

Abstract
Efforts to define precisely the role of 5-HT2B receptors in normal and disease processes have been hindered by the absence of selective antagonists. To address this deficiency, we developed a series of naphthylpyrimidines as potentially useful 5-HT2B receptor antagonists.
RS-127445 (2-amino-4-(4-fluoronaphth-1-yl)-6-isopropylpyrimidine) was found to have nanomolar affinity for the 5-HT2B receptor (pKi=9.5±0.1) and 1,000 fold selectivity for this receptor as compared to numerous other receptor and ion channel binding sites.
In cells expressing human recombinant 5-HT2B receptors, RS-127445 potently antagonized 5-HT-evoked formation of inositol phosphates (pKB=9.5±0.1) and 5-HT-evoked increases in intracellular calcium (pIC50=10.4±0.1). RS-127445 also blocked 5-HT-evoked contraction of rat isolated stomach fundus (pA2=9.5±1.1) and (±)α-methyl-5-HT-mediated relaxation of the rat jugular vein (pA2=9.9±0.3). RS-127445 had no detectable intrinsic activity in these assays.
In rats, the fraction of RS-127445 that was bioavailable via the oral or intraperitoneal routes was 14 and 60% respectively. Intraperitoneal administration of RS-127445 (5 mg kg−1) produced plasma concentrations predicted to fully saturate accessible 5-HT2B receptors for at least 4 h.
In conclusion, RS-127445 is a selective, high affinity 5-HT2B receptor antagonist suitable for use in vivo. The therapeutic potential of this molecule is being further evaluated.
Keywords: 5-HT, serotonin, 5-HT2B receptor, RS-127445
Introduction
The 5-HT2B receptor is a G-protein coupled receptor that was first identified in rat stomach fundus (Clineschmidt et al., 1985; Baez et al., 1990). Initially termed a 5-HT1-like or 5-HT2F receptor (Kursar et al., 1992), its subsequent cloning and functional expression revealed structural and transduction properties similar to 5-HT1C and 5-HT2 receptors (Foguet et al., 1992; Kursar et al., 1994). This led to a reorganization of the 5-HT2 receptor family with this receptor being designated as the 5-HT2B receptor (Hoyer et al., 1994; Baxter et al., 1995; Hoyer & Martin, 1997).
5-HT2B receptors are widely expressed in peripheral tissues of various species. 5-HT2B receptor mRNA or protein immunoreactivity has been found throughout the gastrointestinal tract including smooth muscle of the stomach fundus, oesophagus, small intestine and colon. The receptor has also been found in the placenta, uterus, lung and prostate. 5-HT2B receptors are present in many vascular beds and have been localized to both vascular smooth muscle and vascular endothelial cells (Schmuck et al., 1994; Kursar et al., 1994; Bonhaus et al., 1995; Ullmer et al., 1995; 1996; Choi & Maroteaux, 1996).
Since activation of 5-HT2B receptors has been shown to contract stomach and intestinal smooth muscle (Vane, 1959; Baxter et al., 1994; Borman & Burleigh, 1995), it is likely that a primary function of these receptors is to mediate contractile responses to 5-HT in those smooth muscle tissues in which it is expressed. However, recent findings suggest that 5-HT2B receptors in vascular endothelium may mediate a nitric oxide-dependent relaxation (Glusa & Richter, 1993; Bodelsson et al., 1993; Ellis et al., 1995). Thus while the predominant function of peripheral 5-HT2B receptors in most smooth muscle tissues may be to mediate contraction, their role in vascular tissues may be more complex, depending on the specific vascular bed examined and the pathological state of the tissue (Watts et al., 1996).
Efforts to obtain a more complete understanding of peripheral 5-HT2B receptors in normal and pathological states have been hampered by the absence of suitable high affinity, selective ligands. Most antagonists commonly used to characterize the function of 5-HT2 receptors, such as mianserin, ritanserin, mesulergine and methysergide, fail to discriminate between 5-HT2 receptor subtypes. Recently, several 5-HT2B/5-HT2C selective antagonists, with low affinity for the 5-HT2A receptor, have been described (Forbes et al., 1995; Nozulak et al., 1995; Kennett et al., 1996). However, most of these ligands fail to adequately discriminate between the 5-HT2B and 5-HT2C receptor subtypes (Baxter, 1996). Recently several 5-HT2B selective ligands have emerged. These include SB 204741 (N-(1-methyl-1H-indol-5-yl)N′-(3-methyl-5-isothiazoyl) urea) which has 100 fold selectivity for the human recombinant 5-HT2B receptor, albeit with relatively low affinity (Bonhaus et al., 1995; Baxter, 1996) and a series of compounds, based on a β-carboline structure, (Audia et al., 1996). These ligands (e.g., LY-23728: 1-(3,4-dimethoxy-benzyl)-2,3,4,9-tetrahydro-1H-β-carboline) are selective for the 5-HT2B receptor as compared to the 5-HT2A and 5-HT2C receptors and have relatively high affinity, but their overall receptor specificity has not been reported nor their suitability for in vivo studies.
To further define the function of 5-HT2B receptors, and ultimately to test the clinical utility of 5-HT2B antagonists in various disease states, we initiated a programme aimed at developing selective, high affinity 5-HT2B receptor antagonists suitable for in vivo applications. These efforts yielded a series of naphthylpyrimidines having high affinity and selectivity for the 5-HT2B receptor. The in vitro pharmacology and in vivo pharmacokinetic properties of one of these compounds, RS-127445(2-amino-4- (4 - fluoronaphth-1-yl) - 6 - isopropylpyrimidine) is described in Figure 1.
Figure 1.
