) 1 μM, were stimulated with increasing concentrations of UTP (10 nM–10 μM) for 15 min. We observed a slow increase of the level of intracellular cyclic AMP leading to a 30% augmentation when stimulated by UTP as compared to the control level (Figure 6A). The P2Y2 expressing cells were also stimulated for various time (2–15 min) by low UTP concentrations (10, 30 and 100 nM), in the presence of FK 1 μM. No inhibition of the cyclic AMP accumulation could be observed (Figure 6B). The effect of nucleotides (i.e. UTP, ATP and AP4A at 10 μM) was also tested in absence of FK. UTP generated a 2 fold increase of the cyclic AMP level after a 15 min incubation. AP4A and especially ATP displayed a much stronger stimulation. A significant part of this response is likely to be due to the degradation of adenine nucleotides into adenosine acting on A2 receptors (Communi et al., 1997). These experiments did not reveal any significant difference between the two polymorphic receptors (data not shown).
Figure 6.
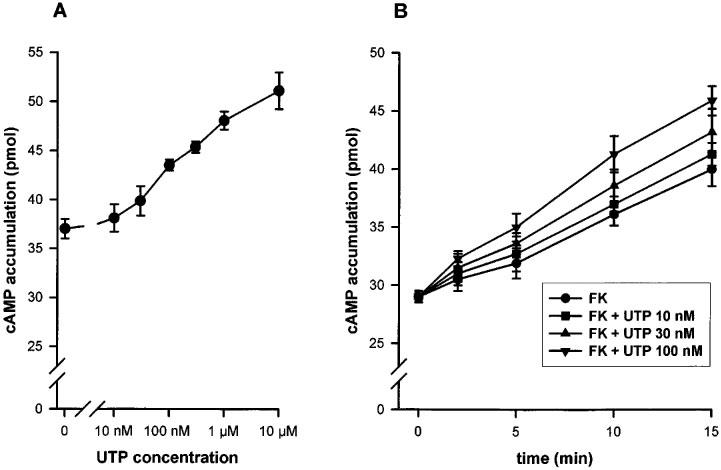
(A) Concentration-response curve of the UTP effect on cyclic AMP accumulation in 1321N1 cells expressing the human P2Y2 R variant (similar results were observed for the C variant). The P2Y2 expressing 1321N1 cells were stimulated 15 min with increasing concentrations of UTP. (B) Time-course of the UTP effect on cyclic AMP accumulation. 1321N1 cells expressing the R variant were stimulated by low concentrations of UTP (10, 30 or 100 mM) for various periods (2, 5, 10 or 15 min). For both experiments, prior to stimulation, the cells were preincubated 30 min with rolipram 25 μM, then 10 min with FK 1 μM and rolipram 25 μM before the addition of UTP, supplemented by FK and rolipram. The data represent the mean±s.d. of triplicate points and are representative of three independent experiments.
Discussion
The P2 receptors are known to be activated by extracellular nucleotides and to mediate many physiological and pharmacological responses in different cells and organs (Dubyak & El-Moatassim, 1993). The P2Y2 receptor, which is activated by UTP, ATP and also AP4A (Lazarowski et al., 1995), belongs to that family. Recently, the human P2Y2 receptor, cloned from the human airway epithelial cell line CF/T43 (Parr et al., 1994), has acquired an increasing importance because of its role in the treatment of respiratory diseases such as cystic fibrosis (Donaldson & Boucher, 1998).
In the process of cloning the human P2Y2 receptor coding sequence in order to establish a stable recombinant astrocytoma 1321N1 cell line as a tool for drug screening, we identified two forms of the P2Y2 receptor. One corresponded to the sequence published by Parr et al. (1994), except for a single base difference (1049G–1049A) leading to the replacement of glycine 350 by glutamic acid: as the difference was found in all the DNA analysed, it has to correspond to a sequencing error in the published sequence. The second was characterized by the replacement of arginine 334 into a cysteine and was found only in a minority of the samples analysed indicating the existence of a gene polymorphism. Sequence analysis performed on 100 European genomic DNA samples randomly taken showed that 66% of individuals are homozygotes R/R, 29% are heterozygotes R/C and 5% are homozygotes C/C. These data are in complete agreement with the Hardy-Weinberg equilibrium (R2+C2+2RC=1; R2= homozygotes R/R frequency, C2= homozygotes C/C frequency and 2RC = heterozygotes R/C; R=0.8 and C=0.2). The experimental result fits the theoretical equilibrium, indicating that the two alleles of the P2Y2 gene are segregated in a Mendelian manner.
