

Abstract
The rostral ventromedial medulla (RVM) is thought to play a crucial role in the antinociceptive actions of cannabinoids. This study examined the actions of the cannabinoid receptor agonist, WIN55,212-2, on membrane properties and GABAergic synaptic transmission in RVM neurons using whole cell patch clamp recordings in brain slices.
WIN55,212-2 (3 μM) had no effect on membrane K+ conductance of primary or secondary RVM neurons. Primary neurons responded to the κ-opioid receptor agonist U69,593 (300 nM–1 μM). Secondary neurons responded to the μ,δ-opioid receptor agonist met-enkephalin (10 μM).
WIN55,212-2 reduced the amplitude of electrically evoked (GABAergic) inhibitory postsynaptic currents (IPSCs) in all neurons (58%, pEC50=6.2±0.1). The inhibition was reversed by the CB1 receptor selective antagonist, SR141716 (3 μM). WIN55,212-2 also produced relative facilitation of the second IPSC to paired evoked IPSCs.
WIN55,212-2 and met-enkephalin reduced the rate of spontaneous miniature IPSCs in all cells (44 and 53%), but had no effect on their amplitude distributions or kinetics.
These results suggest that the antinociceptive actions of cannabinoids within RVM are primarily due to presynaptic inhibition of GABAergic neurotransmission. The neuronal substrates of cannabinoid actions in RVM therefore differ from those of opioids, which have both pre- and postsynaptic inhibitory actions.
Keywords: Cannabinoid, opioid, antinociception, rostral ventromedial medulla, nucleus raphe magnus, potassium current, synaptic transmission, presynaptic inhibition, GABAergic neurotransmission
Introduction
Cannabinoid receptor agonists produce antinociception and synergistically enhance the analgesic actions of opioids after either systemic or central administration (Smith et al., 1998; see Howlett, 1995 for review). Microinjection studies have implicated similar supraspinal sites in the antinociceptive activity of both opioids and cannabinoids (Lichtman et al., 1996; Martin et al., 1998; Meng et al., 1998). The rostral ventromedial medulla (RVM) is thought to be critical for the supraspinal antinociceptive actions of both opioids and cannabinoids (Fields et al., 1991; Martin et al., 1998; Meng et al., 1998). The RVM, particularly the nucleus raphe magnus, forms a component of a descending inhibitory network that modulates nociceptive neurotransmission at the level of the dorsal horn of the spinal cord (Fields et al., 1991). μ-Opioid agonists have no direct actions on, but inhibit GABAergic synaptic transmission onto primary RVM cells in vitro, which are thought to represent serotonergic neurons that project to the dorsal horn of the spinal cord (Pan et al., 1990; 1993; 1997). In contrast, μ-Opioid receptor agonists directly hyperpolarize secondary RVM cells by increasing a K+ conductance (Pan et al., 1990; 1997). These observations have led to the hypothesis that μ-opioids disinhibit descending antinociceptive neurons in the RVM via direct somatic inhibition of secondary, GABAergic interneurons.
Microinjections of cannabinoid agonists into the RVM produce antinociception (Martin et al., 1998). The antinociceptive activity of systemically injected cannabinoids is also dependent on functional integrity of the RVM, suggesting that this is a necessary site for the antinociceptive actions of these drugs (Meng et al., 1998). However, the cellular mechanisms of action of cannabinoids within CNS regions which mediate antinociception are unknown. The present study examined the effects of cannabinoid receptor agonists on primary and secondary cells, and on GABAergic synaptic transmission in the RVM.
Methods
Sprague-Dawley rats (12–24 days old) were anaesthetized with halothane, decapitated and coronal brain slices were cut (250 μm) at the level of the facial nerve in ice cold artificial cerebrospinal fluid (ACSF). The slices were maintained at 34°C in a submerged chamber containing ACSF equilibrated with 95% O2 and 5% CO2, and then transferred to a superfusing chamber (32°C) for recording (Vaughan et al., 1997). The ACSF contained (mM): NaCl, 126; KCl, 2.5; NaH2PO4, 1.4; MgCl2, 1.2; CaCl2, 2.4; glucose, 11; NaHCO3, 25.
