

Abstract
The combination of interleukin-2 (IL-2) and IL-4 reduces the inhibitory effects of glucocorticoids on granulocytemacrophage colonystimulating factor (GM-CSF) production, in agreement with the hypothesis that this combination causes glucocorticoid resistance. Whether a general cytokine resistance to glucocorticoids is induced by IL-2 and IL-4 has not been reported.
Mononuclear blood cells from healthy individuals were pretreated with IL-2, IL-4, or IL-2+ IL-4 (31.3–500 U ml−1) for 48 h, prior to lipopolysaccharide (LPS; 10 ng ml−1; 20 h) and budesonide addition. Cytokine levels in the supernatants were analysed using specific immunoassays. DNA content was analysed to estimate cell numbers.
GM-CSF production was totally inhibited by budesonide at 10−8 M in vehicle treated cultures, while IL-10 was inhibited to 33.4±4.3% of control. IL-2, IL-4, or IL-2+IL-4 reduced the inhibitory effects of budesonide on GM-CSF to similar levels (23.7±6.7, 31.6±8.5 and 35.1±4.3% of control, respectively). IL-2, IL-4, or IL-2+IL-4 also reduced the inhibitory effects of budesonide on IL-10 production (46.5±6.6, 55.9±7.3%, and 68.3±9.9% of control, respectively). In contrast, IL-8, IL-12 and TNF-α production did not become resistant to budesonide.
Thus, glucocorticoid resistance induced by IL-2 and IL-4 is not general at the cytokine production level. While the glucocorticoid sensitivity of GM-CSF and IL-10 production decreased, the sensitivity of IL-8, IL-12 or TNF-α production was unchanged. Also, the mixture of IL-2 and IL-4 is not crucial for induction of glucocorticoid resistance of GM-CSF production.
Keywords: Glucocorticoid resistance, budesonide, IL-2, IL-4, GM-CSF, IL-8, IL-10, IL-12, TNF-α, mononuclear blood cells
Introduction
Since the introduction of glucocorticoids for the treatment of inflammatory and autoimmune diseases, increasing evidence point to the existence of glucocorticoid resistance in some patients (Schwarz et al., 1968; Chikanza & Panayi, 1993; Frieri & Madden, 1993). Glucocorticoid resistance has mostly been studied in asthmatics where it is characterized by failure of glucocorticoids to suppress eosinophil numbers and activated T cells (Schwarz et al., 1968; Carmichael et al., 1981; Leung et al., 1995). Several investigators have also reported data on defective glucocorticoid responses of mononuclear blood cells which correlate with clinical glucocorticoid resistance (Kay et al., 1981; Poznansky et al., 1984; Wilkinson et al., 1989; Corrigan et al., 1991; Alvarez et al., 1992).
Leung and colleagues recently proposed that increased production of interleukin-2 (IL-2) and IL-4 causes glucocorticoid resistance. They reported increased expression of IL-2 and IL-4 mRNA in bronchoalveolar lavage cells of glucocorticoid-resistant asthmatics (Leung et al., 1995). In addition, increased IL-2 and IL-4 mRNA expression was found in endomyocardial biopsies of patients that developed glucocorticoid-resistant cardiac allograft rejection (Bann et al., 1996). Furthermore, there are several reports showing that the combination of IL-2 and IL-4 induces defective glucocorticoid receptor properties in normal mononuclear blood cells, similar to those in cells of glucocorticoid-resistant asthmatics (Kam et al., 1993; Sher et al., 1994; Klemm et al., 1996; Spahn et al., 1996). The IL-2 and IL-4 hypothesis is further supported by our previous data that the combination of IL-2 and IL-4 induces resistance of granulocyte-macrophage colony-stimulating factor (GM-CSF) production to glucocorticoids (Larsson et al., 1997). We demonstrated that the combination of IL-2 and IL-4 has functional consequences at the cytokine level that might contribute to the reduced anti-inflammatory effects of glucocorticoids in glucocorticoid-resistant patients.
However, there are several independent reports that IL-2 alone abrogates glucocorticoid inhibition of T cell proliferation (Walker et al., 1987; Hazcku et al., 1994). These results suggest that IL-2 alone may cause glucocorticoid resistance. Except for studies on glucocorticoid receptor affinity there are no further studies assessing whether a combination of IL-2 and IL-4 is needed for induction of glucocorticoid resistance. Hence, in the present study we have investigated the effects of IL-2 or IL-4, alone or in combination, on GM-CSF production and on its inhibition by budesonide. In order to study whether a general glucocorticoid resistance at the cytokine level could be induced, we also compared the effects of IL-2 or IL-4 alone, or in combination, on the production of IL-8, IL-10, IL-12 and tumour necrosis factor-α (TNF-α).
