

Abstract
In electrophysiological measurements the β-carboline ethyl 6-benzyloxy-β-carboline-3-carboxylate (ZK 91085) acts as a positive allosteric modulator on rat recombinant α1β2γ2 GABAA receptors and binds with high affinity (IC50=1.5 nM) to the [3H]-flunitrazepam site. Flumazenil was able to partially counteract the current modulation. These observations indicate an action of ZK 91085 at the benzodiazepine binding site.
At the dual subunit combination α1β2, which lacks the γ subunit required for benzodiazepine modulation, we still observed a potentiation of GABA currents. Thus ZK 91085 acts via an additional site on the channel. At the subunit combination α1β1, ZK 91085 potentiation is strongly reduced as compared to α1β2. In binding studies, ZK 91085 was able to decrease [35S]-TBPS binding in α1β2γ2 and α1β2 but not in α1β1. This selectivity of ZK 91085 for receptors containing the β2 isoform over those containing the β1 isoform is reminiscent of the action of loreclezole.
To identify amino acid residues important for the second type of modulation, we functionally compared wild type α1β2 and mutant receptors for stimulation by ZK 91085. The mutation β2N265S, that abolishes loreclezole effects, also abolishes ZK 91085 stimulation. The mutation β2Y62L increased stimulation by ZK 91085 3–4 fold, locating an influencing entity of the second type of action of ZK 91085 at an α/β subunit interface.
Structural intermediates of ZK 91085 and the β-carboline abecarnil, the latter of which only slightly potentiated GABA currents in α1β2, were analysed to determine structural requirements for modulation.
ZK 91085 thus allosterically stimulates the GABAA receptor through two sites of action: the benzodiazepine site and the loreclezole site in contrast to classical β-carbolines, that confer negative allosteric modulation through the benzodiazepine site.
Keywords: GABAA receptor, ZK 91085, β-carbolines, DMCM, loreclezole
Introduction
The GABAA receptor (γ-aminobutyric acid type A receptor) is one of the major inhibitory ion channels in the central nervous system. So far six isoforms of its subunits (α, β, γ, δ, ε, π) and 15 subtypes have been discovered (Burt & Kamatchi, 1991; Macdonald & Olsen, 1994; Dunn et al., 1994; Rabow et al., 1995; Davies et al., 1997; Hedblom & Kirkness, 1997). The transmembrane subunits of the GABAA receptor show homology to the subunits of the nicotinic acetylcholine receptor, glycine receptor and the serotonin type 3 receptor and belong to the same superfamily of ligand gated ion channels. It is believed that the GABAA receptor forms a pentameric structure (Nayeem et al., 1994) and the major isoform consists of α1, β2, and γ2 subunits (McKernan & Whiting, 1996). A stoichiometry of two α, two β and one γ has been proposed (Chang et al., 1996; Tretter et al., 1997). Many classes of drugs interact with this channel, including benzodiazepines, barbiturates and neurosteroids (Sieghart, 1995). The GABAA receptor has been targeted for pharmacological control of anxiety, sleep and epilepsy. Expression of different recombinant subunit combinations results in channels displaying different pharmacological and electrophysiological properties (Sigel et al., 1990; Rabow et al., 1995). In situ hybridization techniques demonstrate that different receptor subunits are expressed in distinct regions of the brain (Wisden et al., 1992; Laurie et al., 1992). Thus, drugs which exhibit a subunit selectivity in vitro may consequently act on GABAA receptor subtypes of specific regions in the brain.
β-carbolines are known to bind with high affinity to the benzodiazepine binding site of the GABAA receptor. One of the best characterized β-carbolines is DMCM, which acts as a negative allosteric modulator by reducing the frequency of channel opening (Rogers et al., 1994). At low concentrations (nM range) DMCM inhibits GABA currents, whereas at high concentrations (μM range) the dose-response curve interestingly reverses and DMCM stimulates submaximal GABA responses (Sigel & Baur, 1988; Sigel et al., 1990; Im et al., 1995). Recently, this potentiation by DMCM in the μM range has been ascribed to a positive allosteric modulation at the loreclezole site (Stevenson et al., 1995). In comparison to the benzodiazepine binding site, the apparent affinity of DMCM for the loreclezole site is three orders of magnitude lower. In vivo DMCM exerts a proconvulsant action. New anticonvulsant drugs with a β-carboline structure acting at both, the benzodiazepine and the loreclezole site, should either be antagonists or positive allosteric modulators at the benzodiazepine site to avoid such a convulsant effect.
Using functional analysis combined with binding studies, we have found that the β-carboline ZK 91085 acts as a positive allosteric modulator at the benzodiazepine site. Furthermore, we observed an additional component of potentiation by ZK 91085 that might be due to an interaction via the loreclezole site. We also show that two point mutations in the β subunit affect this modulation. At least one of the two identified amino acid residues is located at subunit interfaces and is homologous to residues participating in the formation of the agonist site.
Methods
Oocyte expression and electrophysiology
Xenopus laevis oocytes were removed from anaesthetized frogs and manually separated from the ovary by a platinum loop under a light microscope. Prior to defolliculation by a collagenase treatment, the oocytes were injected with 50 nl of 5 mM HEPES-KOH (pH 6.8), containing a mixture of cRNA coding for each of the different subunits. For dual subunit combinations a concentration of 100 nM was used for each transcript. For triple subunit combinations 50 nM of the cRNA coding for α1, 50 nM for β2 and 150 nM for γ2 were injected. A detailed description of the isolation, culturing, injection and defolliculation of the oocytes is given elsewhere (Sigel et al., 1990). The point mutants used were constructed as described previously (Buhr et al., 1997). Numbers of the amino acid residues indicate the position in the corresponding mature rat subunit isoform.
