

Abstract
Actin filament (F-actin) depolymerization leads to the use-dependent rundown of N-methyl-D-aspartate (NMDA) receptor activity in rat hippocampal neurones. Depolymerization is promoted by Ca2+ which enters the cells via NMDA receptor channels. The ras homologue (Rho) GTPases (RhoA, Rac1 and Cdc42) promote actin polymerization and thus control the actin cytoskeleton. We have investigated, by means of the whole-cell patch clamp technique, whether the actin fibres which interact with NMDA receptors are controlled by Rho GTPases.
In the presence of intracellular ATP which attenuates rundown, the C3 toxin from Clostridium (C.) botulinum was used to inactivate RhoA. Indeed, it enhanced the use-dependent rundown of NMDA-evoked inward currents to a level similar to that obtained in the absence of ATP.
Lethal toxin from Clostridium sordellii which inactivates Rac1 and Cdc42 lacked this effect.
We suggest that the function of somatodendritic NMDA receptor channels in rat hippocampal neurones can be modulated by RhoA via its action on F-actin.
Keywords: Hippocampal neurones, NMDA receptor channels, rundown, F-actin, C3 toxin, C2 toxin, lethal toxin, RhoA
Introduction
N-methyl-D-aspartate (NMDA) receptors represent a subtype of glutamate-gated ion channels which are highly Ca2+ permeable. The extent of Ca2+-flux through NMDA receptors is tightly controlled. Protein kinases A and C phosphorylate NMDA receptors and increase their activity (Chen & Huang, 1991; Raman et al., 1996). Inhibition of phosphatases such as calcineurin also has such an effect (Tong et al., 1995). Additional modulations of the activity of NMDA receptors which are tightly linked to Ca2+-dependent processes have been described. Thus, in whole-cell recordings, transient Ca2+-influx through NMDA receptor channels promotes their rapid and reversible inactivation (Ca2+-dependent inactivation, CDI; Legendre et al., 1993). This autoregulation of the receptor complex may be due to two different mechanisms. The first may involve calcineurin which is activated by increased levels of intracellular Ca2+ (see above). The second mechanism may involve the anchoring of NMDA receptors within the cell membrane via F-actin (Allison et al., 1998). Thus, it has been suggested that α-actinin-2 which couples NMDA receptors to F-actin is competitively displaced from the NR1 subunit of the receptors by Ca2+/calmodulin (Ehlers et al., 1996; Wyszynski et al., 1997).
During prolonged whole-cell recordings and repetitive stimulation, a use-dependent irreversible inhibition or rundown of NMDA receptor currents can occur (MacDonald et al., 1989; Rosenmund & Westbrook, 1993a) which seems to be due to Ca2+-dependent depolymerization of F-actin (Rosenmund & Westbrook, 1993b). Thus, in cultured rat hippocampal neurones, this Ca2+-dependent rundown of NMDA receptors is attenuated if actin filaments are stabilized with high concentrations of intracellular ATP which act independently of protein phosphorylation (Rosenmund & Westbrook, 1993a,1993b). Moreover, stabilization of F-actin with phalloidin prevented, whereas destruction of these fibers with cytochalasin D or G promoted the rundown of NMDA channels, even in the presence of intracellular ATP. Likewise, the C2 toxin of C. botulinum which destroys actin filaments by ADP-ribosylation and inactivation of G-actin (Reuner et al., 1987) also induced the rundown of NMDA channels (Rosenmund & Westbrook, 1993b). Thus, Ca2+-dependent inactivation and NMDA receptor channel rundown have in common that increased intracellular concentrations of Ca2+ disturb the connection between NMDA receptors and F-actin.