Neurons in the RVM were visualized in the triangular midline region dorsal to the pyramidial tracts (Pan et al., 1990) using infra-red Nomarski optics. Whole-cell patch clamp recordings of postsynaptic K+ currents (holding potential −60 mV) were performed using a K+-gluconate based internal solution using patch electrodes (2–5 MΩ) containing (mM): potassium gluconate, 140; NaCl, 15; MgCl2, 1; HEPES, 10; EGTA, 11; MgATP, 2; NaGTP, 0.25. Whole cell voltage clamp recordings of inhibitory postsynaptic currents (IPSCs, holding potential −74 mV) were made using a CsCl based internal solution containing (mM): CsCl, 140; EGTA, 10; HEPES, 5; CaCl2, 2; and MgATP, 2 (both solutions had pH 7.3, osmolarity 270–290 mosmol l−1). Series resistance (<15 MΩ) was compensated by 80% and continuously monitored during experiments. Liquid junction potentials of −12 mV for K+-gluconate and −4 mV for CsCl based internal solutions were corrected.
Electrically-evoked evoked IPSCs (eIPSCs) were elicited via bipolar tungsten stimulating electrodes placed 100–400 μm dorsolateral to the recording electrode (rate 0.05–0.067 Hz, stimuli: 5–70 V, 20–400 μs), in the presence of CNQX (3 μM). For paired pulse experiments, two stimuli of identical strength were applied with an inter-stimulus interval of 50 ms. Spontaneous miniature IPSCs (mIPSCs) were obtained in the presence of TTX (0.3 μM) and CNQX (3 μM), and recorded on video tape (via a SONY PCM501). Evoked and miniature IPSCs were filtered (1 kHz low-pass filter) and sampled at 5 kHz for on-line and later off-line analysis (Axograph 4.0, Axon). The amplitudes of eIPSCs were calculated to construct time plots of eIPSC amplitude. Miniature IPSCs above a preset threshold (three to five standard deviations above baseline noise) were automatically detected by a sliding template algorithm, then manually checked off-line. The mIPSCs were counted in 20–60 s epochs to construct time plots of mIPSC rate and were ranked by amplitude to construct probability density functions.
Stock solutions of all drugs were diluted to working concentrations using ACSF immediately before use and applied by superfusion. Stock solutions of cannabinoids were prepared in dimethylsulphoxide and diluted using ACSF to a final concentration of 0.03–0.1% dimethylsulphoxide and 0.05% bovine serum albumin to decrease adsorption to the perfusion system. The superfusion system was dismantled and rinsed with ethanol after each recording that involved superfusion of a cannabinoid. Stock solutions of all other drugs were made in distilled water, or added directly to the ACSF. Met-enkephalin (methionine-enkephalin), bicuculline methiodide, and serotonin (5-hydroxytryptamine) were obtained from Sigma (Sydney, Australia); CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Pen-Thr-NH2) was donated by the National Institute on Drug Abuse (U.S.A.); CNQX (6-cyano-7-nitroquinoxaline-2,3-dione) was from Tocris Cookson (Bristol, U.K.); naloxone hydrochloride, nor-BNI (nor-binaltorphimine dihydrochloride), WIN55,212 mesylate and U69,593 were from Research Biochemicals Inc. (Natick, MA, U.S.A.); TTX (tetrodotoxin) was from Alomone (Jerusalem, Israel); SR141716 (N-piperidino-5-(4-chlorophenyl)-1-(2,4-di chlorophenyl)-4-methyl-3-pyrazole-carboxamide) was donated by Sanofi Recherche. All pooled data are expressed as means±s.e.mean, and statistical comparisons made using paired t-tests.
Results
RVM neurons were voltage clamped at −60 mV and classified as being primary or secondary cells on the basis of their postsynaptic responses to μ-opioid and κ-opioid receptor activation (Pan et al., 1990; 1997). In primary neurons, superfusion of the κ-opioid receptor agonist, U69,593 (300 nM–1 μM), produced an outward current which was reversed by the κ-opioid receptor antagonist nor-BNI (300 nM, n=4, Figure 1a and c). Superfusion of the selective μ,δ-opioid receptor agonist met-enkephalin (10 μM) produced no change in membrane current in any primary neurons (Figure 1a and c, n=4). In secondary neurons, superfusion of met-enkephalin (10 μM, n=8) produced an outward current which was reversed by the μ-opioid receptor antagonist CTAP (1 μM, n=7). Superfusion of U69,593 (300 nM–1 μM) produced no change in membrane current in any secondary neurons (Figure 1b and c, n=8).