Oocytes were placed on a nylon grid in a 0.4 ml bath and perfused by a gravity-driven system throughout the experiment at 6 ml min−1 with a modified Barth's medium consisting of (in mM) NaCl 90, KCl 1, MgCl2 1 CaCl2 1, HEPES-NaOH 5, pH 7.4. Cells were impaled with two 0.3–1 MΩ resistance microelectrodes containing 3 M KCl and the membrane potential was clamped at −80 mV (two-electrode voltage-clamp). All experiments were performed at room temperature (22–29°C). Allosteric modulation depends on the GABA concentration that is coapplied with the modulating drug. Therefore modulation was quantified at a GABA concentration eliciting alone approximately 5% (EC5) of the maximal current amplitude measured at 10 mM GABA. EC5 was established by determining Imax first and subsequently titrating with GABA until the desired relative response was obtained. A full GABA concentration-response curve was not determined for each of the analysed subunit combinations. But our measurements of the respective EC5 of different subunit combinations are in line with previous observations that dual subunit combinations have an about 7 fold higher apparent affinity of GABA compared to triple combinations (Sigel et al., 1990). Unless indicated otherwise, experiments were carried out using a new oocyte for every compound analysed to prevent contamination. Drugs were bath applied 30–40 s at least until the peak of the response was observed. Between two applications, oocytes were perfused with the modified Barth's medium (see above) for at least 3 min to ensure full recovery from desensitization. Concentration-response curves were fitted to the logistic equation: I=Imax/(1+(EC50/c)), where c is the drug concentration, EC50 is the concentration of drug eliciting half maximal response and Imax is the maximal response. All β-carbolines were a kind gift from Dr Schneider (Schering, Germany), loreclezole was obtained from Janssen and all other compounds were from Sigma or Fluka.
Binding assays
HEK-293 cells (American type of Culture Collection, Rockville, MD, U.S.A., CRL 1573) were maintained in Dulbecco's modified Eagle's medium (GIBCO-BRL, Grand Island, NY, U.S.A.) supplemented with 10% foetal calf serum (JHR Biosciences, Lenexa, KS, U.S.A.), 2 mM glutamine, 50 μM β-mercaptoethanol, 100 units ml−1 penicillin G and 100 μg ml−1 streptomycin in 75-cm2 Petri dishes by using standard cell culture techniques.
3×106 HEK-293 cells were transfected with a total of 21 μg cDNA encoding for the rat α1-, β2-, and γ2-subunits (ratio 1 : 1 : 1), or with 20 μg cDNA encoding for α1- and β2-, or α1- and β1- subunits (ratio 1:1) subcloned individually into pCDM8 expression vectors, using the calcium phosphate precipitation method (Chen & Okayama, 1988). The medium was changed 20 h after transfection and the cells were harvested 48 h after transfection by scraping into phosphate buffered saline. Cells were centrifuged at 12,000×g for 10 min and the cell pellet was homogenized in 50 mM Tris-citrate buffer, pH 7.4 by using an Ultraturrax, followed by three centrifugation (200,000×g for 20 min) resuspension cycles, and were then used for ligand binding studies or were stored at −20°C.
For binding studies, membranes were centrifuged and resuspended in 50 mM Tris-citrate buffer, pH 7.4, at a protein concentration of about 1 mg ml−1 as measured by the BCA-protein assay kit of Pierce Chem. Co. with bovine serum albumin as standard. Membranes (0.5 ml) were then incubated in a total of 1 ml of a solution containing 50 mM Tris-citrate buffer, pH 7.4, 150 mM NaCl and 2 nM of [3H]-flunitrazepam in the absence or presence of 10 μM diazepam or various concentrations of ZK 91085 for 90 min at 4°C (Zezula et al., 1996). For [35S]-TBPS binding studies, membranes were incubated in a total of 1 ml of a solution containing 50 mM Tris-citrate buffer, pH 7.4, 200 mM NaBr and 2 nM [35S]-TBPS in the absence or presence of 10 μM picrotoxin or various concentrations of ZK 91085 for 3 h at room temperature (Zezula et al., 1996). Membranes were then filtered through Whatman GF/B filters. For [3H]-flunitrazepam binding, the filters were rinsed twice with 5 ml of ice-cold 50 mM Tris-citrate buffer. When [35S]-TBPS binding was investigated, the filters were rinsed three times with 3.5 ml of this buffer. Filters were transferred to scintillation vials and subjected to scintillation counting after addition of 3.5 ml Hydrofluor (National Diagnostics, NJ, U.S.A.) scintillation fluid. Non-specific binding determined in the presence of 10 μM diazepam or 10 μM picrotoxin was subtracted from total [3H]-flunitrazepam or [35S]-TBPS binding, respectively, to result in specific binding.
Results
Expression of wild type and mutant GABAA receptors in Xenopus oocytes and in HEK 293 cells
Wild type α1β2γ2, α1β2, α1β1 and mutant α1β2Y62Lγ2, α1Y209Aβ2γ2, α1β2γ2F77L, α1β2Y62L, α1Y209Aβ2, α1F64Lβ2, α1T206Aβ2, α1β2N265S rat recombinant GABAA receptors were functionally expressed in Xenopus laevis oocytes. All of the wild type and mutant receptors produced chloride inward currents of comparable amplitude in response to application of GABA. GABA concentrations required to elicit EC5 indicate no significant change in the apparent GABA affinity for most of the mutated dual and triple combinations relative to wild type receptors. Exceptions were α1F64Lβ2, α1β2Y62L and α1T206Aβ2, which displayed a shift amounting to about 60-, 3- and 3 fold lower affinities, respectively. α1β2γ2, α1β2 and α1β1 rat recombinant GABAA receptors were also expressed in HEK 293 cells for binding studies.