The cysteine 334 located in the carboxy terminal tail close to the seventh transmembrane region constitutes a potential site of palmitoylation, a process known to modulate G-coupled receptors function (Mumby, 1997). This possibility reinforced our interest in comparing the pharmacological properties of the two P2Y2 receptors. Both variants were separately transfected into 1321N1 cells which were cloned by limiting dilution. Expression was documented by Northern blotting (data not shown) and functional assays. Time-course and concentration-response experiments were performed. The InsP3 accumulation induced by UTP, ATP or ATPγS stimulation, in the absence of LiCl, was characterized by a very fast onset, followed by a slower increase, reaching a plateau before decreasing to the basal level. Interestingly, despite its resistance to hydrolytic enzymes, the decay of the ATPγS response was more rapid than that of a comparable concentration of UTP. The second phase of InsP3 accumulation in response to nucleotides was slightly slower with the 334C variant as compared to the 334R form. The two variants were more clearly discriminated using ATPγS, a non-degradable agonist: with the 334R form, the InsP3 response had a more rapid onset and was also more transient, as compared to the 334C variant. The concentration-response curves characterizing the stimulation by UTP, ATP, AP4A and ATPγS were similar for both receptors and the EC50 were almost identical. We also studied the effects of antagonists of P2Y receptors (suramin, PPADS and RB2) on the InsP3 formation. Under the experimental conditions used (30 s preincubation with the antagonist before the nucleotide addition), our results showed that suramin and PPADS were weak antagonists as previously described (Charlton et al., 1996). Interestingly, we found that RB2 was the most active inhibitor of the P2Y2 receptor. Obviously that rank order of potency might have been affected by the short preincubation time, since some of these agents are characterized by a slow equilibration (Leff et al., 1990). These experiments did not reveal new differences between the two variants.
Moreover, in this work, we investigated the sensitivity to PTX and the interaction of the human P2Y2 receptor with the adenylyl cyclase pathway. These experiments did not reveal additional differences between the two forms of receptor, but they shed new light on the coupling of the human P2Y2 receptor to G proteins. In previous studies, the PTX sensitivity of the recombinant human P2Y2 receptor was studied only at maximal agonist concentrations and shown to be partial: about 25% in the human P2Y2 transfected 1321N1 cells (Parr et al., 1994) and 35% in the mouse P2Y2 transfected K-562 cells (Erb et al., 1993). The inhibitory effect of PTX on the responses mediated by natively expressed P2Y2 receptors was variable: partial in HL-60 cells (Dubyak et al., 1988) and complete in BAEC (bovine aortic endothelial cells) (Motte et al., 1993). In this study, we showed that the PTX sensitivity of the P2Y2 receptor is critically dependent on the agonist concentration: the inhibition was very strong or complete at low nucleotide concentration but progressively disappeared at concentrations producing a maximal effect. We also showed that PTX sensitivity was greater at early times during the stimulation and disappeared at later times in parallel with a shift of the concentration-response curves to higher agonist concentrations. This shift is unlikely to be due to nucleotide degradation, since it was of the same magnitude for UTP and ATPγS. It might be due to a reduction in the number of receptors present on the surface of the cell or of the available pool of G protein able to couple to the receptor. These experiments suggest that at least two G-proteins are involved. At low concentrations of nucleotides, the coupling via Gq,11 could be enhanced by the simultaneous activation of a Gi,0 protein. This is reminiscent of the study performed by Baltensperger & Porzig (1997) in HEL cells: they found that the activation of the phospholipase C pathway by the endogenous P2Y2 receptor was abolished by a Gα16 antisense but was also partially inhibited by PTX. The coupling to a Gi,0 protein is consistent with the presence of a threonine residue at the end of the third intracytoplasmic loop of the human P2Y2 receptor. This residue is a unique feature of Gi,0-coupled receptors (Liu et al., 1995). The modulation of adenylyl cyclase by the human P2Y2 receptor was also studied in this paper. After 15 min stimulation, we observed a slight enhancement of the level of cyclic AMP reached under forskolin stimulation, likely to result from a crosstalk between phospholipase C and adenylyl cyclase pathways (Haslauer et al., 1998). The adenylyl cyclase of the P2Y2 receptors expressing cells was not inhibited even at low agonist concentrations (10, 30 and 100 nM) for shorter stimulation periods (2, 5 and 10 min). This suggests that the G protein inhibited by PTX is likely a G0 subtype.