Figure 1.
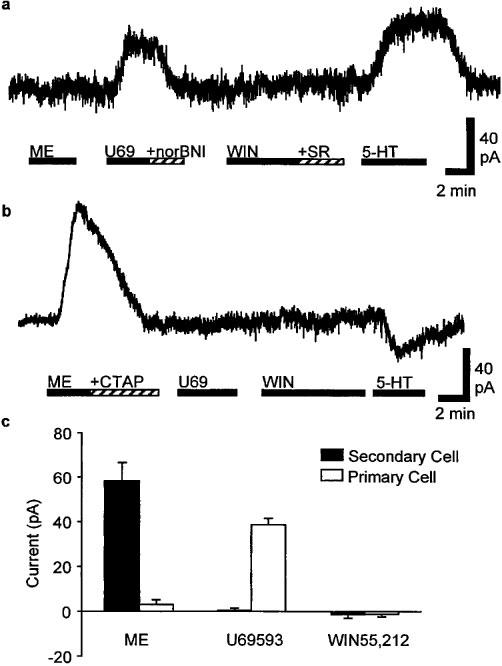
WIN55,212-2 does not affect postsynaptic membrane currents in primary or secondary RVM neurons. Representative membrane current responses of (a) primary and (b) secondary RVM neurons during superfusion of U69,593 (300 nM, U69), nor-BNI (300 nM), met-enkephalin (10 μM, ME), CTAP (1 μM), WIN55,212-2 (3 μM, WIN), SR141716 (3 μM, SR) and serotonin (3 μM, 5-HT). Neurons were voltaged clamped at −60 mV. (c) Pooled membrane currents produced by met-enkephalin (10 μM, ME), U69,593 (1 μM) and WIN55,212-2 (3 μM) in primary (open columns, n=4) and secondary neurons (filled columns, n=8).
Superfusion of the cannabinoid receptor agonist, WIN55,212-2 (3 μM) did not produce an outward current in any of the primary (n=4) or secondary (n=8) cells tested (Figure 1a–c). Subsequent addition of the cannabinoid CB1 receptor antagonist SR141716 (3 μM) also produced no effects on membrane currents (n=6, Figure 1a). Both primary and secondary neurons responded to serotonin (3–10 μM) with either outward (123±77 pA, n=4), or inward currents (−95±26 pA, n=5) (Figure 1a and b).
Electrical stimulation in the presence of CNQX (3 μM) evoked bicuculline (30 μM) sensitive inhibitory postsynaptic currents (eIPSCs) in RVM neurons (Figure 2b). These experiments were carried out using Cs+ filled pipettes to block post-synaptic K+ currents activated by opioid agonists, thus the cells could not be readily classified as primary or secondary cells. WIN55,212-2 (3 μM) reduced the amplitude of eIPSCs by an average of 58±6% in all cells tested (n=7, P<0.005, Figure 2a and b). The WIN55,212-2 induced reduction in eIPSC amplitude was reversed by addition of the CB1 selective antagonist SR141716 (3 μM, reversed to 89±6% of control, n=6, P>0.05, Figure 2b). The reduction in eIPSC amplitude produced by WIN55,212-2 was dose-dependent, with a pEC50 of 6.2±0.1 (Figure 2c). Application of WIN55,212-2 had no effect on the membrane current, or the conductance of the neurons at −74 mV (Cs+ filled electrodes used). Met-enkephalin (10 μM) reduced the amplitude of eIPSCs by an average of 61±6% in all cells tested (n=8, P<0.05). This inhibition was reversed by the addition of CTAP (1 μM, n=4), or by naloxone (1 μM, n=4) (pooled reversed to 92±7% of control, P>0.05, Figure 2b).
Figure 2.