Modulation by ZK 91085 via the benzodiazepine site
Displacement of the ligand [3H]-flunitrazepam by ZK 91085 (Figure 1) indicated an α subunit specific affinity for the benzodiazepine site (data on file, Schering, Germany). Functional studies with ZK 91085 were undertaken to confirm this finding. They demonstrated a stimulation by various concentrations of ZK 91085 of currents elicited by GABA in oocytes expressing α1β2γ2, α2β2γ2 and α5β2γ2 rat recombinant GABAA receptors (representative current traces for the subunit combination α1β2γ2 are shown in Figure 2), but we failed to observe a subunit specificity (not shown). To understand this apparent contradiction, we further characterized the action of ZK 91085 on α1β2γ2 receptors. 10 μM ZK 91085 alone induced currents amounting to less than 0.3% of the maximum current amplitude elicited by GABA, indicating that the observed potentiation is due to allosteric modulation. Dose-response curves of ZK 91085 were bi-phasic (Figure 3) and could be best fitted with a half maximal stimulation of 10±5 nM (mean±s.e.mean of four oocytes from one batch) and the maximal stimulation amounting to 59±3% for the high affinity phase and 1.6±0.7 μM (mean±s.e.mean of four oocytes from one batch) and 694±180% for the lower affinity phase. Solutions containing ZK 91085 at higher concentrations than 10 μM could not be prepared due to solubility problems.
Figure 1.
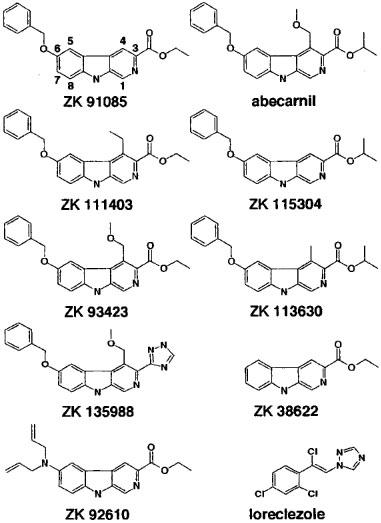
Chemical structure of the β-carbolines used in this study and of the anticonvulsant compound loreclezole.
Figure 2.

Potentiation of GABA responses by ZK 91085 at recombinant rat α1β2γ2, α1β2 and α1β1 GABAA receptors expressed in Xenopus laevis oocytes. Application of GABA alone resulted in approximately 5% of the maximal current amplitude. Increasing concentrations of ZK 91085 were coapplied with GABA. Periods of drug application are indicated by horizontal bars above the current records. Concentrations of ZK 91085 are shown in μM. Three additional experiments for each receptor combination carried out with oocytes from 1–2 batches gave comparable results.
Figure 3.

Concentration response curves for ZK 91085 at recombinant rat α1β2γ2, α1β2 or α1β1 GABAA receptors. Experiments were carried out as described under Figure 2. Values are shown as mean±s.e.mean of four oocytes from 1–2 batches.
We first concentrated on the high affinity phase. Significant stimulation of currents elicited by GABA was first observed at 3 nM ZK 91085. In combination with 1 μM Ro 15-1788 potentiation by 0.1 μM and 0.7 μM ZK 91085 could be reduced by about 70% (Figure 4) and 51% (n=3; not shown), respectively, but the stimulation was not abolished completely. Higher concentrations of ZK 91085 cannot be expected to be inhibited by 1 μM Ro 15-1788 due to the high affinity of ZK 91085 to the benzodiazepine binding site, which is similar to the affinity of Ro 15-1788. Binding studies using [3H]-flunitrazepam as a ligand namely demonstrated that ZK 91085 displaced this ligand with an IC50 value of 1.5 nM (Figure 5). These experiments confirmed a high affinity interaction of ZK 91085 with the [3H]-flunitrazepam binding site.
Figure 4.
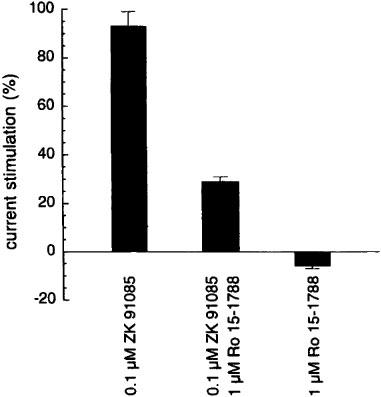
The benzodiazepine antagonist Ro 15-1788 (1 μM) partially inhibits the stimulation by 0.1 μM ZK 91085 at recombinant rat α1β2γ2 GABAA receptors. Results are standardized to control GABA applications that elicited approximately 5% of the maximal current amplitude. Bars show mean±s.e.mean of three oocytes.
Figure 5.

Displacement by ZK 91085 of binding of [3H]-flunitrazepam to membranes from HEK 293 cells transiently transfected with rat recombinant α1β2γ2 GABAA receptors. Bars show mean±s.e.mean of six determinations (two experiments, triplicates each).