Methods
Cell separation and culture
Cell separation and culture were performed as described previously (Larsson et al., 1997). Briefly, venous blood was collected from healthy volunteers into sterile EDTA-containing tubes. Erythrocyte sedimentation was increased by mixing the blood 5 : 1 with saline containing 5% Dextran T500 (Pharmacia Biotech, Uppsala, Sweden) and glucose (30 mg ml−1; BDH Laboratory Supplies, Poole, U.K.). After sedimentation for 30 min at room temperature the plasma fraction was layered on Ficoll Paque-Plus (low endotoxin grade; Pharmacia Biotech, Uppsala, Sweden) and mononuclear cells were isolated. Cell viability was >95%, estimated by the Trypan blue exclusion test. Granulocyte content was <2%, estimated by cell counts in Türks solution and later confirmed by May-Grünwald-Giemsa staining of cytospin cells. The cytospin preparations (n=17) contained 43.9±2.9% monocytes and 53.5±3.0% lymphocytes. In the whole study 11 different healthy blood donors were used, some of them were used twice, i.e. a total of 17 cell separations were performed. Cells from the same donor were not used twice in the same type of experiment. In each experiment six different cell separations (from six different blood donors) were used.
The mononuclear cells were washed with sterile phosphate buffered saline (PBS) and resuspended in sterile RPMI 1640 with HEPES buffer (25 mM) and L-glutamine (Gibco, Paisley, U.K.). Foetal calf serum (1%, Gibco, Paisley, U.K.), benzylpenicillin (0.1 mg ml−1; Astra AB, Södertälje, Sweden) and streptomycin sulphate (0.1 mg ml−1; Sigma, St Louis MO, U.S.A.) were added. Aliquots of 2×106 were cultured in 24-well culture plates at 37°C, 5% CO2 98% rH in the presence and absence of IL-2 and IL-4 (31.3–500 U ml−1; R&D Systems Europe, Abingdon, U.K.). The cytokines were reconstituted in sterile PBS with the addition of bovine serum albumin (0.1%, low endotoxin BSA; Sigma, St Louis MO, U.S.A.). After 48 h, lipopolysaccharide (10 ng ml−1, LPS; E. coli 0.26 : B6; Difco Laboratories, Detroit, U.S.A.) and various concentrations of budesonide (10−11–10−7 M; Astra Draco AB, Lund, Sweden) were added as aliquots of 10 μl. After another 20 h of incubation the whole culture plates were centrifuged at 200×g for 10 min at 4°C. The cell-free supernatants were transferred to plastic tubes and stored at −70°C until analysis.
Cytokine analysis
GM-CSF, IL-8, IL-10 and IL-12 (p70) levels were assayed using ELISA-kits (R&D Systems Europe, Abingdon, U.K.; lower detection limit 1.5, 10, 1.5 and 5 pg ml−1, respectively). TNF-α was analysed using an antibody pair (R&D Systems Europe, Abingdon, U.K.; lower detection limit 5 pg ml−1). Briefly, MAB610 (4 μg ml−1 in PBS) was coated overnight to 96-well ELISA-plates. The plates were washed three times in PBS with 0.05% Tween-20, blocked with 1% BSA+5% sucrose in PBS for 1 h and washed again. The plates were incubated 2 h with 100 μl samples or recombinant standards, washed and incubated 2 h with the biotinylated detection antibody BAF210 (200 ng ml−1 in Tris-buffered saline with 0.1% BSA). After washing, streptavidin-horse radish peroxidase (Zymed Laboratories, San Francisco, CA, U.S.A.) and tetramethylbenzidine-peroxidase substrate (Kirkegaard and Perry Laboratories, Gaithersburg, MD, U.S.A.) were used for detection. All values are expressed as mean±standard error of the mean (s.e.mean). Cytokine (percent of control) was calculated as percent cytokine in a culture with budesonide addition compared with a similarly pre-treated culture without budesonide. Thus, a ‘value of 0% of control' means total inhibition by budesonide and a ‘value of 100% of control' means that budesonide had no effect.
DNA-analysis
After the cell plates were centrifuged and the culture supernatants collected, DNA-content was analysed using a fluorochrome protocol modified from Blaheta (Blaheta et al., 1991). Aliquots of 600 μl PBS and 400 μl Hoechst compound 33342 (10 μg ml−1 in PBS; B-2261, Sigma, St Louis MO, U.S.A.) were added to the culture wells. The plates were covered and incubated for 30 min in a dark place, at room temperature. Fluorescence was measured using a fluorometer with a 360 nm excitation filter and a 460 nm emission filter (Cytofluor 2350, Millipore, U.K.). DNA-content was calculated using a standard curve of calf thymus DNA in PBS.
Statistics
All values are expressed as means±standard error of the mean (s.e.mean). Values were compared using analysis of variance (ANOVA) with experiment and treatment as factors, followed by pairwise comparisons. Parametric tests were used. P values <0.05 were considered significant.
Results
Effects of IL-2, IL-4, and IL-2+IL- 4 on GM-CSF production
The vehicle used in all presented experiments was cell culture medium RPMI 1640 supplemented with FCS (1%), benzylpenicillin (0.1 mg ml−1), streptomycin sulphate (0.1 mg ml−1) and LPS (10 ng ml−1). The LPS was added first after 48 h of incubation as 10 μl aliquots of a stock solution in RPMI 1640 medium. No GM-CSF was detected in medium cultures in the absence of LPS.