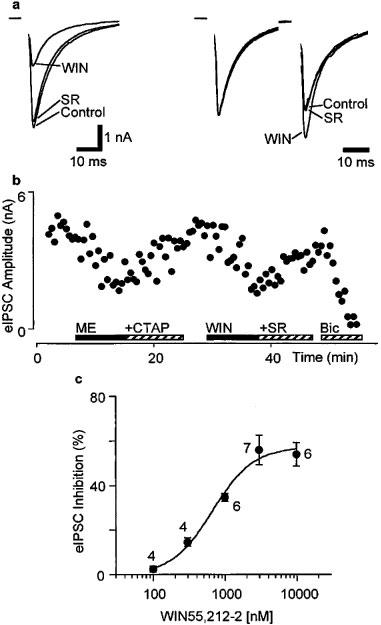
Evoked IPSCs in RVM neurons are inhibited by WIN55,212-2. (a) The left panel shows the evoked IPSCs (average of eight IPSCs in a single neuron) in the presence of CNQX (3 μM) before drug application (Control), in the presence of WIN55,212-2 (3 μM, WIN) and then after addition of SR141716 (3 μM, SR, superimposed trace). In the right panel, the first of a pair of evoked IPSCs has been normalized to the same amplitude in the presence and absence of drugs to demonstrate relative facilitation of the second IPSC in the presence of WIN55,212-2. Stimulus artefacts have been blanked. (b) Time course of the inhibition of the evoked IPSC amplitude in the same neuron as in (a) during application of met-enkephalin (10 μM, ME) and addition of CTAP (1 μM), then during application of WIN55,212-2 (3 μM, WIN) and addition of SR141716 (3 μM, SR). The evoked IPSC was completely abolished by superfusion of bicuculline (30 μM, Bic). Each point is the mean of two consecutive eIPSCs. (c) Concentration-response relationship for percentage inhibition of evoked IPSC amplitude produced by WIN55,212-2. Each point shows the mean±s.e.mean of responses of several different neurons, with the number of neurons indicated adjacent to each point. A logistic function was fitted (Kaleidograph, Synergy Software) to determine the pEC50 (6.2±0.1, slope factor=1.5±0.4).
Under control conditions, the mean ratio of the amplitude of paired eIPSCs (inter-stimulus interval 50 ms) was 1.25±0.13, with both paired-pulse facilitation and depression being observed (S2/S1 range=0.89–1.64, n=6). Superfusion of WIN55,212-2 (3 μM) produced a significant increase in the mean ratio of S2/S1 (132±14% of control, P<0.05), which was reversed by the addition of SR141716 (3 μM, 99±5% of control, P>0.05) (n=5, Figure 2a).
Spontaneous miniature IPSCs (mIPSCs) were readily observed during whole-cell voltage clamp recordings in the presence of CNQX (3 μM) and TTX (0.3 μM). This concentration of TTX prevented Na+-dependent action potentials and evoked postsynaptic currents (data not shown). Superfusion of WIN55,212-2 (3 μM) reduced the frequency of mIPSCs (Figure 3a and b), but had no effect on their amplitude distributions or kinetics (Figure 3c and d). On average, the mean mIPSC frequency was reduced by 44±2% during superfusion of WIN55,212-2 (P<0.005), whereas the mean amplitude was 101±3% of control in these neurons (P>0.05, n=7). The reduction in mIPSC rate produced by WIN55,212-2 was reversed by the addition of SR141716 (3 μM, 101±4% of control, P>0.05, n=6, Figure 3a). In the same neurons, superfusion of met-enkephalin (10 μM) reduced the mean mIPSC frequency by 53±6% (P<0.005) and the mean mIPSC amplitude by 8±6% (P>0.05, n=5, Figure 3a). The met-enkephalin induced reduction in mIPSC rate was reversed by the addition of naloxone (1 μM, 96±6% of control, P>0.05, n=5).
Figure 3.