In summary, we described here the first polymorphism affecting a P2Y family member, the human P2Y2 receptor, and we analysed its distribution in a European population. Although the polymorphism generates a potential palmitoylation site, no marked pharmacological difference was observed between the two polymorphic receptors for the natural agonists we tested (UTP, ATP and AP4A). However, there were slight differences in time-course, especially when ATPγS was used as agonist. The observation of such differences is interesting in view of the potential use of long-acting phosphorothioate nucleotides in the treatment of cystic fibrosis and chronic bronchitis (Donaldson & Boucher, 1998). Furthermore, the distribution of the two allelic variants should be investigated in clinical subsets of lung diseases, in order to evaluate if this polymorphism may act as disease modifier. Moreover, this study has revealed new pharmacological features of the human P2Y2 receptor and added evidence that this receptor is coupled to at least two G proteins, probably a G0 protein when the receptor is stimulated by low concentration of ligand and for a short period. The second G protein implicated is pertussis toxin insensitive, likely a Gq,11 protein: this coupling predominates when the P2Y2 receptor is activated by high agonist concentration and/or for long duration.
Acknowledgments
This work was supported by an Action de Recherche concertée of the Communauté Française de Belgique, by the Belgian Programme on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister's Office, Federal Service for Science, Technology and Culture, by a grant of the Fonds de la Recherche Scientifique Médicale, by the Fondation Médicale Reine Elisabeth, and by Boehringer Ingelheim. R. Janssens is a fellow of the Fonds pour la Formation à la Recherche dans l'Industrie et l'Agriculture. We also thank Valérie Dubois and Anne Renard for expert technical assistance, Dr V. De Maertelaer, Dr Graham A. Place from Bayer, Dr D. Communi, Dr G. Vassart and Dr J. E. Dumont for helpful advice and discussions.
Abbreviations
- AP4A
P1,P4-di(adenosine-5′) tetraphosphate
- ATPγS
adenosine 5′-O(3-γ-thio)triphosphate
- cAMP
cyclic AMP
- DMEM
Dulbecco's modified Eagle's medium
- FK
forskolin
- InsP3
inositol trisphosphates
- PCR
polymerase chain reaction
- PPADS
pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid
- PTX
pertussis toxin
- RB2
reactive blue 2
- T50
time for InsP3 response to decline to 50% of its maximum
- T95
time to reach 95% of maximal InsP3 response
References
- ABBRACHIO M.P., BURNSTOCK G. Purinoceptors: are there families of P2X and P2Y purinoceptors. Pharmacol. Ther. 1994;64:445–475. doi: 10.1016/0163-7258(94)00048-4. [DOI] [PubMed] [Google Scholar]
- AKBAR J.M.K., DASARI V.R., WEBB T.E., AYYANATHAN K., PILLARISETTI K., SANDHU A.K., ATHWAL R.S., DANIEL J.L., ASHBY B., BARNARD E.A., KUNAPULI S.P. Molecular cloning of a novel P2 purinoceptor from human erythroleukemia cells. J. Biol. Chem. 1996;271:18363–18367. doi: 10.1074/jbc.271.31.18363. [DOI] [PubMed] [Google Scholar]
- BALTENSPERGER K., PORZIG H. The P2U purinoceptor obligatorily engages the heterotrimeric G protein G16 to mobilize intracellular Ca2+ in human erythroleukemia cells. J. Biol. Chem. 1997;272:10151–10159. doi: 10.1074/jbc.272.15.10151. [DOI] [PubMed] [Google Scholar]