Additional modulation by ZK 91085
The lacking α subunit specificity, the bi-phasic stimulation of α1β2γ2 receptors and their only partial blockade by Ro 15-1788 all indicated an interaction of ZK 91085 via an additional site. To separate the two effects, we functionally expressed receptors lacking a γ2 subunit, which is required for benzodiazepine modulation (Pritchett et al., 1989; Sigel et al., 1990; Güenther et al., 1995). Representative current traces obtained with α1β2 are shown in Figure 2. Concentration-response curves for ZK 91085 at receptors consisting of either α1β2γ2 or α1β2 subunits are compared in Figure 3. A potentiation by ZK 91085 was still present at α1β2 receptors but smaller as compared to the triple subunit combination. The EC50 value at α1β2 was >1.8 μM. Additionally, 10 μM ZK 91085 induced an approximately 5 fold shift to the left of the GABA dose-response curve, without affecting the maximal current amplitude (data not shown, two experiments each). As expected, stimulation by 1 μM ZK 91085 amounting to 136±13% (mean±s.e.mean of 22 oocytes from nine batches) proved to be insensitive to 1 μM of the benzodiazepine antagonist Ro 15-1788 at α1β2 receptors (142±11%, mean±s.e.mean of four oocytes from one batch). 10 μM ZK 91085 alone induced currents amounting to maximally 0.1% of the maximum current amplitude elicited by GABA, demonstrating that the observed stimulation is due to a positive allosteric modulation.
At α1β1 receptors, the current stimulation by 10 μM ZK 91085 amounted to only 24% of that at the subunit combination α1β2 (Figure 3). This selectivity of ZK 91085 for receptors containing the β2 isoform as compared to those containing the β1 isoform is reminiscent of the loreclezole action (Wafford et al., 1994; Wingrove et al., 1994). The point mutant β2N265S, that replaces the amino acid residue found in the β2 subunit by the corresponding residue in the β1 subunit, and results in the removal of loreclezole potentiation also abolished stimulation by ZK 91085 (Table 1). We tested also whether the potentiation by ZK 91085 is additive with that of loreclezole. Representative current traces are shown in Figure 6. A saturating concentration of 10 μM loreclezole (Wafford et al., 1994; Stevenson et al., 1995; determined here) stimulated GABA currents by 786±35% (mean±s.e.mean of four oocytes from two batches) at α1β2 receptors. No additional potentiation was observed when loreclezole was coapplied with 10 μM ZK 91085 (767±36%, mean±s.e.mean of four oocytes from two batches).
Table 1.
Effect of point mutations on current stimulation by 1 μM ZK 91085
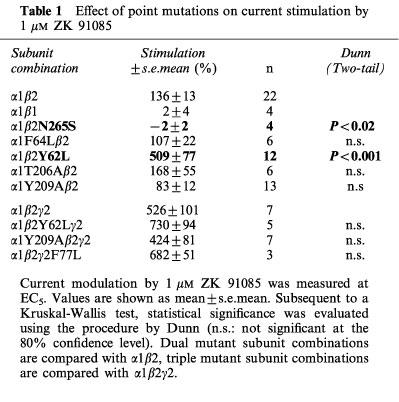
Figure 6.
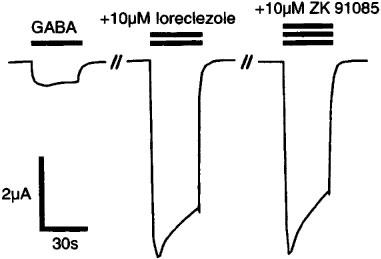
Potentiation of GABA currents by loreclezole and ZK 91085 is not additive at recombinant rat α1β2 GABAA receptors. The concentration of GABA eliciting approximately 5% of the maximal current amplitude was determined first. Ten μM loreclezole strongly stimulated these GABA currents. When 10 μM ZK 91085 was coapplied with 10 μM loreclezole, no additional stimulation was observed. Bars above the current records indicate the periods of drug application. Three additional experiments carried out with oocytes of two separate batches gave comparable results.
Recently, loreclezole was demonstrated to inhibit [35S]-TBPS binding to cells expressing α1β2γ2 and α1β3γ2 receptors, but not to those expressing the α1β1γ2 receptors (Wafford et al., 1994). Similarly, ZK 91085 also decreased [35S]-TBPS binding to membranes from HEK 293 cells transiently transfected with α1β2 or α1β2γ2 receptors (Figure 7). The IC50 value was decreased about 3 fold at α1β2γ2 receptors as compared to α1β2 channels. At the dual subunit combination α1β1, ZK 91085 failed to decrease [35S]-TBPS binding.
Figure 7.
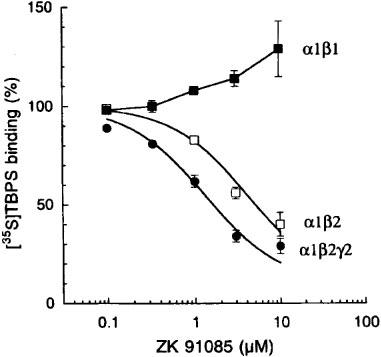
Effect of ZK 91085 on [35S]-TBPS binding to transiently transfected HEK 293 cells expressing recombinant rat α1β2, α1β1 or α1β2γ2 GABAA receptors. Bars show mean±s.e.mean of six determinations (two experiments, triplicates each).
Effects of the drug structure on current stimulation
Stimulation of GABA currents by 10 μM ZK 91085 amounted to 271±52% (mean±s.e.mean of four oocytes from one batch). 10 μM of the β-carboline abecarnil was over 10 fold less potent than ZK 91085 at α1β2 receptors (Figure 8). Both β-carbolines are structurally closely related. Thus, it was interesting to see which structural entities are required for modulation. We therefore measured concentration response curves of the structural intermediates available to us (Figures 1 and 8). Maximal stimulation could not be determined as solutions containing these drugs at higher concentrations than 10 μM could not be prepared due to solubility problems. EC50 values could therefore not be determined. As an exception, ZK 135988 and ZK 38622 were soluble up to 100 μM. However, the concentration response curves of these compounds were not saturated at 100 μM either.
Figure 8.