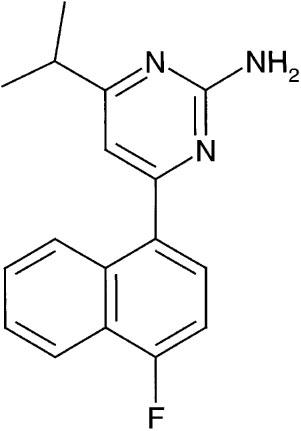
Structure of RS-127445 (2-amino-4-(4-fluoronaphth-1-yl)-6-isopropylpyrimidine).
Methods
Cell culture
HEK-293 cells stably expressing human recombinant 5-HT2B receptors were maintained in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum and 250 μg ml−1 G-418. CHO-K1 cells stably expressing the human 5-HT2A, 5-HT2B or 5-HT2C receptors were maintained in Ham's F-12 supplemented with 10% newborn calf serum and 300 μg ml−1 G-418. All cells were cultured in a humidified atmosphere at 37°C.
Radioligand binding
CHO-K1 cells expressing human 5-HT2A, 5-HT2B or 5-HT2C receptors were harvested using 2 mM EDTA in phosphate buffered saline. Cell membranes were prepared by four cycles of homogenization (Brinkman P10 disrupter) and centrifugation (48,000×g for 15 min). As previously described, each assay was established so as to achieve steady state conditions and to optimize specific binding (Bonhaus et al., 1995). For the 5-HT2A receptor, membranes from 1×106 cells were incubated with 0.2 nM [3H]-ketanserin at 32°C for 60 min. Nonspecific binding was determined using 10 μM methysergide. For the 5-HT2B receptor, membranes from 1.5×106 cells were incubated with 0.2 nM [3H]-5-HT at 4°C for 120 min. Nonspecific binding was determined using 10 μM 5-HT. For the 5-HT2C receptor, membranes from 3×105 cells were incubated with 0.5 nM [3H]-mesulergine at 32°C for 60 min. Nonspecific binding was determined using 10 μM methysergide. Assays were terminated by vacuum filtration through glass fibre filters (GF/B) which had been pretreated with 0.1% polyethyleneimine. Total and bound radioactivity was determined by liquid scintillation counting. Greater than 90% specific binding was achieved in each of these assays.
The selectivity of RS-127445 for 5-HT2B receptors was further examined by testing the compound for affinity at over 100 additional ion channel or receptor binding sites. These studies were conducted by Cerep (Celle L'Evescault France) using standard protocols.
Inhibition of 5-HT evoked formation of inositol phosphates
HEK-293 cells expressing the human 5-HT2B receptor were incubated with [3H]-myoinositol (1.67 μCi ml−1) in 162 cm2 flasks overnight at 37°C in an inositol free Ham's F12 medium containing 10% dialyzed foetal bovine serum. The cells were harvested, washed five times with phosphate buffered saline and resuspended in inositol free Ham's F12 media at density of approximately 3×106 cells ml−1. RS-127445 (10 μM) was initially dissolved in 10% (v v−1) DMSO with 90% inositol free Ham's F12 medium. Subsequent dilutions were made with inositol free Ham's F12 medium. 5-HT was dissolved in inositol free Ham's F12 medium containing 100 mM LiCl and 1 mM ascorbate. RS-127445, vehicle or other antagonists were pre-incubated with 240 μl of cell suspension at 37°c for 20 min. The reactions were initiated by addition of 5-HT. Sixty minutes later, the reactions were terminated by adding 50 μl of ice-cold 20% perchloric acid, chilled in an ice-water bath for 10 min and then neutralized with 160 μl of 1 N KOH. Each sample was diluted with 2 ml of 50 mM Tris-HCl, pH 7.4 at room temperature. The aqueous portion (2.2 ml) was transferred onto Dowex AG1X8 columns (1 ml, 1 : 1, w v−1) which had been washed with 5 ml of distilled water. The columns were then washed with 18 ml of distilled water and the inositol phosphates were eluted with 3 ml of 1 N HCl. The eluted radioactivity was determined by liquid scintillation spectroscopy using a Packard 1900CA analyzer.
Inhibition of 5-HT evoked increases in intracellular calcium
HEK-293 cells expressing human 5-HT2B receptors were seeded in 98-well poly-D-lysine coated plates at a density of 50,000 cells well−1 in Dulbecco's modified Eagle's medium containing 10% (v v−1) dialyzed foetal bovine serum and 250 μg ml−1 G-418. After an overnight incubation, the cells were washed three times with Hank's buffered saline solution containing 2 mM CaCl2, 10 mM HEPES and 2.5 mM probenecid. The cells were incubated with 2 μM Fluo-3 AM in Hank's buffered saline solution for 60 min at 37°C. The Fluo-3 loaded cells were then washed four times with Hank's buffered saline solution.
Antagonists were added at least 40 min prior to the addition of agonist. Agonist induced changes in intracellular calcium concentrations were determined at room temperature using a Fluorometric Imaging Plate Reader (Molecular Devices Corporation, Sunnyvale, CA, U.S.A.).
Inhibition of 5-HT evoked contraction of the isolated rat stomach fundus
Functional responses in rat isolated stomach were measured as described by Baxter et al. (1994). Male Sprague-Dawley rats were euthanized by CO2 asphyxiation. Longitudinal stomach fundus muscle, devoid of the mucosal layer, was mounted in the longitudinal plane under 1 g tension in a 10 ml organ bath containing Tyrode's physiological salt solution (pH 7.4 at 37°C). Antagonists were equilibrated with the tissue for at least 1 h.
Concentration-response curves to 5-HT were then generated in the presence of 0.1 mM pargyline, 30 μM cocaine and 30 μM corticosterone.