LPS-stimulated GM-CSF production in vehicle-treated mononuclear cell cultures from six healthy individuals was 11.2±3.7 pg ml−1 (Figure 1). Pre-treatment of mononuclear blood cells with IL-2 (31.3-500 U ml−1) for 48 h prior to LPS treatment (10 ng ml−1, 20 h) potently stimulated GM-CSF production. Pretreatment with the combination of IL-2 and IL-4 (31.3–500 U ml−1 of each) for 48 h prior to LPS treatment (10 ng ml−1; 20 h) also had stimulatory effects on GM-CSF production (P<0.01 for all doses compared with vehicle). Pre-treatment with IL-4 (31.3–500 U ml−1) for 48 h prior to LPS treatment (10 ng ml−1; 20 h) had no effect on GM-CSF production. No dose-response relationships were observed. IL-2 alone stimulated GM-CSF production more than IL-2 and IL-4 in combination.
Figure 1.
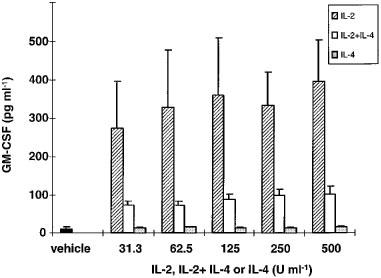
GM-CSF production by mononuclear blood cells pre-treated with IL-2, IL-4 or IL-2+IL-4 for 48 h prior to LPS-stimulation (10 ng ml−1 for 20 h). The vehicle was RPMI 1640 supplemented with FCS (1%), benzylpenicillin (0.1 mg ml−1), streptomycin sulphate (0.1 mg ml−1) and LPS (10 ng ml−1; 20 h). Results are expressed as mean±s.e.mean of six separate experiments. Mononuclear cells were isolated from six healthy blood donors.
Induction of glucocorticoid resistance on GM-CSF production
Since new batches of IL-2 and IL-4 and LPS were used, different doses of a mixture of the cytokines were tested. A single, high dose of budesonide (10−8 M), which normally inhibits GM-CSF completely, was used in these experiments. We found that all tested doses of IL-2+IL-4 partly reversed the inhibitory effects of budesonide on GM-CSF production (P<0.01). A tendency for a dose-response relationship was observed (Figure 2). Resistance of GM-CSF to budesonide inhibition was inducible in all donors, but to a varying degree. The maximal reversal of budesonide inhibition for each individual was found after pre-treatment with 250 or 500 U ml−1 each of IL-2+IL-4. Hence, in the following experiments 500 U ml−1 each of IL-2+IL-4 were used.
Figure 2.
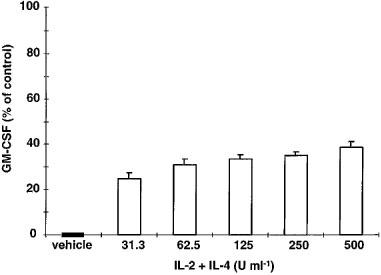
Effects of IL-2 and IL-4 pre-treatment (48 h) on budesonide (10−8 M) inhibition of LPS-stimulated GM-CSF production by mononuclear blood cells. The vehicle was RPMI 1640 supplemented with FCS (1%), benzylpenicillin (0.1 mg ml−1), streptomycin sulphate (0.1 mg ml−1) and LPS (10 ng ml−1 for 20 h). GM-CSF (percent of control) was calculated as percent GM-CSF in a culture with budesonide addition compared with a similarly pre-treated culture without budesonide. A ‘value of 0% of control' means total inhibition by budesonide and a ‘value of 100% of control' means lack of budesonide effect. Results are expressed as mean±s.e.mean of six separate experiments. Mononuclear cells were isolated from six healthy blood donors. All doses of IL-2+IL-4 counteracted the inhibitory effects of budesonide on GM-CSF production (P<0.01).
Effects of IL-2, IL-4, and IL-2+IL- 4 on cell proliferation
DNA-content in the cell cultures was assayed to study whether IL-2, IL-4, or IL-2+IL-4 had any effects on cell proliferation. DNA was analysed in cell cultures (n=6) which were incubated with or without cytokines (500 U ml−1) for 48 h and then stimulated with LPS (10 ng ml−1) for 20 h. DNA-content after IL-2 pre-treatment was 106.3±7.4% of vehicle treatment. DNA-content after IL-4 pre-treatment was 108.5±5.0% of vehicle treatment and DNA-content after IL-2+IL-4 pre-treatment was 122.2±14.0% of vehicle treatment. There were no significant differences in the effects of IL-2, IL-4, or IL-2+IL-4 on DNA-content.
Budesonide effects on GM-CSF production after IL-2, IL-4, or IL-2+IL-4 pre-treatment
GM-CSF production in cultures without cytokine pre-treatment was totally inhibited by budesonide (10−8 M) (Table 1 and Figure 3). IL-2, IL-4, or IL-2+IL-4 pre-treatment (500 U ml−1) reduced the inhibitory effect of budesonide on GM-CSF production (P<0.01). GM-CSF production was 23.7±6.7% of control after budesonide addition to IL-2 pre-treated cultures. In IL-4 or IL-2+IL-4 pre-treated cultures GM-CSF production after budesonide treatment was 31.6±8.5 and 35.1±4.3% of control, respectively. IL-2 or IL-4 alone counteracted the inhibitory effect of budesonide on GM-CSF production without significant difference compared with the combination of IL-2 and IL-4 (Table 1 and Figure 3).