In recent years, the small GTPases of the Rho family RhoA, Rac1 and Cdc42 have been found to regulate actin polymerization, as shown by the formation of stress fibers, lamelli and filopodia. In the areas of focal adhesions and focal contacts, actin fibers of the cytoskeleton connect via protein complexes to the membrane (Hall, 1994, 1998; Nobes & Hall, 1995). Based on this evidence, we have studied in cultured hippocampal neurones whether Rho proteins can modulate the function of NMDA channels. To this end, the rundown of NMDA-evoked currents was measured with the whole-cell patch clamp technique. To elucidate the role of the small GTPases, clostridial toxins were used. Thus, the C3 toxin of C. botulinum inactivates RhoA (Aktories et al., 1989), while the lethal toxin of C. sordellii inactivates Rac and Cdc42 (Genth et al., 1996). Our results suggest that actin fibers which interact with somatodendritic NMDA receptors are under the control of the small GTPase RhoA.
Methods
Mixed neuronal-astroglial cell cultures were prepared from hippocampi of newborn Wistar rats as described previously (Benz et al., 1998). Dissociated cells were seeded on laminin coated coverslips with DMEM/F12 medium supplemented with insulin, selenite and transferrin as well as B27 supplement and incubated at 37°C in a humidified athmosphere of 95% air and 5% CO2. Experiments were performed in whole-cell patch clamp recordings on neurons cultured for 8–10 days. The external (bath) solution contained (mM): NaCl 162, KCl 2.4, CaCl2 1.2, HEPES 10, glycine 0.01 and glucose 11 (320 mosmol, pH 7.3 with NaOH). Tetrodotoxin (0.5 μM), picrotoxine (100 μM) and strychnine (2 μM) were added to inhibit spontaneous activity as well as ligand-gated chloride channels. A nominally Ca2+-free medium was prepared by omitting Ca2+. The pipette (internal) solution contained (mM): CsCl 140, CaCl2 1, MgCl2 2, HEPES 10, and EGTA 11 (pH 7.2, 300 mosmol). In some experiments an ATP-regenerating solution (ARS) (MacDonald et al., 1989) consisting of 4 mM K-ATP, 20 mM potassium creatine phosphate, 50 U/ml phosphocreatine kinase and 6 mM MgCl2 was added to the pipette solution; in this case CsCl was reduced to 95 mM. Patch pipettes, prepared from borosilicate capillaries (Science Products GB 150-8P, Hofheim, Germany), had a resistance of 1.5–3 MΩ. Compensation of cell capacitance (Cm) and series resistance (Rs; 60–80%) was achieved with the inbuilt circuitry of the patch amplifier (List EPC-7, Darmstadt, Germany). At the start of the experiments, these values were 23.6±1.4 pF and 10.9±0.4 MΩ, respectively, in the 78 cells used in this study. When measured at the end of the recording period (25 min later), Cm was 21.9±2.1 pF, and Rs was 9.8±0.9 MΩ (P>0.05) indicating that recording conditions did not change during the time course of experiments. NMDA was applied by means of a fast-flow pressurized superfusion system (Adams and List, DAD-12, New York, U.S.A.) at a concentration of 30 μM. This concentration was near to the EC50 value (concentration of half-maximal activation) of 23 μM (Hill coefficient 1.1) as found in preliminary experiments (n=11; data not shown). All other reagents (ATP, toxins) were introduced into the cells by diffusion from the patch pipette. After whole cell access had been achieved, the system was allowed to equilibrate for at least 20 min. Experiments were carried out at room temperature (20–25°C). Membrane currents were filtered at 3 kHz and digitized at 1 KHz (Cambridge Electronic Devices, Cambridge, U.K.). Cells with leak currents greater than −100 pA at the usual holding potential of −70 mV were excluded to prevent nonspecific calcium entry. To analyse rundown, NMDA (30 μM) was applied every 30 s for 3 s over a period of 25 min. For statistical evaluation of rundown, the peak responses at 0.5, 5, 10, 15 and 25 min or at 25 min only were compared to the response at 0 min. Data were expressed as percentage of control ±s.e.mean. If not mentioned otherwise all drugs and agents used were from Sigma (Deisenhofen; Germany). Clostridial toxins were prepared as described previously (Hofmann et al., 1997) and used at concentrations which caused maximum effects on the actin cytoskeleton in CHO cells.