Inhibition of (±) α-methyl-5-HT evoked relaxation of the rat jugular vein
Sprague-Dawley rats were euthanized as described above. Right and left external jugular veins were dissected, cleaned of connective tissues and cut into ring segments approximately 5 mm long. Tungsten hooks (0.125 mm diameter) were inserted through the lumen of the vein and connected to tension transducers. Tissues were kept in 10 ml organ baths containing Kreb's solution supplemented with cocaine (30 μM), corticosterone (30 μM), ketanserin (0.3 μM) and indomethacin (3 μM) at 37°C at a resting tension of 0.5 g. Prior to the initiation of any studies, monamine oxidases were inactivated by a 30 min pre exposure of the tissue to pargyline (0.1 mM). The veins were then exposed to 0.1 μM U46619 (9,11-dideoxy-9 α, 11 aα-methano-epoxy-PGF2α; a thromboxane A2 mimetic) until a stable contraction was attained. Acetylcholine (0.1 μM) was used to verify the integrity of the endothelium and to determine the maximum amount of nitric oxide-dependent relaxation that was achievable. After washout of the acetylcholine and recontraction with U46619, cumulative concentration-response curves to (±)-α-methyl-5-HT were constructed. When maximum relaxation was reached, the baths were rinsed, and the tissues were maintained undisturbed for 2 h. Antagonists were then added to the bath and allowed to equilibrate with the tissue for at least 1 h before a second concentration-response curve to (±)-α-methyl-5-HT was generated.
Pharmacokinetics of RS-127445 in rats
Male Sprague-Dawley rats (200 g) were allowed to acclimatize for at least 2 days. The animals were fasted overnight prior to dosing and for 4 h following dosing. Water was allowed ad libitum.
To compare the plasma kinetics of RS-127445 following different routes of administration, 90 rats were distributed into three treatment groups of 30 rats each. A single dose of RS-127445 (5 mg kg−1) dissolved (2.5 mg ml−1) in ethanol:propylene glycol : water (10 : 50 : 40, v v v−1), was administered to each rat. At 0.08, 0.25, 0.5, 1, 2, 4, 8 and 24 h after dosing, the rats were anaesthetized and blood samples were collected by cardiac puncture.
The relationship between the dose of RS-127445 and the achieved plasma concentration was also examined. Male Sprague-Dawley rats were distributed into three treatment groups. The rats within each group (40) received 1.0, 3.0 or 10.0 mg of RS-127445 by the intraperitoneal route. A blood sample was then collected at 0.5, 1.0, 2.0, 3.0 or 4.0 h following dosing. No more than one blood sample was taken from any one rat. Plasma was stored frozen at −20°C until analysed.
An internal standard was added to aliquots of plasma (0.5 ml) and adjusted to pH 7.2 with phosphate buffer. RS-127445 and the internal standard were extracted into ethyl acetate:hexane (1 : 9, v v−1). The organic extract was evaporated to dryness under a stream of nitrogen at 38°C and dissolved in methanol : phosphate buffer (pH 3.0). An aliquot of the dissolved extract was subjected to reverse-phase HPLC on a 150×4.6 mm, 5 μm C18 column using a mobile phase of 40% acetonitrile in 50 mM sodium phosphate buffer containing 20 mM 1-heptane sulfonic acid (pH 3.0) at a flow rate of 1 ml min−1. RS-127225 was detected at a wavelength of 305 nM. The lower limit of quantification of the assay was 4 ng ml−1.
Methods of data analysis
For competition binding curves IC50 values were obtained using non-linear iterative curve fitting routines and a logistic, four parameter (variable Hill slope) equation:
(Prism–GraphPad Software Inc., San Diego CA, U.S.A.). When Hill slopes were found to be not different than 1.0, estimates of affinity (Ki values) were obtained using the Cheng-Prusoff equation (Cheng & Prusoff, 1973). The data are presented as a negative log of the Ki value (pKi).
For the inositol phosphate studies, EC50 values for 5-HT concentration-response curves were generated. Estimates of affinity (pKB values) were then obtained using Schild regression analysis of the ratios of EC50 values determined in the presence of at least three different concentrations of antagonist. Regression lines with slopes not significantly different from 1.0 were constrained to 1.0.
For the calcium influx studies, IC50 values for antagonist inhibition of 5-HT evoked responses were generated. Since the endpoint of this assay is achieved within seconds of adding the agonist there is no opportunity for the antagonist to re-equilibrate to new steady-state levels of binding. Thus, no attempt was made to generate affinity estimates on the basis of shifts in agonist-response curves.
For the functional studies using rat isolated stomach fundus or jugular vein, agonist concentration-response curves were generated and Schild regression analysis was conducted using at least three different concentrations of antagonist. However, as RS-127445 produced a decrease in the maximum response to 5-HT or (±)α-methyl-5-HT, results from these studies are presented as pA2 rather than pKB values.
Pharmacokinetic calculations were conducted using standard analytical methods. When concentrations of RS-127445 were below the quantification limit of the assay, the values were set to zero. The peak time to maximum plasma concentration (Tmax) and the maximum plasma concentration (Cmax) values were estimated by inspection of the plots of the log plasma concentration versus time. The area under the concentration-time curve from 0–8 h (AUC0–8) was calculated using the linear trapezoidal rule. The plasma half-life (T1/2) was determined using the relationship 0.693/β, where β is the elimination rate constant calculated by linear regression analysis of the terminal phase of the log plasma concentration vs time curve. Bioavailability was calculated by dividing the AUC0–8 obtained following oral or intraperitoneal dosing by the AUC0–8 obtained with intravenous administration of RS-127445.
Materials
[3H]-Mesulergine (75 Ci mmol−1, 99% pure), [3H]-myoinositol (18.6 Ci mmol−1, 99% pure and [3H]-5-HT (91 Ci mmol−1, 99% pure) were obtained from Amersham International (Cardiff, Wales, U.K.). [3H]-Ketanserin (62 Ci mmol−1, 99% pure) was obtained from Dupont-NEN Life Sciences ( Boston, MA, U.S.A.). Ritanserin, mianserin, yohimbine and other ligands were obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.) or Mallinckrodt Baker, Inc (Paris, KT, U.S.A.). LY23728 (1-(3,4-dimethoxy-benzyl)-2,3,4,9-tetrahydro-1H-β-carboline) and RS-127445 were synthesized in the Department of Chemistry, Neurobiology Unit, Roche Bioscience. The fluorescent probe Fluo-3 AM was obtained from Molecular Probes (Eugene, OR, U.S.A.). Analytical solvents were HPLC grade and were purchased from Burdick and Jackson (Muskegon, MI, U.S.A.). The HPLC column used to isolate RS-127445 was a Higgins Analytical HAIsil C18 column (Higgins Analytical, Inc., Mountain View, CA, U.S.A.). Hank's Balanced Salt Solution, Dulbecco's Modified Eagle Medium and other reagents for tissue culture were obtained from Gibco BRL (Grand Island, NY, U.S.A.).