Comparison of the concentration response curves of ZK 91085, abecarnil and structurally related compounds at α1β2 and α1β2N265S receptors. Results are standardized to control GABA applications that elicited approximately 5% of the maximal current amplitude in the same experiments. Bars show mean±s.e.mean of three oocytes.
Specifically, the role of the substituents at position three, four and six were studied. Introduction of an ethyl group at position four of ZK 91085 provides ZK 111403 and reduced stimulation at 10 μM about 5 fold. ZK 93423 has a methoxymethyl group instead of the ethyl group at the same position. This resulted even in a larger reduction in current stimulation. Interestingly, replacement of the ester moiety in ZK 93423 by a triazole group to give ZK 135988 largely restored current stimulation. Introduction of an additional methyl group into the ester moiety at position three of ZK 91085 to yield ZK 115304 interfered less with the current stimulation, but the reduction still amounting to about 2 fold. When an additional methyl group was introduced in ZK 115304 at position four (providing ZK 113630) the potentiation again was nearly abolished.
Replacement of the benzyloxy group at position six of ZK 91085 by the diallylamino group in ZK 92610 resulted only in a small reduction in current stimulation. Compound ZK 38622, lacking a substituent at this position, showed almost the same potentiation as ZK 91085. However, the last two compounds seem to stimulate currents induced by GABA only at much higher concentrations as ZK 91085.
In addition concentration response curves at the dual subunit combination containing the point mutant β2N265S, that abolishes loreclezole effects, were carried out to determine which of the intermediates act similar to loreclezole (Figure 8). In comparison with the wild type, current stimulation of all intermediates was reduced at the mutated receptor with the exception of abecarnil, ZK 93423 and ZK 135988. These three β-carbolines showed a slightly enhanced potentiation at α1β2N265S receptors.
Effects of point mutations on the modulation by ZK 91085
As mentioned previously the mutation β2N265S abolished the current stimulation at α1β2 by ZK 91085. The potentiation of GABA currents by 1 μM ZK 91085 was compared between wild type and further mutant receptors (Table 1). Representative current records for the mutant receptor α1β2Y62L as well as the wild type α1β2 subunit combination are shown in Figure 9. At dual subunit combinations containing the mutant β2Y62L subunit, the potentiation by ZK 91085 was significantly increased. Mutations α1F64L and α1T206A did not result in a significant change of the potentiation by ZK 91085. Channels containing the mutant α1Y209A subunit behaved variably in different batches of oocytes. In two of four injections, the potentiation by ZK 91085 was significantly decreased about 2 fold, but in the other two experiments, potentiation was similar to the wild type control.
Figure 9.
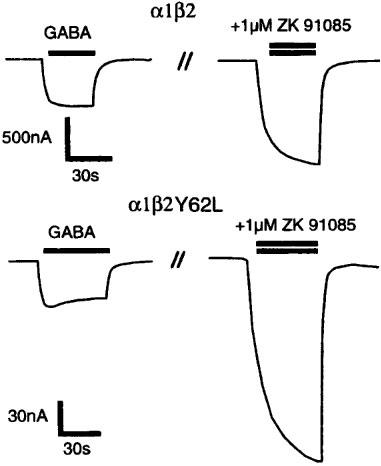
Effect of the β2Y62L point mutation on the current stimulation by 1 μM ZK 91085. After establishment of a GABA concentration eliciting approximately 5% of the maximum current amplitude, this GABA concentration was coapplied with 1 μM ZK 91085. Similar results were obtained in additional experiments for each subunit combination (Table 1). Bars above the current records indicate the periods of drug application.
In triple subunit combinations α1β2γ2 the mutated receptors α1β2Y62Lγ2, α1Y209Aβ2γ2 and α1β2γ2F77L did not result in any significant change as compared to wild type receptors. From the results obtained with the dual subunit combinations, we expected for the mutation β2Y62L a current increase. Intriguingly, such an increase was not found. Possibly a current increase by the mutation is being masked by the large current stimulation by ZK 91085 seen in the triple subunit combination as compared to the dual subunit combination (Figure 3) found prior to mutation.
Figure 10 compares concentration response curves for ZK 91085 in α1β2 and α1β2Y62L channels. As expected, the maximal stimulation by ZK 91085 observed in these experiments was higher for α1β2Y62L as compared to wild type receptors. Due to solubility problems with ZK 91085, the EC50 values could unfortunately not be determined. Current stimulation in α1β2Y62L by 10 μM DMCM (408±44%, mean±s.e.mean of four oocytes from two batches), which stimulates GABA currents via the loreclezole site (Stevenson et al., 1995), was also significantly increased (P<0.025, Mann-Whitney, one-tailed) as compared to the wild type receptor (170±15%, mean±s.e.mean of four oocytes from two batches). Interestingly, the stimulation of the current elicited in α1β2 by 1 μM loreclezole (95±24%, mean±s.e.mean of six oocytes from two batches) was not significantly affected by the mutation in the β subunit (70±28%, mean±s.e.mean of four oocytes from two batches).
Figure 10.

Effect of the mutation β2Y62L on the concentration response curve. GABA eliciting about 5% of the maximal current amplitude was applied either alone or in combination with increasing concentrations of ZK 91085 to wild type α1β2 and mutated α1β2Y62L receptors. Values are shown as mean±s.e.mean of at least four oocytes. Data for the subunit combination α1β2 were taken from Figure 3
Discussion
ZK 91085 stimulates GABAA receptor currents via two different sites
ZK 91085 stimulated currents elicited by GABA in rat recombinant α1β2γ2 channels in a concentration dependent manner in two phases. Action of ZK 91085 via the benzodiazepine site was demonstrated on a functional level by antagonism by Ro 15-1788. About 70% of the current stimulation of 0.1 μM ZK 91085 were inhibited by 1 μM Ro 15-1788 (Figure 4). Binding studies with ZK 91085 also revealed a high affinity for the benzodiazepine site (Figure 5), clearly demonstrating an interaction at this site. In contrast to the classical β-carbolines, which allosterically inhibit GABAA receptor function through this site, ZK 91085 stimulates here. The two-phasic stimulation and the only partial blockade by Ro 15-1788 indicated that ZK 91085 interacts with an additional site on the receptor.