Results
At the holding potential of −70 mV, NMDA (30 μM) evoked inward currents in all 78 hippocampal neurones tested. The response to the first challenge with the agonist (i.e. 20 min after establishing whole-cell conditions) ranged from −370 to −2090 pA. However, these variations reflected the agonist-sensitivity of the different preparations used during the course of the study. Thus, current amplitudes obtained in neurones from the same batch were always very similar, irrespective of the components of the pipette solution (inset Figure 1d; n=6 each, P>0.05).
Figure 1.
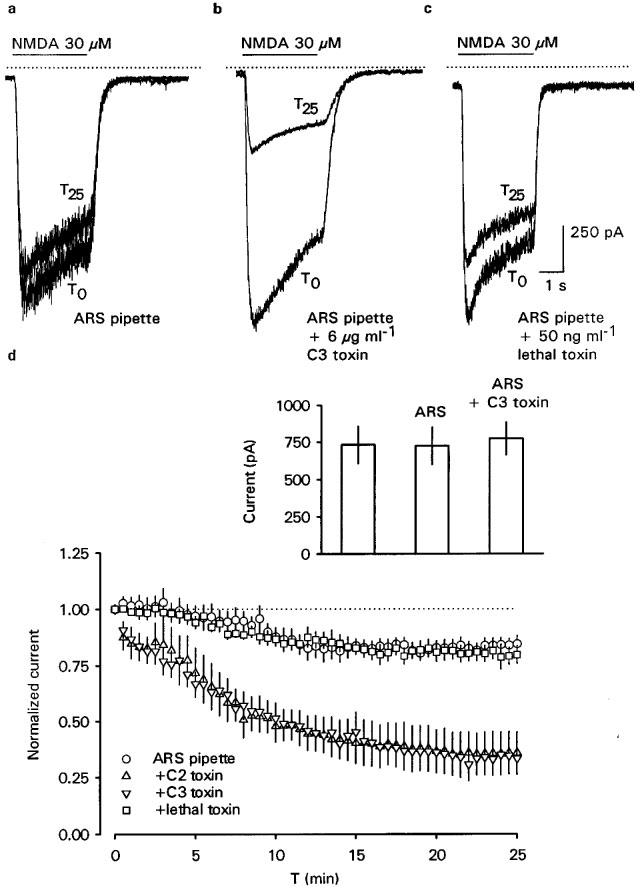
Rundown of NMDA currents can be provoked by the RhoA-inactivating C. botulinum C3 toxin but not by the Rac1- and Cdc42-inactivating lethal toxin of C. sordellii. Inward currents were evoked by 3 s pressure-applications of 30 μM NMDA at 30 s intervals over 25 min. Toxins and an ATP regenerating solution (ARS) were internally microdialyzed via the patch pipette. (a, b and c) Representative inward currents evoked at the holding potential of −70 mV upon the first (T0) and last (T25) application of NMDA are superimposed and were recorded with pipette solutions containing either ARS alone (a), ARS plus C3 toxin (6 μg ml−1; b) or ARS plus lethal toxin (50 ng ml−1; c). The solid, horizontal lines over the current traces indicate the period of NMDA pressure-application, the dotted lines indicate the zero current level. (d) The time course of NMDA current rundown obtained under the same conditions as described in (a). Shown are means±s.e.mean. In addition, the effect of C. botulinum C2 toxin (10 ng ml−1), known to destroy F-actin, on NMDA current rundown is also shown. Currents in the graph were normalized with the first application at T0 (n=6–8). The inset shows NMDA-evoked mean inward current amplitudes obtained when NMDA (30 μM) was applied only once after 20 min of whole-cell dialysis to cells from the same preparation, alternatively using pipette solutions containing no added ARS, ARS or ARS plus C3 toxin (6 μg ml−1; n=6 in each case).