Table 1.
Effects of IL-2, IL-4 and IL-2+IL-4 pre-treatment on budesonide inhibition of LPS-stimulated GM-CSF and IL-10 production by mononuclear blood cells from five healthy blood donors

Figure 3.
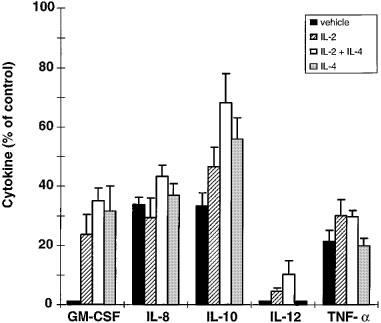
Effects of IL-2, IL-4, or IL-2+IL-4 pre-treatment (48 h; 500 U ml−1) on budesonide (10−8 M) inhibition of LPS-stimulated GM-CSF, IL-8, IL-10, IL-12 and TNF-α production by mononuclear blood cells. The vehicle was RPMI 1640 supplemented with FCS (1%), benzylpenicillin (0.1 mg ml−1), streptomycin sulphate (0.1 mg ml−1) and LPS (10 ng ml−1 for 20 h). Results were calculated as in Figure 2 and are expressed as mean±s.e.mean of five separate experiments. Mononuclear cells were isolated from five healthy blood donors. Cytokine pre-treatment changed the sensitivity of GM-CSF and IL-10 production to budesonide but not that of IL-8, IL-12 or TNF-α production.
Budesonide effects on IL-10 production after IL-2, IL-4, or IL-2+IL-4 pre-treatment
IL-10 was secreted at higher levels than GM-CSF in vehicle treated cultures (Table 1). IL-2+IL-4 pre-treatment (500 U ml−1) inhibited IL-10 production (P<0.01), while IL-2 or IL-4 alone had no significant effects compared with vehicle. Budesonide (10−8 M) decreased IL-10 production to 33.4±4.3% of control in vehicle treated cultures (Table 1, Figure 3). As observed for GM-CSF production, budesonide inhibition of IL-10 was significantly reduced by IL-2+IL-4 pre-treatment (68.3±9.9% of control; P<0.05). IL-2 or IL-4 pre-treatment also tended to decrease the budesonide inhibition of IL-10 production (46.5±6.6% and 55.9±7.3% of control, respectively).
Budesonide effects on IL-8, IL-12 and TNF-α production after IL-2, IL-4, or IL-2+IL-4 pre-treatment
IL-8 was produced in nanogram levels in the cultures without added cytokines (Table 2). Pre-treatment with IL-2 potently stimulated IL-8 production (P<0.01), while IL-4 did not show any stimulating effect. In contrast, the combination of IL-2 and IL-4 reduced IL-8 production compared with vehicle (P<0.01) and compared with IL-4 pre-treatment (P<0.01). Pre-treatment with IL-2, IL-4, or IL-2+IL-4 did not reduce the inhibitory effects of budesonide on IL-8 production (Table 2, Figure 3). Budesonide inhibited IL-8 production to 33.8±2.4% of control in cultures without cytokine pre-treatment, to 29.4±6.6% of control after IL-2 pre-treatment, to 36.9±4.0% of control after IL-4 pre-treatment, and to 43.3±3.8% of control after pre-treatment with IL-2+IL-4. There were no significant differences between the different pre-treatments. Thus, no resistance of IL-8 to budesonide was induced.
Table 2.
Effects of IL-2, IL-4 and IL-2+IL-4 pre-treatment on budesonide inhibition of LPS-stimulated IL-8, IL-12 and TNF-α production by mononuclear blood cells from five healthy blood donors

IL-12 was not detected in cultures without cytokine pre-treatment (Table 2). However, in the cultures pre-treated with IL-4 low IL-12 concentrations were present. Interestingly, pre-treatment with IL-2 potently stimulated IL-12 and the combination of IL-2 and IL-4 tended to stimulate IL-12 even more. Budesonide (10−8 M) totally inhibited IL-12 production in cultures pre-treated with IL-4 (Table 2, Figure 3). In cultures pre-treated with IL-2, or IL-2+IL-4, budesonide inhibited IL-12 to 4.5±1.2 and 10.3±4.6% of control, respectively. The effect of IL-2 did not differ significantly from the effect of IL-2+IL-4.
TNF-α production was potently stimulated by pre-treatment with IL-2, IL-4, or IL-2+IL-4 (P<0.01; Table 2). IL-2 stimulated TNF-α production more than IL-4 (P<0.01) or IL-2+IL-4 (P<0.01). IL-2+IL-4 stimulated TNF-α production more than IL-4 alone (P<0.01). Pre-treatment with IL-2, IL-4 or IL-2+IL-4 did not reduce the inhibitory effect of budesonide on TNF-α production (Table 2, Figure 3). Budesonide decreased TNF-α production to 21.5±3.7% of control in cultures without cytokine pre-treatment. TNF-α production was reduced by budesonide to 30.1±5.4% of control after IL-2 pre-treatment, to 19.9±2.6% of control after IL-4 pre-treatment, and to 29.7±2.1% of control after pre-treatment with IL-2+IL-4. There were no significant differences between the different pre-treatments. Thus, no resistance of TNF-α production to budesonide was induced.