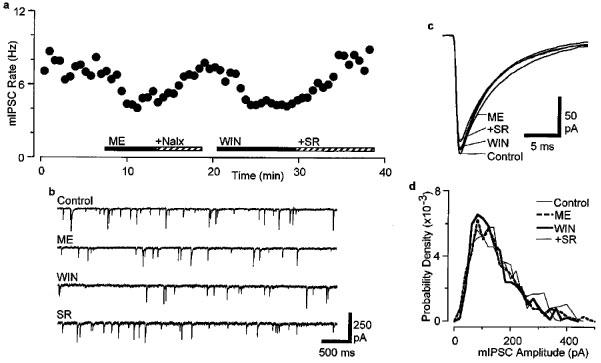
WIN55,212-2 decreases the frequency of spontaneous mIPSCs. (a) Time course of mIPSC rate during superfusion of met-enkephalin (10 μM, ME) and addition of naloxone (1 μM, Nalx), then during superfusion of WIN55,212-2 (3 μM, WIN) and addition of SR141716 (3 μM, SR). Recordings were carried out in the presence of CNQX (3 μM) and TTX (0.3 μM). (b) Raw current traces of mIPSCs before (Control) and during superfusion of met-enkephalin (ME) and WIN55,212-2 (WIN), then after addition of SR141716 (SR). (c) Averaged traces of mIPSCs before and during superfusion of met-enkephalin and WIN55,212-2, then after addition of SR141716 (number of events=778, 523, 434, 766 for 120 s epochs of Control, ME, WIN and SR, respectively). (d) Probability density distributions (bin width=20 pA) of mIPSC amplitude for the epochs indicated in (c). (a–d) are taken from one neuron.
Discussion
This study demonstrates that CB1 cannabinoid receptor activation inhibits GABAergic synaptic transmission in rat RVM. Like μ-opioids, cannabinoids act presynaptically to reduce the probability of transmitter release from GABAergic terminals within the RVM. However, unlike μ- and κ-opioids, cannabinoids do not produce a postsynaptic inhibitory increase in K+ conductance in RVM neurons.
Cannabinoids acted via CB1 cannabinoid receptors to inhibit GABAergic synaptic transmission in rat RVM, as has been demonstrated for GABAergic and glutamatergic synaptic transmission in other brain regions (Shen et al., 1996; Chan et al., 1998; Levenes et al., 1998; Szabo et al., 1998). Inhibition of GABAergic synaptic transmission produced by the cannabinoid agonist WIN55,212 was reversed by the CB1 specific antagonist SR141716 (3 μM). These results are consistent with the presence of cannabinoid receptor binding sites in the RVM, which have been previously defined by autoradiography using the cannabinoid radioligand [3H]-CP55,940 (Herkenham et al., 1991), as well as the presence of CB1 mRNA using in situ hybridization (Matsuda et al., 1993).
The potency of WIN55,212 (pEC50=6.2) was similar to that previously observed in other studies using brain slices (Levenes et al., 1998; Chan et al., 1998; Szabo et al., 1998), hippocampal cultures (Deadwyler et al., 1993) and oocytes (Henry & Chavkin, 1995). However, the potency of WIN55,212 was much greater in other studies using hippocampal cultures (Shen et al., 1996; Twitchell et al., 1997) and transfected cells (Mackie et al., 1995). The difference in agonist potency between slices, cultures and transfected cells may be due to the lipophilic nature of cannabinoids which results in adsorption to the perfusion system and reduced penetration into the slice and access to the synaptic cleft. This precludes any meaningful comparison of the actual concentration of cannabinoids reaching cells between brain slice preparations, isolated cells and functional analgesia studies. The differences may also be due to overexpression of cannabinoid receptors in transfected cells, variations in receptor reserve between different brain regions particularly given the relatively low levels of cannabinoid binding sites in the RVM (Herkenham et al., 1991), and differences receptor/effector coupling.
A number of observations demonstrated that the cannabinoid induced inhibition of GABAergic synaptic transmission was likely to be mediated by a presynaptic mechanism, as previously shown for GABAergic transmission in the basal ganglia (Chan et al., 1998; Szabo et al., 1998) and for glutamatergic transmission in hippocampal pyramidal cells (Shen et al., 1996) and cerebellar purkinje cells (Levenes et al., 1998). Firstly, WIN55,212 and met-enkephalin reduced the rate of spontaneous miniature IPSCs without having any effect on their amplitude distributions or kinetics, which provides direct evidence for a presynaptic site of action. Secondly, the inhibition of evoked IPSCs by WIN55,212 was associated with relative facilitation of the second IPSC to paired stimuli. This paired-pulse facilitation is a presynaptic process, arising from an increase in the probability of transmitter release (e.g. Isaacson & Walmsley, 1995). Lastly, cannabinoids had no postsynaptic actions on RVM neurons (see below). Thus, activation of presynaptic CB1 and μ-opioid receptors (Pan et al., 1990) both act to reduce the probability of transmitter release from GABAergic terminals within the RVM.