In medium containing 2 mM extracellular Ca2+, repeated stimulation with 30 μM NMDA (every 30 s over 25 min=2/min) caused a rundown of current peak amplitudes (Table 1). When the cells were stimulated only twice, i.e., 20 and 45 min after gaining whole-cell access, current amplitudes were stable (Table 1) indicating that the rundown was use-dependent and not due to the washout of cytosolic components during the recordings. As already reported (Rosenmund & Westbrook, 1993b), rundown was largely prevented in nominally Ca2+-free bath medium (Table 1). Internal microdialysis of the cells with the ATP-regenerating solution (ARS) also prevented the rundown despite the presence of extracellular Ca2+ (Figure 1a and d and Table 1). For the subsequent experiments, we reduced the rundown by including ARS and 2 mM Ca2+ in the bath and pipette solution, respectively. Under these conditions, the inclusion in the pipette solution of C. botulinum C2 toxin (10 ng ml−1), which inhibits actin polymerization and thereby destroys actin filaments (Reuner et al., 1987), promoted a pronounced rundown within 25 min (Figure 1d and Table 1). Thus, intact F-actin was indeed important for the function of NMDA receptor channels (Rosenmund & Westbrook, 1993b).
Table 1.
Effects of clostridial toxins on the rundown of NMDA receptor channels in rat hippocampal neurones

The possible role of small Rho GTPases in rundown was investigated with clostridial toxins. Inactivation of RhoA with intracellularly applied C. botulinum C3 toxin (6 μg ml−1) caused a strong and time-dependent rundown (Figure 1b and d and Table 1). While this rundown was very similar to that found in the absence of ARS, it was significantly greater than the residual rundown in the presence of intracellular ATP (Table 1). Moreover, the extent of C3 toxin-induced rundown was very similar to that caused by C2 toxin (Table 1). This similar inhibition was in agreement with our hypothesis that the actions of both toxins resulted in the destruction of F-actin. In contrast, inhibition of Rac1 and Cdc42 with C. sordellii lethal toxin (50 ng ml−1) did not enhance the residual rundown observed in the presence of ARS (Figure 1c and d and Table 1).
Discussion
Our results unequivocally demonstrate that the GTPase RhoA participates in the regulation of somatodendritic NMDA-receptor function in rat hippocampal neurones. RhoA is a member of a family of small GTPases which act as molecular switches in intracellular signal-transduction pathways and are involved in the regulation of the actin cytoskeleton (Hall, 1998). RhoA can be selectively inactivated by ADP ribosylation induced by C. botulinum C3 toxin (Aktories et al., 1989). In our experiments, this toxin induced a rundown of NMDA receptor channels, whereas inactivation of the GTPases Rac1 and Cdc42 by C. sordellii lethal toxin was without any effect. Of the known effects exerted by RhoA its promotion of F-actin assembly (Paterson et al., 1990; Ridley & Hall, 1992) is most likely involved, since destruction of F-actin with C. botulinum C2 toxin (Reuner et al., 1987) also caused rundown (Rosenmund & Westbrook, 1993b and our study). C3 as well as the C2 toxin caused a rundown to approximately 35% of the NMDA-evoked control current. This reduction seemed to be maximal as was indicated by the time-course which plateaued already after 20 min. In this context, it is noteworthy that, in single channel recordings, uncoupling of the NMDA channels from F-actin by calcium/calmodulin caused a reduction in open probability to 25% (Ehlers et al., 1996). Taken together, these data indicate that even the complete uncoupling of NMDA receptors from F-actin does not cause a complete rundown.
Thus, our data show that RhoA regulates the actin fibres which seem to serve as a scaffold for the anchoring of somatodendritic NMDA receptors in hippocampal neurones. In addition to the Ca2+-dependent regulation of NMDA receptor function (which have been outlined in the Introduction), our finding indicates an additional level of regulation of NMDA receptor activity, namely via the activity of RhoA.