To characterize this second site of action, we analysed the dual subunit combination α1β2. This receptor lacks the γ2 subunit, which is required for benzodiazepine modulation (Pritchett et al., 1989; Sigel et al., 1990; Güenther et al., 1995). ZK 91085 still stimulated GABA responses at the dual subunit combination α1β2, although at higher concentrations only (Figures 2 and 3). The β-carboline DMCM has previously been described to not only interact with the benzodiazepine site but also with the loreclezole binding site (Stevenson et al., 1995). To test whether the present stimulation of α1β2 receptors by the β-carboline ZK 91085 can also be ascribed to an interaction at the loreclezole site, we investigated the modulation at a dual subunit combination containing the β1 isoform, which shows a strongly reduced loreclezole stimulation (Wafford et al., 1994). Indeed, ZK 91085 stimulation at α1β1 receptors was also nearly abolished (Figures 2 and 3) and this selectivity for the β2 over the β1 subunit could be confirmed by displacement studies of [35S]-TBPS by ZK 91085 (Figure 7).
The β-carboline DMCM and the general anaesthetic etomidate display the same β subunit specificity (Stevenson et al., 1995; Hill-Venning et al., 1997). Site-directed mutagenesis studies previously demonstrated, that the β isoform specificity of loreclezole, DMCM and etomidate is determined by a single asparagine residue in β2 and β3 (β2 Asn-265 and β3 Asn-265), the corresponding amino acid in β1 being a serine (Stevenson et al., 1995; Belelli et al., 1997; Wingrove et al., 1994). Mutation β2N265S also determined the observed β subunit specificity for ZK 91085.
Functional competition experiments showed, that GABA currents potentiated by loreclezole are further stimulated by coapplication of either zolpidem, pentobarbitone or the steroid 5α-pregnan-3α-ol-20-one, suggesting separate binding sites (Wafford et al., 1994). In contrast, such experiments with loreclezole and ZK 91085 did not show any additional stimulation by ZK 91085, in line with an interaction of both ligands at the same binding site (Figure 6). In summary, our results are consistent with the hypothesis, that potentiation by ZK 91085 at α1β2γ2 receptors is actually an addition of the modulation at the benzodiazepine and most probably the loreclezole binding site.
Structural features of β-carbolines important for modulation
Due to solubility problems, only a limited concentration range of the β-carboline derivates could be studied (Figure 8). It is not entirely clear from our data whether potency or efficacy is affected by the structural changes. Therefore we compare here in first instance the extent of current stimulation at 10 μM.
The structurally related abecarnil only slightly stimulated GABA currents at α1β2 receptors as compared to ZK 91085 (Figure 8). Thus, it was interesting to determine structural entities required for this modulation. An additional methyl group in the ethyl ester moiety (ZK 115304) reduced current stimulation about 2 fold and additional substituents at position four (ZK 111403, ZK 93423 and ZK 113630) strongly interfered with current stimulation. Interestingly, replacement of the ester by a triazole group in abecarnil (ZK 135988) restored stimulation for the most part in spite of the methoxymethyl substituent present. A triazole group is also present in loreclezole. Whether or not this fact is important for the current stimulation is not known. Replacement of the ester by a triazole group in ZK 91085 may enhance its stimulatory effect.
When the benzyloxy group of ZK 91085 at position six is replaced or even omitted (ZK 92610 and ZK 38622), the extent of potentiation at 10 μM was not much affected. However, stimulation was only evident at high concentrations of the substances, such that the benzyloxy substituent may be important for apparent affinity of a substance.
To see whether the observed potentiation displays similar properties as the current stimulation by loreclezole, we also tested all these compounds for modulation at the channel containing the point mutant β2N265S, which strongly interferes with loreclezole potentiation. All compounds studied showed indeed a behaviour reminiscent of the action of loreclezole with the exception of abecarnil, ZK 93423 and ZK 135988. Interestingly, all three compounds carry a methoxymethyl group at position four. This substituent may abolish the sensitivity of current stimulation to the point mutation β2N265S.
ZK 91085 was tested for an anticonvulsant action in mice (Hollinshead et al., 1990). This β-carboline displayed no effect at 40 mg kg−1. In the same study ZK 91085 revealed an IC50 value of 8.9 nM for the benzodiazepine site, which is in agreement with our results (Figure 5). These authors discussed an ester hydrolysis in vivo as responsible for the lack of activity of ZK 91085 in vivo.
β2Y62L influences ZK 91085 modulation of α1β2
Amino acid residues located at subunit interfaces affect GABA and benzodiazepine action (Sigel & Buhr, 1997). We were interested to see whether residues homologous or identical to these residues affect the modulation of ZK 91085. The dual subunit combinations carrying the point mutation β2Y62L markedly increased potentiation by ZK 91085 (Table 1, Figures 9 and 10). It is not clear from our data, whether the binding site for ZK 91085 is directly affected or whether the conformational changes occurring after binding are altered by the mutation. A general change in channel conformation in the mutant receptor α1β2Y62L appears unlikely, as this mutant showed only a small effect on the apparent affinity of GABA (see results). DMCM stimulation also was markedly increased in α1β2Y62L as compared to the wild type (see results). In contrast, loreclezole action was unaffected in α1β2Y62L receptors (see results). This finding is surprising, as all results discussed so far for ZK 91085 clearly paralleled the behaviour of loreclezole. This apparent contradiction may be explained by partially overlapping binding sites of ZK 91085 (or DMCM) and loreclezole, where β2Y62 is located in the binding pocket for ZK 91085 (or DMCM) but not that for loreclezole (Figure 1).