In contrast to μ- and κ-opioids, WIN55,212-2, at a concentration which produced maximal inhibition of evoked IPSCs, did not increase an inwardly rectifying K+ conductance in either primary or secondary RVM neurons. The absence of postsynaptic actions of cannabinoid agonists on RVM neurons is consistent with the relatively low density of cannabinoid receptor binding sites and CB1 mRNA in the region (Herkenham et al., 1991; Matsuda et al., 1993). These findings also suggest that presynaptic inhibition of GABAergic transmission by cannabinoids may be confined to GABAergic terminals rather than direct somatic actions on RVM neurons. However, cannabinoid receptor stimulation has been reported to both increase K+ conductance and decrease Ca2+ conductance when CB1 receptors are expressed in other cells (Mackie et al., 1995), and cannabinoid actions on Ca2+ channels were not examined in the present study.
While both cannabinoids and μ-opioids produce antinociception within the RVM, their actions appear to differ at the cellular level. In the present study it was found that cannabinoid agonists act via presynaptic CB1 receptors to inhibit transmitter release from GABAergic terminals, but have no postsynaptic actions. In contrast, μ-opioids directly inhibit secondary, presumably GABAergic RVM interneurons via an increased K+ conductance, in addition to a presynaptic inhibition of GABA release (Pan et al., 1990; 1997). μ-Opioids are thought to produce antinociception in RVM, at least in part, via disinhibition of neurons which descend to the dorsal horn of the spinal cord (Fields et al., 1991; Pan et al., 1997). The present results suggest that cannabinoid agonists may also produce antinociception via a disinhibitory mechanism, although it is unclear whether or not the same populations of GABAergic nerve terminals are affected.
Although the role of primary and secondary cells (see Introduction) in the descending antinociceptive actions of opioids has not been fully resolved, direct inhibition of one population of RVM neurons by μ-opioids (‘on' cells which display increased action potential activity immediately before and during tail-flick responses) and disinhibition of another population (‘off' cells which display decreased action potential activity prior to and during tail-flick responses) is associated with opioid antinociception in the RVM (Fields et al., 1991). Like μ-opioids, systemically administered cannabinoid agonists inhibit on-cell and disinhibit off-cell activity in RVM (Meng et al., 1998). The present findings suggest that systemically administered WIN55,212-2 affects on-cell and perhaps partly affects off-cell activity in RVM in vivo indirectly via modulation of afferents arising from other cannabinoid sensitive brain regions such as the periaqueductal gray (Lichtman et al., 1996). This distinct mechanism of action could contribute to the synergistic analgesic interactions between cannabinoids and opioids because different populations of neurons are affected by each drug class but both mechanisms are likely to contribute to antinociception.