Results
Radioligand binding
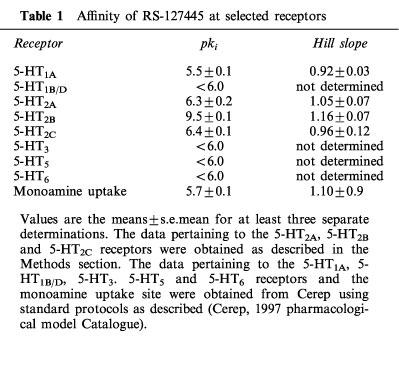
RS-127445 potently displaced [3H]-5-HT from human recombinant 5-HT2B receptors expressed in CHO-K1 cells. The affinity (pKi value) of RS-127445 for the 5-HT2B receptor was 9.5±0.1 (n=9). RS-127445 was selective for the 5-HT2B receptor, having approximately 1000 fold lower affinity for the human recombinant 5-HT2A, 5-HT2C, 5-HT5, 5-HT6 and 5-HT7 receptors, a 5-HT1A receptor in rat brain membranes, a 5-HT1B/D receptor in bovine caudate, and a monoamine uptake site in rabbit platelets (Table 1).
Table 1.
Affinity of RS-127445 at selected receptors
RS-127445 (10 μM tested in duplicate) had no appreciable affinity (less than 50% inhibition of specific binding) at the following binding sites: adenosine A1 and A2A receptors; adrenergic α1A, α1B, α2A, α2B, β1, β2 and β3 receptors; adenosine, choline, dopamine and serotonin uptake sites; angiotensin AT1 receptors; Atrial Natriuretic Factor receptor; bombesin and; bradykinin B1 and B2 receptors; Calcitonin Gene Related Peptide receptor; the benzothiazepine, dihydropyridine and phenylalkylamine binding sites on L-type Ca2+ channels; a conotoxin (GVIA) binding site on N-type Ca2+ channels; cannabinoid CB1 and CB2 receptors; the cholecystokinin CCKA and CCKB receptors; dopamine D1, D2L, D2S, D4.2, D4.4, D4.7 and D5 receptors; endothelin ETA and ETB receptors; an EGF binding site; an estrogen receptor; the GABA, benzodiazepine and picrotoxin binding sites on the GABA receptor; a peripheral benzodiazepine binding site; the galanin and glucocorticoid receptors; the AMPA and kainate glutamate receptors; the NMDA, glycine and phencyclidine binding sites on the NMDA receptor; the strychnine-sensitive glycine receptor; histamine H1, H2 and H3 receptors; the inositol trisphosphate binding site; an insulin receptor; interleukin 1α, 2, 6 and 8 receptors; the leukotriene B4 and D4 receptors; muscarinic M1, M2, M3 and M4 receptors; neurokinin NK1 receptors; neuropeptide Y1 and Y2 receptors; a neurotensin receptor, a central nicotinic acetylcholine receptor, δ, κ and μ opiate receptors; a phorbol ester binding site; a platelet activating factor receptor; the platelet derived growth factor receptor; the KA, KATP, KV and SKCa potassium channels; the progesterone and purinergic P2X receptors; sigma σ1 and σ2 binding sites; the batrachotoxin binding site on sodium channels; the somatostatin, testosterone and thromboxane A2 receptors and, binding sites on the TRH, TGF-β, TNF-α, VIP1 and vasopressin V1 receptors.
Effect of RS-127445 on 5-HT evoked increases in the formation of inositol phosphates
5-HT concentration-dependently increased the rate of formation of inositol phosphates in HEK-293 cells expressing the human recombinant 5-HT2B receptor (pEC50 value of 8.2±0.1). By contrast, RS-127445, at concentrations up to 100 μM, had no stimulatory effect on inositol phosphate production (Figure 2).
Figure 2.
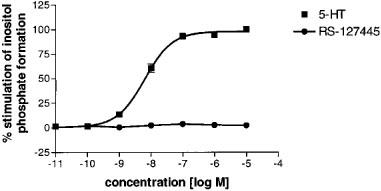
Effect of 5-HT and RS-127445 on the formation of [3H]-inositol phosphates in HEK-293 cells expressing the human 5-HT2B receptor. Each data point is the means±s.e.mean of three determinations (error bars are not shown when smaller than the symbol).
RS-127445 potently inhibited the 5-HT evoked increase in the formation of inositol phosphates in cells expressing 5-HT2B receptors, producing parallel rightward shifts in the concentration response curves of 5-HT (Figure 3A). Schild regression analysis generated a pKB value of 9.5±0.1 with the slope of the Schild regression being not different from 1.0 (0.95±0.23) (Figure 3B).
Figure 3.
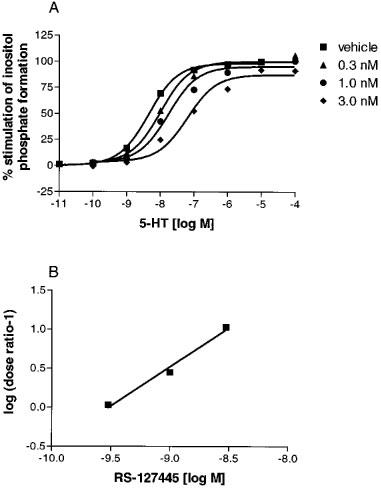
(A) 5-HT mediated stimulation of inositol phosphate formation in the presence of ascending concentrations of RS-127445. Each data point is the means±s.e.mean of 12 different determinations. (B) Schild regression of the data in A.