Discussion
The present study indicates that IL-2 and IL-4 do not induce a general glucocorticoid resistance at the cytokine production level. Although IL-2, IL-4, or IL-2 and IL-4 in combination, reduce the budesonide sensitivity of GM-CSF and IL-10 production, the budesonide sensitivity of TNF-α, IL-8 or IL-12 production does not change. The present study also indicates that the combination of IL-2 and IL-4 is not crucial for induction of glucocorticoid resistance of GM-CSF production. Either IL-2 or IL-4 alone reduces the glucocorticoid sensitivity of GM-CSF production to the same degree as IL-2 and IL-4 in combination.
Previous reports indicated that a mixture of IL-2 and IL-4 is necessary to mimic the reduced glucocorticoid receptor affinity and the reduced glucocorticoid anti-proliferative effects in mononuclear blood cells of glucocorticoid-resistant asthmatics (Kam et al., 1993; Sher et al., 1994). However, when binding affinity studies were repeated in glucocorticoid-resistant asthmatics with the more potent glucocorticoid budesonide instead of dexamethasone, no reduction of the glucocorticoid receptor affinity was observed (Spahn et al., 1996). Still, lymphocyte proliferation was inhibited less by budesonide in the glucocorticoid-resistant asthmatics than in the glucocorticoid-sensitive patients (Spahn et al., 1996). Mechanisms other than decreased glucocorticoid receptor affinity seem therefore to be of more importance in glucocorticoid resistance.
We recently reported that IL-2 and IL-4 in combination induce a functional resistance to glucocorticoids (Larsson et al., 1997). GM-CSF production became resistant to three glucocorticoids, which differed in structure as well as in potency. DNA-measurements using a vital dye in the present study, suggest that IL-2 and IL-4 indeed change the functional cell response to glucocorticoids rather than influence cell growth. This is in agreement with data reported by Kam et al. (1993). When using the [3H]-thymidine assay they could not detect any effects of IL-2 and IL- 4 on proliferation of mitogen stimulated mononuclear blood cells. Thus, the reduced anti-inflammatory effects of glucocorticoids in the resistant patients might be explained by a reduced functional cell response to glucocorticoids, i.e. reduced cytokine glucocorticoid sensitivity, due to increased IL-2 and IL-4 production.
In the present study we found that either IL-2 or IL-4 alone reduce the glucocorticoid sensitivity of GM-CSF production to the same degree as IL-2 and IL-4 in combination. There are several independent reports supporting our data indicating that IL-2 alone may cause glucocorticoid resistance. Exogenous IL-2 counteracted glucocorticoid inhibition of T cell proliferation in mononuclear blood cells as well as glucocorticoid mediated apoptosis in a cell line (Walker et al., 1987; Hazcku et al., 1994; Guizani et al., 1996). In this cell line (MLA-E7T; gibbon ape T cell line), the IL-2 induced glucocorticoid resistance correlated with induced expression of the transcription factor activator protein-1 (AP-1). Interestingly, AP-1 levels were increased in mononuclear blood cells of glucocorticoid-resistant asthmatics and the binding of the glucocorticoid receptor to DNA was reduced (Adcock et al., 1995a,1995b). Adcock and colleagues therefore suggested that increased levels of AP-1 cause glucocorticoid resistance by interfering with the glucocorticoid receptor and thereby reduce glucocorticoid repression of cytokine genes. A similar reduction of the glucocorticoid receptor-DNA binding was in fact induced by IL-2 and IL-4 (Leung et al., 1997). However, IL-2 or IL-4 were not studied separately nor were effects on transcription factors (Leung et al., 1997). In the present study, IL-4 alone also reduced budesonide inhibition of GM-CSF production. However, in the cell line (MLA-E7T) discussed above, IL-4 did not induce expression of AP-1 or rescue the cells from glucocorticoid mediated apoptosis (Guizani et al., 1996). The similar effects of IL-2, IL-4, or IL-2 and IL- 4 on GM-CSF production in the present study might possibly depend on shared signalling elements of the IL-2 and the IL-4 receptors (Kondo et al., 1993).
The combination of IL-2 and IL-4 reduced IL-10 production in the present study, as previously reported by Irusen (Irusen et al., 1998). Also the budesonide sensitivity of IL-10 production was reduced by IL-2, IL-4 or IL-2 and IL-4 in combination, as observed for GM-CSF production. To our knowledge, effects of IL-2, IL-4, or IL-2+IL-4 on budesonide sensitivity of IL-10 production have not been described previously. IL-10 levels in glucocorticoid-resistant asthmatics have neither been reported. However, the present study together with the reported increase in expression of IL-2 and IL-4 mRNA in glucocorticoid-resistant asthmatics (Leung et al., 1995) might suggest reduced IL-10 production in these patients, caused by increased IL-2 and IL-4 production.