- BOEYNAEMS J.M., PEARSON J.D. P2 purinoceptors on vascular endothelial cells: physiological significance and transduction mechanisms. Trends Pharmacol. Sci. 1990;11:34–37. doi: 10.1016/0165-6147(90)90039-b. [DOI] [PubMed] [Google Scholar]
- BOGDANOV Y., DALE L., KING B.F., WHITTOCK N., BURNSTOCK G. Early expression of a novel nucleotide receptor in the neural plate of Xenopus embryos. J. Biol. Chem. 1997;272:12583–12590. doi: 10.1074/jbc.272.19.12583. [DOI] [PubMed] [Google Scholar]
- BROOKER G., HARPER J.F., TERASEKI W.L., MOYLAN R.D. Radioimmunoassay of cyclic AMP and cyclic GMP. Adv. Cyclic Nucleotides Res. 1979;10:1–33. [PubMed] [Google Scholar]
- CHANG K., HANAOKA K., KUMADA M., TAKUWA Y. Molecular cloning and functional analysis of a novel P2 nucleotide receptor. J. Biol. Chem. 1995;270:26152–26158. doi: 10.1074/jbc.270.44.26152. [DOI] [PubMed] [Google Scholar]
- CHARLTON S.J., BROWN C.A., WEISMAN G.A., TURNER J.T., ERB L., BOARDER M.R. PPADS and suramin as antagonists at cloned P2Y- and P2U-purinoceptors. Br. J. Pharmacol. 1996;118:704–710. doi: 10.1111/j.1476-5381.1996.tb15457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COMMUNI D., GOVAERTS C., PARMENTIER M., BOEYNAEMS J.M. Cloning of a human purinergic P2Y receptor coupled to phospholipase C and adenylyl cyclase. J. Biol. Chem. 1997;272:31969–31973. doi: 10.1074/jbc.272.51.31969. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., PIROTTON S., PARMENTIER M., BOEYNAEMS J.M. Molecular cloning and functional expression of a human uridine nucleotide receptor. J. Biol. Chem. 1995a;270:30849–30852. doi: 10.1074/jbc.270.52.30849. [DOI] [PubMed] [Google Scholar]
- COMMUNI D., RASPE E., PIROTTON S., BOEYNAEMS J.M. Coexpression of P2Y and P2U receptors on aortic endothelial cells. Circ. Res. 1995b;76:191–198. doi: 10.1161/01.res.76.2.191. [DOI] [PubMed] [Google Scholar]
- DONALDSON S.H., BOUCHER R.C.Therapeutic applications for nucleotides in lung disease The P2 Nucleotide Receptors 1998Humana Press Inc.: Totowa, NJ; 413–424.(eds). Turner, J.T., Weisman, G.A. & Fedan, J. pp [Google Scholar]
- DUBYAK G.R., COWEN D.S., MEULLER L.M. Activation of inositol phospholipid breakdown in HL60 cells by P2-purinergic receptors for extracellular ATP. J. Biol. Chem. 1988;263:18108–18117. [PubMed] [Google Scholar]
- DUBYAK G.R., EL-MOATASSIM C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am. J. Physiol. 1993;265:C577–C606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- ERB L., LUSTIG K.D., SULLIVAN D.M., TURNER J.T., WEISMAN G.A. Functional expression and photoaffinity labeling of a cloned P2U purinergic receptor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10449–10453. doi: 10.1073/pnas.90.22.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASLAUER M., BALTENSPERGER K., PORZIG H. Thrombin and phorbol esters potentiate Gs-mediated cAMP formation in intact human erythroid progenitors via two synergistic signaling pathways converging on adenylyl cyclase type VII. Mol. Pharmacol. 1998;53:837–845. [PubMed] [Google Scholar]
- JANSSENS R., BOEYNAEMS J.M., GODART M., COMMUNI D. Cloning of a human heptahelical receptor closely related to the P2Y5 receptor. Biochem. Biophys. Res. Commun. 1997;236:106–112. doi: 10.1006/bbrc.1997.6895. [DOI] [PubMed] [Google Scholar]