Effect of RS-127445 on 5-HT evoked increases in intracellular calcium concentration
5-HT concentration-dependently increased intracellular calcium concentrations in HEK-293 cells expressing 5-HT2B receptors (pEC50 value of 9.2±0.1) while RS-127445, at concentrations up to 100 μM, had no stimulatory effect on calcium influx (Figure 4A).
Figure 4.
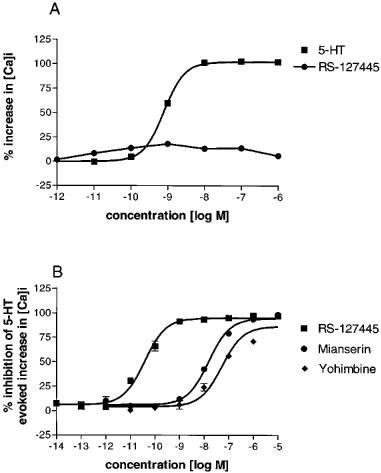
(A) Effects of 5-HT and RS-127445 on intracellular calcium concentrations in HEK-293 cells expressing the human 5-HT2B receptor. Each data point is the means±s.e.mean of four (RS-127445) or eleven (5-HT) different determinations. (B) Inhibition of 5-HT (10 nM) evoked increases in intracellular calcium concentrations by RS-127445, mianserin and yohimbine. Each data point is the means±s.e.mean of seven to eleven different determinations.
RS-127445 potently blocked the 5-HT (10 nM) evoked increases in intracellular calcium concentrations in the HEK-293 cells expressing the 5-HT2B receptor (pIC50 value of 10.4±0.1) (Figure 4B). The rank-order of potencies of a series of 5-HT2 receptor antagonists in inhibiting 5-HT evoked inositol phosphate formation and 5-HT mediated increases in intracellular calcium concentrations were similar (Table 2).
Table 2.
Affinity of selected ligands at human recombinant 5-HT2B receptors
Effect of RS-127445 on 5-HT evoked contraction of the rat stomach fundus
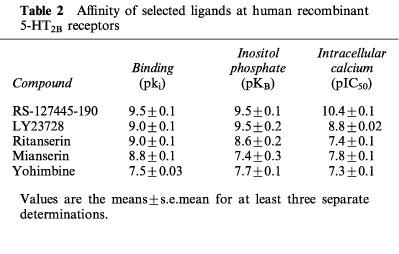
5-HT concentration-dependently contracted the isolated rat stomach fundus with an pEC50 value of 8.3±0.2. By contrast, RS-127445, at concentrations from 0.1–10 μM, had no contractile effect on the isolated fundus.
RS-127445 potently blocked the 5-HT evoked contraction of the isolated fundus. The concentration-response curves to 5-HT were shifted to the right in a concentration-dependent manner. There was also a progressive decrease in the maximum response (Figure 5A). The intercept value on the Schild regression line corresponded to a pA2 value of 9.5±1.1. The slope of the regression was statistically significantly less than unity (0.58±0.09) (Figure 5B).
Figure 5.
(A) Inhibition of 5-HT-evoked contraction of the rat isolated stomach fundus by ascending concentrations of RS-127445. Each data point is the means±s.e.mean of at least three different determinations. (B) Schild regression analysis of the data in A.
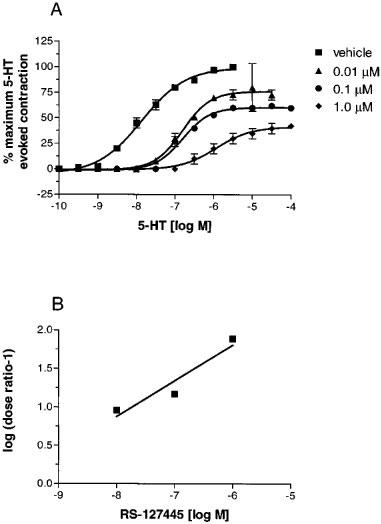
To test whether slow dissociation kinetics may have accounted for the insurmountable nature of the antagonism produced by RS-127445, tissues were exposed to RS-127445 (10 nM) and then washed for varying periods of time. Extensive washing of the isolated stomach fundus resulted in a time-dependent reduction in the block produced by RS-127445. However, even after 60 min of washout, the magnitude of the remaining effect corresponded to 50% of the original concentration of RS-127445 (Figure 6).
Figure 6.
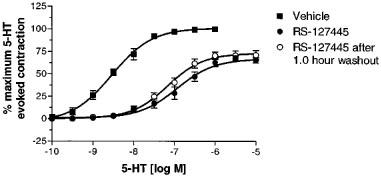
Effect of 1 h of washout on the inhibition of 5-HT-evoked contraction of the rat isolated stomach fundus produced by 0.1 μM RS-127445. Each data point is the means±s.e.mean of at least three different determinations. Similar findings were obtained using 1.0 μM RS-127445.
Effect of RS-127445 on (±)α-methyl-5-HT evoked relaxation of the rat jugular vein
The 5-HT2B receptor agonist (±)α-methyl-5-HT, in a concentration-dependent manner, relaxed rat isolated jugular veins which had been pre-contracted with the U44619. RS-127445, at concentrations up to 10 μM had no relaxant effect on this preparation but blocked the (±)α-methyl-5-HT mediated response.
As was found in the studies with the isolated stomach fundus, RS-127445 produced rightward shifts in the agonist-mediated concentration-response curves with progressive decreases in the maximum response attained by (±)α-methyl-5-HT (Figure 7A). The intercept of the Schild regression line corresponded to a pA2 value of 9.95±0.07 with a slope not statistically different than 1.0 (1.18±0.08) (Figure 7B).