A second surprise was the absence of any functional effect by the β2Y62L mutation in receptors composed of triple subunit combinations. Stimulation by ZK 91085 is much smaller in dual subunit combinations than in triple subunit combinations. It is conceivable that addition of a γ subunit to the dual subunit combination removes a similar constraint as the mutation β2Y62L in the dual subunit combination. This removal of the constraint leads then to a larger current stimulation by ZK 91085 (Table 1).
The residue identified in this work is located at a position directly homologous to regions forming the GABA and the benzodiazepine site in α1β2γ2 receptors. The residue βY62 is directly homologous to αF64, which has been shown to be important for GABA competitive antagonist affinity (Sigel et al., 1992) and can be photolabelled by the agonist muscimol (Smith & Olsen, 1994). It is also directly homologous to γF77, influencing binding of ligands of the benzodiazepine site (Buhr et al., 1997). In the pentamer model of the α1β2γ2 receptor, the two agonist binding sites and the benzodiazepine site are believed to be located at α/β and α/γ subunit interfaces, respectively (Galzi & Changeux, 1994; Sigel & Buhr, 1997). The tyrosine residue identified here as important for ZK 91085 modulation may be located at the remaining α/β subunit interface of the α1β2γ2 receptor, the β2 subunit contributing residue 62. In case our mutation affects the binding site for ZK 91085, this binding site would have to be located at this remaining subunit interface. Of course we cannot exclude an allosteric effect of the mutation β2Y62L on the binding site.
In conclusion, we show that the β-carboline ZK 91085 acts as a positive allosteric modulator at two sites of the GABAA receptor, namely the benzodiazepine site and most probably the loreclezole site. Two point mutations, β2N265S and β2Y62L, markedly affected stimulation by ZK 91085 via the latter site. A structure activity study with structural intermediates of ZK 91085 and abecarnil identified at least positions three and four of β-carbolines as important for the loreclezole like modulation.
Acknowledgments
We would like to thank Dr Schneider (Schering, Germany) for his kind gift of all β-carbolines. This study was supported by Grants 31-37192.93 and 3100-053599.98/1 from the Swiss National Science Foundation and the European Union Grant BIO4-CT96-0585 (BBW 96.0010).
Abbreviations
- DMCM
methyl 6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- GABA
γ-aminobutyric acid
- GABAA
γ-aminobutyric acid type A receptor
- HEK
human embryonic kidney
- TBPS
t-butylbicyclophosphorothionate
- ZK 38622
ethyl β-carboline-3-carboxylate
- ZK 91085
ethyl 6-benzyloxy-β-carboline-3-carboxylate
- ZK 92610
ethyl 6-diallylamino-β-carboline-3-carboxylate
- ZK 93423
ethyl 6-benzyloxy-4-methoxy-methyl-β-carboline-3-carboxylate
- ZK 111403
ethyl 6-benzyloxy-4-ethyl-β-carboline-3-carboxylate
- ZK 113630
isopropyl 6-benzyloxy-4-methyl-β-carboline-3-carboxylate
- ZK 115304
isopropyl 6-benzyloxy-β-carboline-3-carboxylate
- ZK 135988
6-benzyloxy-4-methoxy-methyl-3-(1H-1,2,4-triazol-3-yl)-β-carboline
References
- BELLELI D., LAMBERT J.J., PETERS J.A., WAFFORD K., WHITING P.J. The interaction of the general anaesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUHR A., BAUR R., SIGEL E. Subtle changes in residue 77 of the γ subunit of α1β2γ2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site and suggest the presence of two sites. J. Biol. Chem. 1997;272:11799–11804. doi: 10.1074/jbc.272.18.11799. [DOI] [PubMed] [Google Scholar]
- BURT D.R., KAMATCHI G.L. GABAA receptor subtypes: from pharmacology to molecular biology. FASEB J. 1991;5:2916–2923. doi: 10.1096/fasebj.5.14.1661244. [DOI] [PubMed] [Google Scholar]
- CHANG Y., WANG R., BAROT S., WEISS D.S. Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN C.A., OKAYAMA H. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;7:632–638. [PubMed] [Google Scholar]
- DAVIES P.A., HANNA M.C., HALES T.G., KIRKNESS E.F. Insensitivity to anaesthetic agents conferred by a class of GABA(A) receptor subunit. Nature. 1997;385:820–823. doi: 10.1038/385820a0. [DOI] [PubMed] [Google Scholar]
- DUNN S.M.J., BATESON A.N., MARTIN I.L. Molecular neurobiology of the GABAA receptor. Int. Rev. Neurobiol. 1994;36:51–96. doi: 10.1016/s0074-7742(08)60303-7. [DOI] [PubMed] [Google Scholar]
- GALZI J.L., CHANGEUX J.P. Neurotransmitter-gated ion channels as unconventional allosteric proteins. Curr. Opin. Struc. Biol. 1994;4:554–565. [Google Scholar]
- GUENTHER U., BENSON J., BENKE D., FRITSCHY J.M., REYES G., KNOFLACH F., CRESTANI F., AGUZZI A., ARIGONI M., LANG Y., BLUETHMANN H., MOEHLER H., LUESCHER B. Benzodiazepine-insensitive mice generated by targeted disruption of the gamma 2 subunit gene of gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7749–7753. doi: 10.1073/pnas.92.17.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEDBLOM E., KIRKNESS E.F. A novel class of GABAA receptor subunit in tissues of the reproductive system. J. Biol. Chem. 1997;272:15346–15350. doi: 10.1074/jbc.272.24.15346. [DOI] [PubMed] [Google Scholar]