Acknowledgments
Supported by the National Health & Medical Research Council of Australia and the Medical Foundation of The University of Sydney. Donation of CTAP from the National Institute on Drug Abuse (U.S.A.) is gratefully acknowledged. Donation of SR141716 from Dr Madeleine Mosse, Sanofi Recherche, Montpelier, France is gratefully acknowledged.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- CTAP
D-Phe-Cys-Tyr-D-Trp-Arg-Pen-Thr-NH2
- eIPSC
electrically-evoked inhibitory postsynaptic current
- met-enkephalin
methionine enkephalin
- mIPSC
spontaneous miniature inhibitory postsynaptic current
- nor-BNI
nor-binaltorphimine dihydrochloride
- RVM
rostral ventromedial medulla
References
- CHAN P.K.Y., CHAN S.C.Y., YUNG W.H. Presynaptic inhibition of GABAergic inputs to rat substantia nigra pars reticulata neurons by a cannabinoid agonist. Neuroreport. 1998;9:671–675. doi: 10.1097/00001756-199803090-00020. [DOI] [PubMed] [Google Scholar]
- DEADWYLER S.A., HAMPSON R.E., BENNET B.A., EDWARDS T.A., MU J., PACHECO M.A., WARD S.J., CHILDERS S.R. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors and Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- FIELDS H.L., HEINRICHER M.M., MASON P. Neurotransmitters in nociceptive modulatory circuits. Annu. Rev. Neurosci. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- HENRY D.J., CHAVKIN C. Activation of inwardly rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci. Lett. 1995;186:91–94. doi: 10.1016/0304-3940(95)11289-9. [DOI] [PubMed] [Google Scholar]
- HERKENHAM M., LYNN A.B., JOHNSON M.R., MELVIN L.S., DE COSTA B.R., RICE K.C. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOWLETT A.C. Pharmacology of cannabinoid receptors. Annu. Rev. Neurosci. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- ISAACSON J.S., WALMSLEY B. Counting quanta: direct measurement of transmitter release at a central synapse. Neuron. 1995;15:875–884. doi: 10.1016/0896-6273(95)90178-7. [DOI] [PubMed] [Google Scholar]
- LEVENES C., DANIEL H., SOUBRIE P., CREPEL F. Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar purkinje cells. J. Physiol. (Lond.) 1998;510:867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LICHTMAN A.H., COOK S.A., MARTIN B.R. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J. Pharmacol. Exp. Ther. 1996;276:585–593. [PubMed] [Google Scholar]
- MACKIE K., LAI Y., WESTENBROEK R., MITCHELL R. Cannabinoids activate an inwardly-rectifying potassium conductance and inhibit Q-type voltage-dependent calcium currents in AtT-20 cells transfected with rat brain cannabinoid receptor. J. Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN W.J., TSOU K., WALKER M.J. Cannabinoid recepor-mediated inhibition of the rat tail-flick reflex after microinjection into the rostral ventromedial medulla. Neurosci. Lett. 1998;242:33–36. doi: 10.1016/s0304-3940(98)00044-5. [DOI] [PubMed] [Google Scholar]
- MATSUDA L.A., BONNER T.I., LOLAIT S.J. Localization of cannabinoid receptor mRNA in rat brain. J. Comp. Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- MENG I.D., MANNING B.H., MARTIN W.J., FIELDS H.L. An analgesia circuit activated by cannabinoids. Nature (Lond.) 1998;395:381–383. doi: 10.1038/26481. [DOI] [PubMed] [Google Scholar]
- PAN Z.Z., TERSHNER S.A., FIELDS H.L. Cellular mechanism for anti-analgesic action of agonists of the κ-opioid receptor. Nature (Lond.) 1997;389:382–385. doi: 10.1038/38730. [DOI] [PubMed] [Google Scholar]
- PAN Z.Z., WESSENDORF M.W., WILLIAMS J.T. Modulation by serotonin of the neurons in rat nucleus raphe magnus in vitro. Neuroscience. 1993;54:421–429. doi: 10.1016/0306-4522(93)90263-f. [DOI] [PubMed] [Google Scholar]
- PAN Z.Z., WILLIAMS J.T., OSBORNE P.B. Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. J. Physiol. (Lond.) 1990;427:519–532. doi: 10.1113/jphysiol.1990.sp018185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN M., PISER T.M., SEYBOLD V.S., THAYER S.A. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J. Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH F.L., CICHEWICZ D., MARTIN Z.L., WELCH S.P. The enhancement of morphine antinociception in mice by Δ9-tetrahydrocannabinol. Pharmacol. Biochem. Behav. 1998;60:559–566. doi: 10.1016/s0091-3057(98)00012-4. [DOI] [PubMed] [Google Scholar]
- SZABO B., DORNER L., PFREUNDTNER C., NORENBERG W., STARKE K. Inhibition of GABAergic inhibitory postsynaptic currents in rat corpus striatum. Neuroscience. 1998;85:395–403. doi: 10.1016/s0306-4522(97)00597-6. [DOI] [PubMed] [Google Scholar]
- TWITCHELL W., BROWN S., MACKIE K. Cannabinoids inhibit N- and P/Q-type calcium currents in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- VAUGHAN C.W., INGRAM S.L., CHRISTIE M.J. Actions of ORL1 receptor ligand, nociceptin on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. J. Neurosci. 1997;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]