An open question remains. How was RhoA activated in the hippocampal neurons? In this context one should note that members of the Rho-family (RhoA, Rac1 and Cdc42) can be activated by a wide array of external stimuli such as serum, thrombin, growth factors, bradykinin, bombesin and lysophosphatidic acid which all act via membrane bound receptors. Hence, it cannot be excluded, that RhoA activation was due to constituents of our cell culture medium. More interestingly, modulation of RhoA activity via membrane receptors may provide a pathway, by which external stimuli can regulate NMDA receptor function in rat hippocampal neurones.
Acknowledgments
We thank Prof. M. Kohlhardt for important discussions. This work was supported by grants of the Deutsche Forschungsgemeinschaft (SFB 505/B4).
Abbreviations
- ARS
ATP-regenerating-solution
- C.
clostridium
- F-actin
actin filament
- NMDA
N-methyl-D-aspartate
- Rho
ras homologue
References
- AKTORIES K., BRAUN U., RÖSENER S., JUST I., HALL A. The rho gene product expressed in E. coli is a substrate of botulinum ADP-ribosyltransferase C3. Biochem. Biophys. Res. Commun. 1989;158:209–213. doi: 10.1016/s0006-291x(89)80199-8. [DOI] [PubMed] [Google Scholar]
- ALLISON D.W., GELFAND V.I., SPECTOR I., CRAIG A.M. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J. Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENZ I., MEYER D.K., KOHLHARDT M. Properties and the cytoskeletal control of Ca++-independent large conductance K+ channels in neonatal rat hippocampal neurons. J. Membr. Biol. 1998;161:275–286. doi: 10.1007/s002329900334. [DOI] [PubMed] [Google Scholar]
- CHEN L., HUANG L. Y. Sustained potentiation of NMDA receptor-mediated glutamate responses through activation of protein kinase C by a mu opioid receptor. Neuron. 1991;7:319–326. doi: 10.1016/0896-6273(91)90270-a. [DOI] [PubMed] [Google Scholar]
- EHLERS M.D., ZHANG S., BERNHADT J.P., HUGANIR R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- GENTH H., HOFMANN F., SELZER J., REX G., AKTORIES K., JUST I. Difference in protein substrate specificity between hemorrhagic toxin and lethal toxin from Clostridium sordellii. Biochem. Biophys. Res. Commun. 1996;229:370–374. doi: 10.1006/bbrc.1996.1812. [DOI] [PubMed] [Google Scholar]
- HALL A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Ann. Rev. Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- HALL A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- HOFMANN F., BUSCH C., PREPENS U., JUST I., AKTORIES K. Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J. Biol. Chem. 1997;272:11074–11078. doi: 10.1074/jbc.272.17.11074. [DOI] [PubMed] [Google Scholar]
- LEGENDRE P., ROSENMUND C., WESTBROOK G.L. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J. Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACDONALD J.F., MODY I., SALTER M.W. Regulation of N-methyl-D-aspartate receptors revealed by intracellular dialysis of murine neurones in culture. J. Physiol. 1989;414:17–34. doi: 10.1113/jphysiol.1989.sp017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOBES C.D., HALL A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- PATERSON H.F., SELF A.J., GARRETT M.D., JUST I., AKTORIES K., HALL A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J. Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMAN I.M., TONG G., JAHR C.E. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- REUNER K.H., PRESEK P., BOSCHEK C.B., AKTORIES K. Botulinum C2 toxin ADP-ribosylates actin and disorganizes the microfilament network in intact cells. Eur. J. Cell Biol. 1987;43:134–140. [PubMed] [Google Scholar]
- RIDLEY A.J., HALL A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- ROSENMUND C., WESTBROOK G.L. Rundown of N-methyl-D-aspartate channels during whole-cell recording in rat hippocampal neurons: role of Ca2+ and ATP. J. Physiol. (Lond) 1993a;470:705–729. doi: 10.1113/jphysiol.1993.sp019884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENMUND C., WESTBROOK G.L. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993b;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- TONG G., SHEPHERD D., JAHR C.E. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- WYSZYNSKI M., LIN J., RAO A., NIGH E., BEGGS A.H., CRAIG A.M., SHENG M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]