Figure 7.
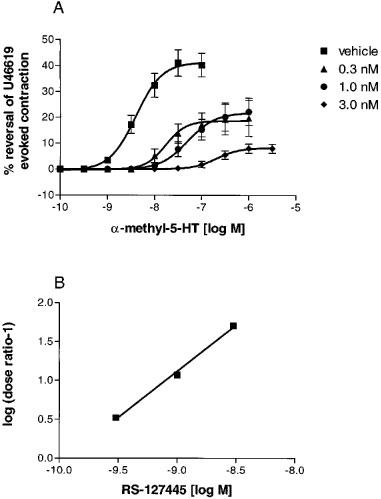
(A) α-methyl-5HT mediated relaxation of precontracted rat jugular vein in the presence of ascending concentrations of RS-127445. Each data point is the means±s.e.mean of at least 4–12 different determinations. (B) Schild regression of the data in Figure 6A.
In vivo studies with RS-127445
RS-127445 (5 mg kg−1) was administered to rats by oral, intraperitoneal and intravenous routes. Peak plasma concentrations were rapidly achieved with the highest concentrations being found at the first time-point measured following intravenous and intraperitonael administration (0.08 h) and by 0.25 h following dosing by the oral route of administration. RS-127445 was cleared from plasma with an estimated terminal elimination half-life of approximately 1.7 h. The bioavailability of RS-127445, when administered by the oral and intraperitoneal routes was approximately 14 and 62% of that obtained by intravenous administration (Figure 8A). To test whether plasma levels were proportional to the dose administered, RS-127445 was given by the intraperitoneal route at doses of 1, 3 and 10 mg kg−1. Increasing the dose of RS-127445 resulted in proportional increases in its concentration in the plasma (Figure 8B).
Figure 8.
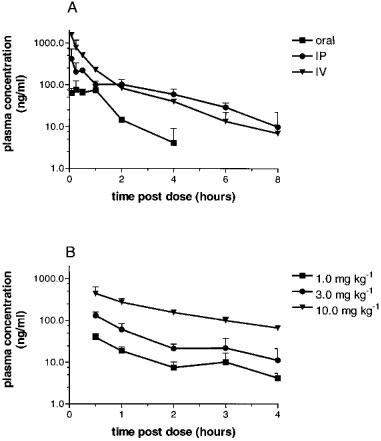
(A) Plasma concentrations of RS-127445 following dosing (5 mg kg−1) via various routes. Each data point represents the means±s.e.mean for three animals. The peak plasma concentrations of RS-127445 for these routes were 75.7±48.1, 417±298 and 1550±220 ng ml−1 respectively. This entire experiment has been repeated at least three times with similar results. (B) Plasma concentrations of RS-127445 following intraperitoneal dosing with different doses of RS-127445. Each data point is the means±s.e.mean of eight animals.
Discussion
RS-127445 (2-amino-4-(4-fluoronaphth-1-yl)-6-isopropylpyrimidine) was discovered during the course of studies aimed at developing bioavailable, high affinity and selective 5-HT2B receptor antagonists. RS-127445 was found to have nM affinity and 1000 fold selectivity for the 5-HT2B receptor. RS-127445 is thus among the highest affinity, most selective 5-HT2B receptor ligands to be described (Audia et al., 1996; Baxter, 1996).
The possibility that RS-127445 had agonist activity at 5-HT2B receptors was evaluated using cell-based assays and HEK-293 cells expressing human recombinant 5-HT2B receptors. Transfected HEK-293 cells were used for these functional assays because the 5-HT2B receptor mediated responses were quantitatively greater in this cell line than in the transfected CHO-K1 cells. Both the phospholipase C-mediated conversion of myoinositol into inositol phosphates and the subsequent elevation in intracellular calcium concentrations were used as indices of 5-HT2B receptor activation (Foguet et al., 1992; Schmuck et al., 1994). In HEK-293 cells expressing human recombinant 5-HT2B receptors, 5-HT potently stimulated the formation of inositol phosphates and increased intracellular calcium concentrations. Whereas, RS-127445, at concentrations up to 10 μM, had no stimulatory effect on the formation of inositol phosphates or on intracellular calcium concentrations. Thus, regardless of the endpoint used to measure receptor activation, RS-127445 had no detectable agonist activity at these human recombinant 5-HT2B receptors.
Consistent with it being a high affinity antagonist, RS-127445 potently blocked the 5-HT evoked increase in inositol phosphate formation. The results of Schild regression analysis were consistent with RS-127445 acting in a competitive manner and having nM affinity for the receptor. Also consistent with it being a high affinity antagonist, RS-127445 blocked the 5-HT evoked increases in intracellular calcium concentrations with a potency 1000 times greater than that of yohimbine.
Since species-species differences in the pharmacological properties of 5-HT2B receptors have been reported (Wainscott et al., 1996), the potency and activity of RS-127445 at endogenous rat 5-HT2B receptors was also examined. The most extensively characterized 5-HT2B receptor mediated functional response, and indeed the response that was first used to define the 5-HT2B receptor, is contraction of the isolated rat stomach fundus (Clineschmidt et al., 1985 ; Baxter et al., 1994; Cox & Cohen, 1995). Consistent with it being a ‘silent' antagonist RS-127445 produced no detectable response in the isolated stomach fundus but, potently antagonized the contractile response produced by 5-HT. A second functional response which has been attributed to 5-HT2B receptors is relaxation of the rat jugular vein (Bodelsson et al., 1993; Ellis et al., 1995). As was found in the studies using isolated stomach fundus, RS-127445 had no detectable intrinsic activity but acted as an antagonist, potently blocking the relaxation produced by the 5-HT2B receptor agonist α-methyl-5-HT. These findings demonstrate that RS-127445 is a potent antagonist, devoid of intrinsic activity at both endogenous rat and human recombinant 5-HT2B receptors.