We also investigated the glucocorticoid effects on TNF-α, IL-8 and IL-12 production for comparisons with findings in glucocorticoid-resistant asthmatics. In glucocorticoid-resistant and glucocorticoid-sensitive asthmatics the in vitro glucocorticoid sensitivity of TNF-α production by monocytes does not differ (Lane et al., 1993). In agreement, the glucocorticoid sensitivity of TNF-α production did not change with IL-2, IL-4, or IL-2 and IL-4 pre-treatment in the present study. In contrast, IL-8 mRNA in mononuclear blood cells of glucocorticoid-resistant asthmatics was resistant to in vitro glucocorticoid treatment (Adcock et al., 1995b) while IL-8 production in the present study did not become glucocorticoid-resistant with IL-2, IL-4, or IL-2 and IL-4 treatment. This discrepancy between our in vitro model and the glucocorticoid resistant patients could possibly depend on methodological differences. In the asthmatic patients IL-8 was studied at the mRNA level while we measured secreted IL-8 protein. It is also possible that the glucocorticoid resistance induced by IL-2, IL-4, or IL-2 and IL-4 in vitro differs from the resistance in asthmatic patients. Furthermore, in glucocorticoid resistant asthmatics the number of cells in bronchial biopsies expressing mRNA of the IL-12 subunit p40 was increased and did not decrease after glucocorticoid therapy (Naseer et al., 1997). In the present study, we also demonstrated IL-12 stimulation by IL-2, IL-4 or the combination of IL-2 and IL-4. Although IL-12 was not detected in vehicle treated cultures, we demonstrated in a previous study that IL-12 production is totally inhibited by budesonide (Larsson & Linden, 1998). Thus, it seems as if IL-2, IL-4 or IL-2 and IL-4 in combination did not induce resistance of IL-12 production to budesonide. In the present study, bioactive IL-12 protein (heterodimeric p70) was studied while only mRNA of one IL-12 subunit (p40) was studied in the asthmatics (Naseer et al., 1997). Monitoring p40 mRNA following corticosteroid therapy might not reflect the actual change in production of bioactive IL-12 and may explain the discrepant results. Still, IL-12 is one of the key cytokines in the Th1-Th2 balance and a possible increase of IL-12 in glucocorticoid-resistant asthma is intriguing and needs to be studied further.
IL-2 and IL-4 had different effects, alone and in combination, on the various cytokines assayed in the present study. While IL-2 potently stimulated GM-CSF, IL-8, IL-12 and TNF-α production, IL-10 was weakly decreased. IL-4 alone had no effects on GM-CSF, IL-8 and IL-10 production but stimulated IL-12 and TNF-α production. As in the present study, stimulation of IL-12 and TNF-α production by IL-4 has been observed when mononuclear cells were pretreated with IL-4 for more than 20 h before LPS-stimulation. (D'Andrea et al., 1995). The combination of IL-2 and IL-4 stimulated IL-12 production even more than IL-2 alone, while IL-8 and IL-10 production was decreased. GM-CSF and TNF-α production was more stimulated by the combination than with IL-4 alone, but less than with IL-2 alone. The present results are in agreement with previously reported data on inhibitory effects of IL-4 on IL-2 stimulated cytokine production by mononuclear blood cells (Bello-Fernandez et al., 1991). Of the two cytokines that became glucocorticoid resistant, GM-CSF was stimulated by IL-2 and IL-4 while IL-10 was decreased. Of the non-resistant cytokines, IL-8 was decreased and both IL-12 and TNF-α were stimulated. Thus, the induced resistance seemed not to depend on the level of cytokine stimulation.
To our knowledge there is no information on IL-2 and IL-4 protein levels in the airways of glucocorticoid-resistant asthmatics, only information on increased IL-2 and IL-4 mRNA expression (Leung et al., 1995). Studies in glucocorticoid-sensitive asthmatics show however that mononuclear blood cells have the capacity to produce high local concentrations of both IL-2 and IL-4 (Nimmagadda et al., 1997). Thus, the reduced anti-inflammatory effects of glucocorticoids in the glucocorticoid-resistant asthmatics might possibly be explained by reduced glucocorticoid sensitivity of key cytokines such as GM-CSF, due to increased IL-2 and IL-4 production.
In conclusion, the present study indicates that the glucocorticoid resistance of cytokine production induced by IL-2, IL-4, or IL-2 and IL-4 in combination, is not general at the cytokine production level. GM-CSF and IL-10 production became resistant to budesonide inhibition, while the glucocorticoid sensitivity of TNF-α, IL-8 and IL-12 production did not change. Further, we found no evidence that the combination of IL-2 and IL-4 is needed for induction of glucocorticoid resistance of GM-CSF production. Either IL-2 or IL-4 alone counteracts the inhibitory effect of budesonide on GM-CSF production to the same degree as the combination of IL-2 and IL-4.
Acknowledgments
We thank Suzanne Glans and Inger Forsbrant for the blood sampling and Per Larsson for the statistical analysis. We also thank the Swedish Heart and Lung Foundation for grants.