- LAZAROWSKI E.R., WATT W.C., STUTTS M.J., BOUCHER R.C., HARDEN T.K. Pharmacological selectivity of the cloned human P2U-purinoceptor: potent activation by diadenosine tetraphosphate. Br. J. Pharmacol. 1995;116:1619–1627. doi: 10.1111/j.1476-5381.1995.tb16382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFF P., WOOD B.E., O'CONNOR S.E. Suramin is a slowly-equilibrating but competitive antagonist at P2x-receptors in the rabbit isolated ear artery. Br. J. Pharmacol. 1990;101:645–649. doi: 10.1111/j.1476-5381.1990.tb14134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI Q., SCHACHTER J.B., HARDEN T.K., NICHOLAS R.A. The 6H1 orphan receptor, claimed to be the P2Y5 receptor, does not mediate nucleotide-promoted second messenger responses. Biochem. Biophys. Res. Commun. 1997;236:455–460. doi: 10.1006/bbrc.1997.6984. [DOI] [PubMed] [Google Scholar]
- LIU J., CONKLIN B.R., BLIN J., YUN J., WESS J. Identification of a receptor/G-protein contact site critical for signaling specificity and G-protein activation. Proc. Natl. Acad. Sci. U.S.A. 1995;92:11642–11646. doi: 10.1073/pnas.92.25.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUSTIG K.D., SHIAU A.K., BRAKE A.J., JULIUS D. Expression cloning of an ATP receptor from mouse neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5113–5117. doi: 10.1073/pnas.90.11.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOTTE S., PIROTTON S., BOEYNAEMS J.M. Heterogeneity of ATP receptors in aortic endothelial cells: involvement of P2Y and P2U receptors in the inositol phosphate response. Circ. Res. 1993;72:504–510. doi: 10.1161/01.res.72.3.504. [DOI] [PubMed] [Google Scholar]
- MUMBY S.M. Reversible palmitoylation of signaling proteins. Curr. Opin. Cell Biol. 1997;9:148–154. doi: 10.1016/s0955-0674(97)80056-7. [DOI] [PubMed] [Google Scholar]
- PARR C.E., SULLIVAN D.M., PARADISO A.M., LAZAROWSKI E.R., BURCH L.H., OLSEN J.C., ERB L., WEISMAN G.A., BOUCHER R.C., TURNER J.T. Cloning and expression of a human P2U nucleotide receptor, a target for cystic fibrosis pharmacotherapy. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VELU T.J., BEGUINOT L., VASS W.C., ZHANG K., PASTAN I., LOWRY D.R. Retroviruses expressing different levels of the normal epidermal growth factor receptor: biological properties and new bioassay. J. Cell. Biochem. 1989;39:153–166. doi: 10.1002/jcb.240390207. [DOI] [PubMed] [Google Scholar]
- WEBB T.E., HENDERSON D., KING B.F., WANG S., SIMON J., BATESON A.N., BURNSTOCK G., BARNARD E.A. A novel G protein-coupled P2 purinoceptor (P2Y3) activated preferentially by nucleoside diphosphates. Mol. Pharmacol. 1996a;50:258–265. [PubMed] [Google Scholar]
- WEBB T.E., KAPLAN M.G., BARNARD E.A. Identification of 6H1 as a P2Y purinoceptor: P2Y5. Biochem. Biophys. Res. Commun. 1996b;219:105–110. doi: 10.1006/bbrc.1996.0189. [DOI] [PubMed] [Google Scholar]
- WEBB T.E., SIMON J., KRISHEK B.J., BATESON A.N., SMART T.G., KING B.F., BURNSTOCK G., BARNARD E.A. Cloning and functional expression of a brain G protein-coupled ATP receptor. FEBS Lett. 1993;324:26–30. doi: 10.1016/0014-5793(93)81397-i. [DOI] [PubMed] [Google Scholar]
- YOKOMIZO T., IZUMI T., CHANG K., TAKUWA Y., SHIMIZU T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387:620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]