One anomalous finding to arise from the functional studies using rat stomach fundus or jugular vein was that the inhibitory actions of RS-127445 could not be fully surmounted with increasing concentrations of agonist. This raised the possibility that RS-127445 may be acting by a noncompetitive mechanism. However, a more plausible explanation is that the experimental conditions did not allow sufficient time for steady-state conditions to be achieved following addition of the agonist (Kenakin, 1993). This possibility is supported by the finding that inhibitory actions of RS-127445 were not fully reversed even after washing the tissues in drug-free buffer for an hour.
To evaluate the suitability of RS-127445 for studying the role of 5-HT2B receptors in vivo, its pharmacokinetic properties in the rat were characterized. RS-127445 was rapidly absorbed following both oral and intraperitoneal administration with peak plasma concentrations being achieved within 15 min of dosing. Moreover, the plasma concentrations of RS-127445 that were achieved were proportional to the administered dose. There were no obvious dose or route-dependent limitations to the absorption of RS-127445. Approximately 60% of an intraperitoneal dose and 14% of the oral dose of RS-127445 (5 mg kg−1) was bioavailable. The plasma concentrations of RS-127445 that were generated (following intraperitoneal administration of 5 mg kg−1) exceeded 20 nM for at least 4 h following dosing. Thus plasma concentrations of RS-127445 which would be predicted to fully saturate accessible 5-HT2B receptors can be readily achieved and maintained in the rat.
In summary RS-127445 is a novel high affinity, selective 5-HT2B receptor antagonist devoid of detectable intrinsic activity. Moreover, the compound is readily absorbed by various routes of administration. Thus RS-127445 may prove useful in further in vivo studies of 5-HT2B receptor mediated processes and will be further evaluated as a potential therapeutic agent for the treatment of disorders in which activation of 5-HT2B receptors may play a pathological role.
Abbreviations
- RS-127445
(2-amino-4-(4-fluoronaphth-1-yl)-6-isopropylpyrimidine)
- SB 204741
(N-(1-methyl-1H-indol-5-yl)N′-(-benzyl)-2,3,4,9-tetrahydro-1H-β-carboline)
- U46619
(9,11-dideoxymethyl-5-isothiazolyl) urea)
- LY-23728
(1-(3,4-dimethoxy3-9 α, 11 aα-methano-epoxy-PGF2α)
- maximum plasma concentration
(Tmax)
- maximum plasma concentration
(Cmax)
- The plasma half-life
(T½)
- Elimination rate constant
(β)
- The area under the concentration-time curve from 0–8 hours
(AUC0–8)
- HEK
(human embryonic kidney)
- CHO
(Chinese hamster ovary)
References
- AUDIA J.E., EVARD D.A., MURDOCH G.R., DROSTE J.J., NISSEN J.S., SCHENCK K.W., FLUDZINSKI P., LUCAITES V.L., NELSON D.L., COHEN M.L. Potent, selective tetrahydro-B-carboline antagonists of the serotonin2B (5HT2B) contractile receptor in the rat stomach fundus. J. Med. Chem. 1996;39:2773–2780. doi: 10.1021/jm960062t. [DOI] [PubMed] [Google Scholar]
- BAEZ M., YU L., COHEN M.L. Pharmacological and molecular evidence that the contractile response to serotonin in rat stomach fundus is not mediated by activation of the 5-hydroxytryptamine1C receptor. Mol. Pharmacol. 1990;38:31–37. [PubMed] [Google Scholar]
- BAXTER G., KENNETT G., BLANEY F., BLACKBURN T. 5-HT2 receptor subtypes: a family re-united. Trends Pharmacol. Sci. 1995;16:105–110. doi: 10.1016/s0165-6147(00)88991-9. [DOI] [PubMed] [Google Scholar]
- BAXTER G.S. Novel discriminatory ligands for 5-HT2B receptors. Behav. Brain Res. 1996;73:149–152. doi: 10.1016/0166-4328(96)00087-3. [DOI] [PubMed] [Google Scholar]
- BAXTER G.S., MURPHY O.E., BLACKBURN T.P. Further characterization of 5-hydroxytryptamine receptors (the putative 5-HT2B) in rat stomach fundus longitudinal muscle. Br. J. Pharmacol. 1994;112:323–331. doi: 10.1111/j.1476-5381.1994.tb13072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BODELSSON M., TORNEBRANDT K., ARNEKLO-NOBIN B. Endothelial relaxing 5-hydroxytryptamine receptors in the rat jugular vein: similarity with the 5-hydroxytryptamine1C receptor. J. Pharmacol. Exp. Ther. 1993;264:709–716. [PubMed] [Google Scholar]
- BONHAUS D.W., BACH C., DESOUZA A., SALAZAR F.H., MATSUOKA B.D., ZUPPAN P., CHAN H.W., EGLEN R.M. The pharmacology and distribution of human 5-hydroxytryptamine2B (5-HT2B) receptor gene products: comparison with 5-HT2A and 5-HT2C receptors. Br. J. Pharmacol. 1995;115:622–628. doi: 10.1111/j.1476-5381.1995.tb14977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORMAN R.A., BURLEIGH D.E. Functional evidence for a 5-HT2B receptor mediating contraction of longitudinal muscle in human small intestine. Br. J. Pharmacol. 1995;114:1525–1527. doi: 10.1111/j.1476-5381.1995.tb14935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;92:881–894. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CHOI D.S., MAROTEAUX L. Immunohistochemical localisation of the serotonin 5-HT2B receptor in mouse gut, cardiovascular system, and brain. FEBS Letters. 1996;391:45–51. doi: 10.1016/0014-5793(96)00695-3. [DOI] [PubMed] [Google Scholar]