Abbreviations
- AP-1
activator protein-1
- BSA
bovine serum albumin
- ELISA
enzyme linked immuno sorbent assay
- GM-CSF
granulocytemacrophage colonystimulating factor
- IL-2
interleukin-2
- IL-4
interleukin-4
- IL-8
interleukin-8
- IL-10
interleukin-10
- IL-12
interleukin-12
- LPS
lipopolysaccharide
- PBS
phosphate buffered saline
- TNF-α
tumour necrosis factor-α
References
- ADCOCK I.M., LANE S.J., BROWN C.R., LEE T.H., BARNES P.J. Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. J. Exp. Med. 1995a;182:1951–1958. doi: 10.1084/jem.182.6.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADCOCK I.M., LANE S.J., BROWN C.R., PETERS M.J., LEE T.H., BARNES P.J. Differences in binding of glucocorticoid receptor to DNA in steroid-resistant asthma. J. Immunol. 1995b;154:3500–3505. [PubMed] [Google Scholar]
- ALVAREZ J., SURS W., LEUNG D.Y.M., IKLÉ D., GELFAND E.W., SZEFLER S.J. Steroid-resistant asthma: Immunologic and pharmacologic features. J. Allergy Clin. Immunol. 1992;89:714–721. doi: 10.1016/0091-6749(92)90379-g. [DOI] [PubMed] [Google Scholar]
- BAAN C.C., NIESTERS H.G.M., BALK A.H.M.M., MOCHTAR B., ZONDERVAN P.E., VAN GELDER T., WEIMAR W. Steroid resistance in clinical heart transplantation: the role of simultaneous IL-2 and IL-4 mRNA expression. Transplantation Proceedings. 1996;28:3239–3240. [PubMed] [Google Scholar]
- BLO-FERNANDEZ C., OBLAKOWSKI P., MEAGER A., DUNCOMBE A.S., RILL D.M., HOFFBRAND A.V., BRENNER M.K. IL-4 acts as a homeostatic regulator of IL-2 induced TNF and IFN-γ. Immunology. 1991;72:161–166. [PMC free article] [PubMed] [Google Scholar]
- BLAHETA R.A., FRANZ M., AUTH M.K.H., WENISCH H.J.C., MARKUS B.H. A rapid non-radioactive fluorescence assay for the measurement of both cell number and proliferation. J. Immunol. Methods. 1991;142:199–206. doi: 10.1016/0022-1759(91)90107-q. [DOI] [PubMed] [Google Scholar]
- CARMICHAEL J., PATERSON I.C., DIAZ P., CROMPTON G.K., KAY A.B., GRANT I.W.B. Corticosteroid resistance in chronic asthma. Brit. Med. J. 1981;282:1419–1422. doi: 10.1136/bmj.282.6274.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHIKANZA I.C., PANAYI G.S. The effects of hydrocortisone on in vitro lymphocyte proliferation and interleukin-2 and -4 production in corticosteroid sensitive and resistant subjects. Eur. J. Clin. Invest. 1993;23:845–850. doi: 10.1111/j.1365-2362.1993.tb00740.x. [DOI] [PubMed] [Google Scholar]
- CORRIGAN C.J., BROWN P.H., BARNES N.C., TSAI J.-J., FREW A.J., KAY A.B. Glucocorticoid resistance in chronic asthma: Peripheral blood T lymphocyte activation and comparison of the T lymphocyte inhibitory effects of glucocorticoids and cyclosporin A. Am. Rev. Respir. Dis. 1991;144:1026–1032. doi: 10.1164/ajrccm/144.5.page. [DOI] [PubMed] [Google Scholar]
- D'ANDREA A., MA X., TE-AMEZAGA M., PAGANIN C., TRINCHIERI G. Stimulatory and inhibitory effects of interleukin-4 and -13 on the production of cytokines by human peripheral blood mononuclear cells. J. Exp. Med. 1995;181:537–546. doi: 10.1084/jem.181.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRIERI M., MADDEN J. Chronic steroid-resistant urticaria. Ann. Allergy. 1993;70:13–20. [PubMed] [Google Scholar]
- GUIZANI L., PEIN-WOLFF M., BREARD J., BINETRUY B., BERTOGLIO J. Mechanisms in interleukin-2 protection against glucocorticoid-induced apoptosis: Regulation of AP-1 and glucocorticoid receptor transcriptional activities. J. Interferon. Cyt. Res. 1996;16:601–609. doi: 10.1089/jir.1996.16.601. [DOI] [PubMed] [Google Scholar]
- HACZKU A., ALEXANDER A., BROWN P., ASSOUFI B., BAIQING L., KAY A.B., CORRIGAN C. The effect of dexamethasone, cyclosporin and rapamycin on T-lymphocyte proliferation in vitro: Comparison of cells from patients with glucocorticoid-sensitive and -resistant chronic asthma. J. Allergy Clin. Immunol. 1994;93:510–519. doi: 10.1016/0091-6749(94)90361-1. [DOI] [PubMed] [Google Scholar]
- IRUSEN I., CHUNG K.F., BARNES P.J., ADCOCK I.M. P38 mitogen activated protein kinase mediates IL-2 and IL-4 mediated changes in glucocorticoid receptor affinity. Eur. Resp. J. 1998;12 suppl. 28, 116s:P837. [Google Scholar]