- HILL-VENNING C., BELELLI D., PETERS J.A., LAMBERT J.J. Subunit-dependent interaction of the general anaesthetic etomidate with the γ-aminobutyric acid type A receptor. Br. J. Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLLINSHEAD S.P., TRUDELL M.L., SKOLNICK P., COOK J.M. Structural requirements for agonist actions at the benzodiazepine receptor: studies with analogues of 6-(Benzyloxy)-4-(methoxymethyl)-β-carboline-3-carboxylic acid ethyl ester. J. Med. Chem. 1990;33:1062–1069. doi: 10.1021/jm00165a028. [DOI] [PubMed] [Google Scholar]
- IM K.I., IM W.B., CARTER D.B., MCKINLEY D.D. Interaction of β-carboline inverse agonists for the benzodiazepine site with another site on GABAA receptors. Br. J. Pharmacol. 1995;114:1040–1044. doi: 10.1111/j.1476-5381.1995.tb13310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAURIE D.J., SEEBURG P.H., WISDEN W. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. J. Neurosci. 1992;12:1063–1076. doi: 10.1523/JNEUROSCI.12-03-01063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACDONALD R.L., OLSEN R.W. GABAA receptor channels. Annu. Rev. Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- MCKERNAN R.M., WHITING P.J. Which GABAA receptor subtypes really occur in the brain. Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- NAYEEM N., GREEN T.P., MARTIN I.L., BARNARD E.A. Quarternary structure of the native GABAA receptor determined by electron microscope image analysis. J. Neurochem. 1994;62:815–818. doi: 10.1046/j.1471-4159.1994.62020815.x. [DOI] [PubMed] [Google Scholar]
- PRITCHETT D.B., SONTHEIMER H., SHIVERS B.D., YMER S., KETTENMAN H., SCHOEFIELD P.R., SEEBURG P.H. Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature. 1989;338:582–585. doi: 10.1038/338582a0. [DOI] [PubMed] [Google Scholar]
- RABOW L.E., RUSSEK S.J., FARB D.H. From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse. 1995;21:189–274. doi: 10.1002/syn.890210302. [DOI] [PubMed] [Google Scholar]
- ROGERS C.J., TWYMAN R.E., MACDONALD R.L. Benzodiazepine and β-carboline regulation of single GABAA receptor channels of mouse spinal neurones in culture. J. Physiol. 1994;475:69–82. doi: 10.1113/jphysiol.1994.sp020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEGHART W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacol. Rev. 1995;47:181–233. [PubMed] [Google Scholar]
- SIGEL E., BAUR R. Allosteric modulation by benzodiazepine receptor ligands of the GABAA receptor channel expressed in Xenopus oocytes. J. Neurosci. 1988;8:289–295. doi: 10.1523/JNEUROSCI.08-01-00289.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIGEL E., BAUR R., KELLENBERGER S., MALHERBE P. Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO. J. 1992;11:2017–2023. doi: 10.1002/j.1460-2075.1992.tb05258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIGEL E., BAUR R., TRUBE G., MOEHLER H., MALHERBE P. The effect of subunit composition of rat brain GABAA receptors on channel function. Neuron. 1990;5:703–711. doi: 10.1016/0896-6273(90)90224-4. [DOI] [PubMed] [Google Scholar]
- SIGEL E., BUHR A. The benzodiazepine binding site on GABAA receptors. Trends Pharmacol. Sci. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- SMITH G.B., OLSEN R.W. Identification of [3H]muscimol photoaffinity substrate in the bovine γ-aminobutyric acidA receptor α-subunit. J. Biol. Chem. 1994;269:20380–20387. [PubMed] [Google Scholar]
- STEVENSON A., WINGROVE P.B., WHITING P.J., WAFFORD K.A. β-carboline γ-aminobutyric acidA receptor inverse agonists modulate γ-aminobutyric acid via the loreclezole binding site as well as the benzodiazepine site. Mol. Pharmacol. 1995;48:965–969. [PubMed] [Google Scholar]
- TRETTER V., EHYA N., FUCHS K., SIEGHART W. Stoichometry and assembly of a recombinant GABAA receptor subtype. J. Neurosci. 1997;17:2728–2737. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAFFORD K.A., BAIN C.J., QUIRK K., MCKERNAN R.M., WINGROVE P.B., WHITING P.J., KEMP J.A. A novel allosteric modulatory site on the GABAA receptor β subunit. Neuron. 1994;12:775–782. doi: 10.1016/0896-6273(94)90330-1. [DOI] [PubMed] [Google Scholar]
- WINGROVE P.B., WAFFORD K.A., BAIN C.J., WHITING P.J. The modulatory action of loreclezole at the γ-aminobutyric acid type A receptor is determined by a single amino acid in the β2 and β3 subunit. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4569–4573. doi: 10.1073/pnas.91.10.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WISDEN W., LAURIE D.J., MONYER H., SEEBURG P.H. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon and mesencephalon. J. Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZEZULA J., SLANY A., SIEGHART W. Interaction of allosteric ligands with GABAA receptors containing one, two or three different subunits. Eur. J. Pharmacol. 1996;301:207–214. doi: 10.1016/0014-2999(96)00066-0. [DOI] [PubMed] [Google Scholar]