- CLINESCHMIDT B.V., REISS D.R., PETTIBONE D.J., ROBINSON J.L. Characterisation of 5-Hydroxytryptamine receptors in rat stomach fundus. J. Pharmacol. Exp. Ther. 1985;235:696–708. [PubMed] [Google Scholar]
- COX D.A., COHEN M.L. 5-Hydroxytryptamine2B receptor signaling in rat stomach fundus: role of voltage-dependent calcium channels, intracellular calcium release and protein kinase C. J. Pharmacol. Exp. Ther. 1995;272:143–150. [PubMed] [Google Scholar]
- ELLIS E.S., BYRNE C., MURPHY O.E., TILFORD N.S., BAXTER G.S. Mediation by 5-hydroxytryptamine2B receptors of endothelium-dependent relaxation in rat jugular vein. Br. J. Pharmacol. 1995;114:400–404. doi: 10.1111/j.1476-5381.1995.tb13240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FOGUET M., HOYER D., PARDO L.A., PAREKH A., KLUXEN F.W., KALKMAN H. O., STUHMER W., LUBBERT H. Cloning and functional characterization of the rat stomach fundus serotonin receptor. EMBO J. 1992;11:3481–3487. doi: 10.1002/j.1460-2075.1992.tb05427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORBES I.T., JONES G.E., MURPHY O.E., HOLLAND V., BAXTER G.S. N-(1-methyl-5-indolyl)-N′-(3-methyl-5-isothiazolyl) urea: a novel, high-affinity 5-HT2B receptor antagonist. J. Med. Chem. 1995;38:855–857. doi: 10.1021/jm00006a001. [DOI] [PubMed] [Google Scholar]
- GLUSA E., RICHTER M. Endothelium-dependent relaxation of porcine pulmonary arteries via 5-HT1C-like receptors. Naunyn-Schmiedebergs Archives of Pharmacology. 1993;347:471–477. doi: 10.1007/BF00166737. [DOI] [PubMed] [Google Scholar]
- HOYER D., CLARKE D.E., FOZARD J.R., HARTIG P.R., MARTIN G.R., MYLECHARANE E.J., SAXENA P.R., HUMPHREY P.P.A. VII. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol. Rev. 1994;46:157–204. [PubMed] [Google Scholar]
- HOYER D., MARTIN G. 5-HT receptor classification and nomenclature: Towards a harmonization with the human genome. Neuropharmacology. 1997;36:419–428. doi: 10.1016/s0028-3908(97)00036-1. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Pharmacologic analysis of drug-receptor interaction. New York: Raven Press, Ltd; 1993. [Google Scholar]
- KENNETT G.A., WOOD D.M., BRIGHT F., CILIA J., PIPER D.C., GAGER T., THOMAS D., BAXTER G.S., FORBES I.T., HAM P., BLACKBURN T.P. In vitro and in vivo profile of SB 206553, a potent 5-HT2C/5-HT2B receptor antagonist with anxiolytic-like properties. Br. J. Pharmacol. 1996;117:427–434. doi: 10.1111/j.1476-5381.1996.tb15208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURSAR J.D., NELSON D.L., WAINSCOTT D.B., BAEZ M. Molecular cloning, functional expression, and mRNA tissue distribution of the human 5-hydroxytryptamine2B receptor. Mol. Pharmacol. 1994;46:227–234. [PubMed] [Google Scholar]
- KURSAR J.D., NELSON D.L., WAINSCOTT D.B., COHEN M.L., BAEZ M. Molecular cloning, functional expression, and pharmacological characterization of a novel serotonin receptor (5-hydroxytryptamine2B) from rat stomach fundus. Mol. Pharmacol. 1992;42:549–557. [PubMed] [Google Scholar]
- NOZULAK J., KALKMAN H.O., FLOERSHEIM P., HOYER D., SCHOEFFTER P., BUERKI H.R. (+)-cis-4,5,7a,8,9, 10,11,11a-octahydro-7H-10-methylindolo[1,7-bc][2,6]-naphthyridine: a 5-HT2C/2B receptor antagonist with low 5-HT2A receptor affinity. J. Med. Chem. 1995;38:28–33. doi: 10.1021/jm00001a007. [DOI] [PubMed] [Google Scholar]
- SCHMUCK K., ULLMER C., ENGELS P., LUBBERT H. Cloning and functional characterization of the human 5-HT2B serotonin receptor. FEBS Letters. 1994;342:85–90. doi: 10.1016/0014-5793(94)80590-3. [DOI] [PubMed] [Google Scholar]
- ULLMER C., BODDEKE H.G.W.M., SCHMUCK K., LÜBBERT H. 5-HT2B receptor-mediated calcium release from ryanodine-sensitive intracellular stores in human pulmonary artery endothelial cells. Br. J. Pharmacol. 1996;117:1081–1088. doi: 10.1111/j.1476-5381.1996.tb16700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ULLMER C., SCHMUCK K., KALKMAN H.O., LUBBERT H. Expression of serotonin receptor mRNAs in blood vessels. FEBS Letters. 1995;370:215–221. doi: 10.1016/0014-5793(95)00828-w. [DOI] [PubMed] [Google Scholar]
- VANE J.R. The relative activities of some tryptamine analogues on the isolated rat stomach strip preparation. Br. J. Pharmacol. 1959;14:78–98. doi: 10.1111/j.1476-5381.1959.tb00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAINSCOTT D.B., LUCAITES V.L., KURSAR J.D., BAEZ M., NELSON D.L. Pharmacologic characterization of the human 5-hydroxytryptamine2B receptor: Evidence for species differences. J. Pharmacol. Exp. Ther. 1996;276:720–727. [PubMed] [Google Scholar]
- WATTS S.W., BAEZ M., WEBB R.C. The 5-hydroxytryptamine2B receptor and 5-HT receptor signal transduction in mesenteric arteries from deoxycorticosterone acetate-salt hypertensive rats. J. Pharmacol. Exp. Ther. 1996;277:1103–1113. [PubMed] [Google Scholar]