- KAM J.C., SZEFLER S.J., SURS W., SHER E.R., LEUNG D.Y.M. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J. Immunol. 1993;151:3460–3466. [PubMed] [Google Scholar]
- KAY A.B., DIAZ P., CARMICHAEL J., GRANT I.W.B. Corticosteroid-resistant chronic asthma and monocyte complement receptors. Clin. Exp. Immunol. 1981;44:576–580. [PMC free article] [PubMed] [Google Scholar]
- KLEMM J., SURS B.S., SPAHN J.D., SZEFLER S.J., LEUNG D.Y.M. Alterations in glucocorticoid receptor binding to DNA contributes to steroid resistant asthma. J. Allergy Clin. Immunol. 1996;97:A716. [Google Scholar]
- KONDO M., TAKESHITA T., ISHII N., NAKAMURA M., WATANABE S., ARAI K., SUGAMURA K. Sharing of the interleukin-2 (IL-2) receptor γ chain between receptors for IL-2 and IL-4. Science. 1993;262:1874–1877. doi: 10.1126/science.8266076. [DOI] [PubMed] [Google Scholar]
- LANE S.J., WILKINSON J.R.W., COCHRANE G.M., LEE T.H., ARM J.P. Differential in vitro regulation by glucocorticoids of monocyte-derived cytokine generation in glucocorticoid-resistant bronchial asthma. Am. Rev. Respir. Dis. 1993;147:690–696. doi: 10.1164/ajrccm/147.3.690. [DOI] [PubMed] [Google Scholar]
- LARSSON S., BRATTSAND R., LINDEN M. Interleukin-2 and -4 induce resistance of granulocyte-macrophage colony-stimulating factor to corticosteroids. Eur. J. Pharm. 1997;334:256–271. doi: 10.1016/s0014-2999(97)01202-8. [DOI] [PubMed] [Google Scholar]
- LARSSON S., LINDEN M. Effects of a corticosteroid, budesonide, on production of bioactive IL-12 by human monocytes. Cytokine. 1998;10:786–789. doi: 10.1006/cyto.1998.0362. [DOI] [PubMed] [Google Scholar]
- LEUNG D.Y.M., HAMID Q., VOTTERO A., SZEFLER S.J., SURS W., MINSHALL E., CHROUSOS G.P., KLEMM D.J. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor β. J. Exp. Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEUNG D.Y.M., MARTIN R.J., SZEFLER S.J., SHER E.R., YING S., KAY A.B., HAMID Q. Dysregulation of interleukin 4, interleukin 5 and interferon γ gene expression in steroid-resistant asthma. J. Exp. Med. 1995;181:33–40. doi: 10.1084/jem.181.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NASEER T., MINSHALL E.M., LEUNG D.Y.M., LABERGE S., ERNST P., MARTIN R.J., HAMID Q. Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. Am. J. Respir. Crit. Care. Med. 1997;155:845–851. doi: 10.1164/ajrccm.155.3.9117015. [DOI] [PubMed] [Google Scholar]
- NIMMAGADDA S.R., SZEFLER S.J., SPAHN J.D., SURS W., LEUNG D.Y.M. Allergen exposure decreases glucocorticoid receptor binding affinity and steroid responsiveness in atopic asthmatics. Am. J. Respir. Crit. Care. Med. 1997;155:87–93. doi: 10.1164/ajrccm.155.1.9001294. [DOI] [PubMed] [Google Scholar]
- POZNANSKY M.C., GORDON A.C.H., DOUGLAS J.G., KRAJEWSKI A.S., WYLLIE A.H., GRANT I.W.B. Resistance to methylprednisolone in cultures of blood mononuclear cells from glucocorticoid-resistant asthmatic patients. Clin. Sci. 1984;67:639–645. doi: 10.1042/cs0670639. [DOI] [PubMed] [Google Scholar]
- SCHWARZ H.J., LOWELL F.C., MELBY J.C. Steroid resistance in bronchial asthma. Ann. Int. Med. 1968;69:493–499. doi: 10.7326/0003-4819-69-3-493. [DOI] [PubMed] [Google Scholar]
- SHER E.R., LEUNG D.Y.M., SURS W., KAM J.C., ZIEG G., KAMADA A.K., SZEFLER S.J. Steroid-resistant asthma: Cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J. Clin. Invest. 1994;93:33–39. doi: 10.1172/JCI116963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPAHN J.D., LANDWEHR L.P., NIMMAGADDA S., SURS W., LEUNG D.Y.M., SZEFLER S.J. Effects of glucocorticoids on lymphocyte activation in patients with steroid-sensitive and steroid-resistant asthma. J. Allergy Clin. Immunol. 1996;98:1073–1079. doi: 10.1016/s0091-6749(96)80194-1. [DOI] [PubMed] [Google Scholar]
- WALKER K.B., POTTER J.M., HOUSE A.K. Interleukin-2 synthesis in the presence of steroids: a model of steroid resistance. Clin. Exp. Immunol. 1987;68:162–167. [PMC free article] [PubMed] [Google Scholar]
- WILKINSON J.R.W., CREA A.E.G., CLARK T.J.H., LEE T.H. Identification and characterisation of a monocyte-derived neutrophil-activating factor in corticosteroid-resistant bronchial asthma. J. Clin. Invest. 1989;84:1930–1941. doi: 10.1172/JCI114381. [DOI] [PMC free article] [PubMed] [Google Scholar]